Disorders of the posterior pituitary
PHYSIOLOGY OF OSMOTIC AND VOLUME REGULATION
APPROACH TO THE PATIENT: DIFFERENTIAL DIAGNOSIS OF DISORDERS OF WATER METABOLISM
SPECIFIC DISORDERS OF WATER METABOLISM
Hyponatremia with Normal Regulation of Vasopressin
Hyponatremia with Abnormal Regulation of Vasopressin
Other Causes of True and Factitious Hyponatremia
Hypernatremia with Inappropriate Decreased Vasopressin Secretion or Action
Introduction
Maintenance of the tonicity of extracellular fluids within a very narrow range is crucial for proper cell function.1,2 Extracellular osmolality regulates cell shape as well as intracellular concentrations of ions and other osmolytes. Furthermore, proper extracellular ionic concentrations are necessary for the correct function of ion channels, action potentials, and other modes of intercellular communication. Extracellular fluid tonicity is regulated almost exclusively by the amount of water intake and excretion, whereas extracellular volume is regulated by the level of sodium chloride intake and excretion. In children and adults, normal blood tonicity is maintained over a 10-fold variation in water intake by a coordinated interaction among thirst, vasopressin, and renal systems. Dysfunction in any of these systems can result in abnormal regulation of blood osmolality, which if not properly recognized and treated may cause life-threatening dysfunction in neuronal and other cellular activities.
The posterior pituitary, or the neurohypophysis, secretes the nonapeptide hormones vasopressin and oxytocin. Vasopressin controls water homeostasis, and oxytocin regulates smooth muscle contraction during parturition and lactation. Disorders of vasopressin secretion and action lead to clinically important derangements in water metabolism. This chapter summarizes the physiology of water and volume regulation, presents a symptom-based approach to the differential diagnosis of the diseases of water homeostasis, and provides a review of the pathology and treatment of disorders involving these systems. (See Chapter 13 for a discussion of defects in mineralocorticoid regulation that result in disturbances in volume regulation.)
Physiology of osmotic and volume regulation
Osmotic sensor and effector pathways
Vasopressin and oxytocin biochemistry
Vasopressin and oxytocin are evolutionarily related peptides (paralogs), having arisen from gene duplication of a phylogenetically common molecule approximately 450 million years ago.3,4 Both peptides consist of a 6-amino-acid disulfide ring plus a 3-amino-acid tail, with amidation of the carboxy terminus.5,6 As early as 1895, a potent biologic principle—consisting of vascular pressor activity, “birth quickening,” and milk secretory effects—was recognized in neurohypophyseal extracts.7 The sequences of the individual peptides with pressor and antidiuretic capacity (vasopressin) and oxytocic capacity were determined by du Vigneaud and colleagues during the mid-1950s,6 culminating in the synthesis of each hormone in its biologically active form.8,9 In most mammals, vasopressin and oxytocin differ in only two amino acids—one substitution within the ring and one within the tail structure (Figure 11-1). Exploration of the structure-function relationship of specific amino acids within both vasopressin and oxytocin has allowed characterization of molecules with substantial clinical use. Most notably, by replacement of l-arginine with d-arginine at position 8 of the vasopressin molecule, and amino-terminal deamidation, an analog with enhanced, prolonged antidiuretic to pressor activity was found (desmopressin [desamino-d-arginine vasopressin; dDAVP], see Figure 11-1).10 Desmopressin, with an antidiuretic potency more than double that of its parent vasopressin, is now routinely used in clinical practice.
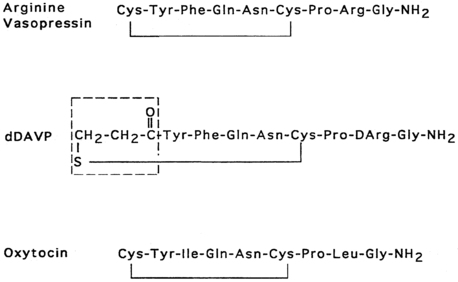
FIGURE 11-1  Structures of vasopressin, dDAVP, and oxytocin. In dDAVP, the de-amidated cysteine is enclosed in the box.
Structures of vasopressin, dDAVP, and oxytocin. In dDAVP, the de-amidated cysteine is enclosed in the box.
The association of vasopressin and oxytocin with specific proteins, the neurophysins, while stored in the neurohypophysis, was apparent as early as 1900.11 Subsequent isolation and characterization of the neurophysins revealed two distinct forms, one type exclusively associated with vasopressin and the other exclusively associated with oxytocin.12,13 Both are single-polypeptide chains of molecular weight 10,000 daltons. Despite extensive biophysical characterization, including crystallography of the oxytocin-neurophysin complex,14,15 the biologic function of the neurophysins remains unclear. Possible roles for the neurophysins include stabilization against degradation during intracellular storage, more efficient packaging within secretory granules, and enhancement of posttranslation processing by the proenzyme convertases.
The common origin of vasopressin and its neurophysin from a single larger precursor was first proposed by Sachs and colleagues,16,17 who showed increased incorporation of 35S cysteine, infused into the canine third ventricle, into vasopressin isolated from the hypothalamus compared with vasopressin isolated from the posterior pituitary. Isolation of the larger precursor from the hypothalamus followed by trypsin digestion produced fragments of size similar to that of vasopressin and its neurophysin, with vasopressin immunoreactivity in the 1000-dalton component.18,19
Since 1990, molecular genetic analyses have furthered the understanding of the synthesis, the processing, and the evolution of the vasopressin and oxytocin preprohormones. Human, mouse, rat, and bovine vasopressin and oxytocin genes each consist of three exons (Figure 11-2).20,21 The first exon encodes the 19-amino-acid signal peptide, followed by vasopressin or oxytocin nonapeptides. This is followed by a 3-amino-acid protease cleavage site leading into the first nine amino acids of neurophysin II (for vasopressin) or neurophysin I (for oxytocin). After interruption of the coding region by an intron, exon 2 continues with neurophysin coding sequences. The third exon completes the sequence of the neurophysin and, for vasopressin only, is followed by coding information for an additional 39-amino-acid glycopeptide (copeptin) whose function is unclear. Preprovasopressin contains 16 cysteines, which likely participate in eight disulfide bridges that determine the tertiary structure of the protein (Figure 11-3). One cysteine pair is present in vasopressin peptide, whereas the rest are in neurophysin.
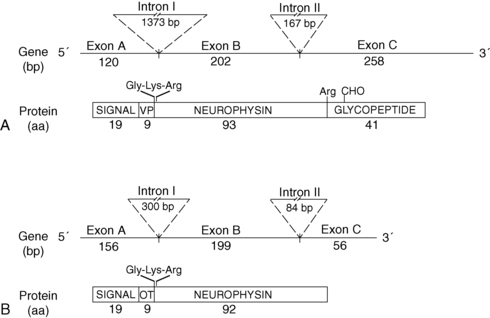
FIGURE 11-2  Structure of the human genes and peptide products of vasopressin (VP) (A) and oxytocin (OT) (B). Shown are the sizes of exons and intron, in nucleotide base pairs (bp) and peptide products in amino acids (aa). Depicted are the amidation-dibasic cleavage signal (Gly-Lys-Arg) at the carboxy terminus of vasopressin and oxytocin and the monobasic cleavage signal at the end of neurophysin. Signal, signal peptide; VP, vasopressin; OT, oxytocin; CHO, carbohydrate.
Structure of the human genes and peptide products of vasopressin (VP) (A) and oxytocin (OT) (B). Shown are the sizes of exons and intron, in nucleotide base pairs (bp) and peptide products in amino acids (aa). Depicted are the amidation-dibasic cleavage signal (Gly-Lys-Arg) at the carboxy terminus of vasopressin and oxytocin and the monobasic cleavage signal at the end of neurophysin. Signal, signal peptide; VP, vasopressin; OT, oxytocin; CHO, carbohydrate.
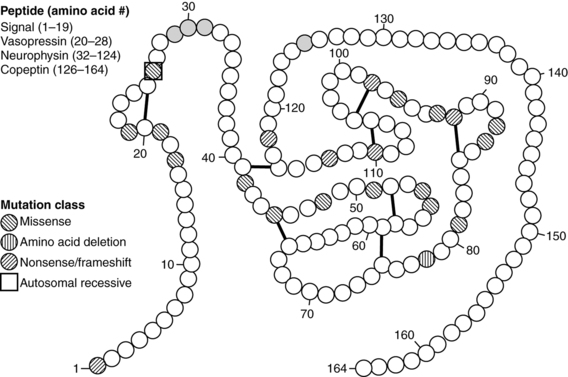
FIGURE 11-3  Structure of preprovasopressin peptide. The 164-amino-qcid preprovasopressin peptide consists of signal peptide, vasopressin, neurophysin, and copeptin. The latter three entities are separated by basic residues (gray), which serve as cleavage sites for proconvertase enzymes. The 16 cysteines are connected by eight putative disulfide bridges. Amino acid mutations are classified as missense, in-frame deletion, or nonsense/frameshift mutations. Most mutations are inherited with an autosomal dominant pattern. The one that has an autosomal recessive pattern is boxed. (Reproduced with permission from Uyeki TM, Barry FL, Rosenthal SM, et al. Successful treatment with hydrochlorothiazide and amiloride in an infant with congenital nephrogenic diabetes insipidus. Pediatr Nephrol 1993; 7:554-6.)
Structure of preprovasopressin peptide. The 164-amino-qcid preprovasopressin peptide consists of signal peptide, vasopressin, neurophysin, and copeptin. The latter three entities are separated by basic residues (gray), which serve as cleavage sites for proconvertase enzymes. The 16 cysteines are connected by eight putative disulfide bridges. Amino acid mutations are classified as missense, in-frame deletion, or nonsense/frameshift mutations. Most mutations are inherited with an autosomal dominant pattern. The one that has an autosomal recessive pattern is boxed. (Reproduced with permission from Uyeki TM, Barry FL, Rosenthal SM, et al. Successful treatment with hydrochlorothiazide and amiloride in an infant with congenital nephrogenic diabetes insipidus. Pediatr Nephrol 1993; 7:554-6.)
In all mammalian species analyzed thus far, oxytocin and vasopressin genes are adjacent in chromosomal location (chromosome 20 in the human22) and linked tail to tail, in opposite transcriptional orientation. In the human, they are separated by 12 kb.22 This likely explains their origin from the ancient duplication of a common ancestral gene.23 Whether this adjacent linkage is of regulatory significance is under investigation.
Vasopressin and oxytocin genes are expressed in the hypothalamic paraventricular and supraoptic nuclei.18,24 The magnocellular components of each of these nuclei are the primary neuronal populations involved in water balance, with vasopressin synthesized in these areas carried by means of axonal transport to the posterior pituitary, its primary site of storage and release into the systemic circulation (Figure 11-4). The bilaterally paired hypothalamic paraventricular and supraoptic nuclei are separated from one another by relatively large distances (approximately 1 cm). Their axons course caudally, converge at the infundibulum, and terminate at different levels within the pituitary stalk and the posterior pituitary gland (see Figure 11-4). Vasopressin is also synthesized in the parvocellular neurons of the paraventricular nucleus, where it has a role in modulating hypothalamic-pituitary-adrenal axis activity. In this site, vasopressin is colocalized in cells that synthesize corticotropin-releasing hormone,25,26 and both are secreted at the median eminence and carried through the portal-hypophyseal capillary system to the anterior pituitary, where together they act as the major regulators of adrenocorticotropic hormone synthesis and release.27 Vasopressin is also present in the hypothalamic suprachiasmatic nucleus, the circadian pacemaker of the body, where its function is unknown.
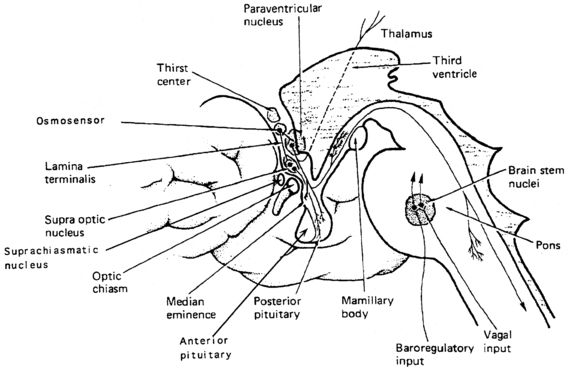
FIGURE 11-4  Vasopressin cells in the hypothalamus. Diagram of vasopressin cell bodies in the supraoptic, paraventricular, and suprachiasmatic hypothalamic nuclei, and axonal termination in the posterior pituitary and median eminence. Because vasopressin axons terminate at different levels in the pituitary stalk and posterior pituitary, the amount of permanent cell loss following neurosurgical insult is determined by the highest level of damage, which will dictate the degree of vasopressin axon transection and retrograde neuronal degeneration. (Modified with permission from reference 507 Baylis, P. H. (1989). Vasopressin and its neurophysin. In L. D. Degroot (Ed.), Endocrinology, (2nd ed.) (p. 213). Philadelphia: WB Saunders.)
Vasopressin cells in the hypothalamus. Diagram of vasopressin cell bodies in the supraoptic, paraventricular, and suprachiasmatic hypothalamic nuclei, and axonal termination in the posterior pituitary and median eminence. Because vasopressin axons terminate at different levels in the pituitary stalk and posterior pituitary, the amount of permanent cell loss following neurosurgical insult is determined by the highest level of damage, which will dictate the degree of vasopressin axon transection and retrograde neuronal degeneration. (Modified with permission from reference 507 Baylis, P. H. (1989). Vasopressin and its neurophysin. In L. D. Degroot (Ed.), Endocrinology, (2nd ed.) (p. 213). Philadelphia: WB Saunders.)
Regulation of vasopressin secretion and thirst
Osmotic regulation.
The work of Verney28 first demonstrated the relationship of increased vasopressin release in response to increasing plasma osmolality, as altered by infusion of sodium chloride or sucrose. At that time, it was postulated that there existed intracranial sensors sensitive to changes in plasma osmolality. Multiple researchers have subsequently confirmed that plasma vasopressin concentration increases in response to increasing plasma tonicity, although the exact nature of the osmosensor has not been defined.29,30 Neurons of the supraoptic nucleus can respond directly to hypertonic stimuli with depolarization and vasopressin secretion,31 but the majority of evidence indicates that osmosensor- and vasopressin-secreting neurons are anatomically distinct.32,33 The osmosensor is likely to reside outside the blood-brain barrier, as implicated by differential vasopressin secretory response to similar changes in plasma osmolality depending on whether the change was induced by salt, sucrose, or urea.28,34 The organ vasculosum of the lamina terminalis (OVLT) and the subfornical organ (SFO), areas of the preoptic hypothalamus outside the blood-brain barrier, are likely sites of osmosensing, because lesions of the OVLT result in impaired vasopressin secretion and hypernatremia.32,33 Also, the site of action of angiotensin II infused intracerebrally or peripherally to produce vasopressin secretion and antidiuresis resides within the OVLT.35–37
The pattern of secretion of vasopressin into blood has been characterized extensively in normal individuals and in those with abnormalities in water homeostasis. Normally, at a serum osmolality of less than 280 mOsm/kg, plasma vasopressin concentration is at or below 1 pg/mL, the lower limit of detection of most radioimmunoassays.29,30 Above 283 mOsm/kg—the normal threshold for vasopressin release—plasma vasopressin concentration increases in proportion to plasma osmolality, up to a maximum concentration of about 20 pg/mL at a blood osmolality of approximately 320 mOsm/kg (Figure 11-5). The osmosensor can detect as little as a 1% change in blood osmolality. Plasma concentrations in excess of 5 pg/mL are also found with nausea, hypotension, hypovolemia, and insulin-induced hypoglycemia, but further increments in urine concentration do not occur, because the peak antidiuretic effect is achieved at 5 pg/mL. The rate of increase of plasma vasopressin concentration, and thus the sensitivity of the osmosensor, exhibits substantial (as much as 10-fold) interindividual variation as plasma osmolality increases.38 The set point for vasopressin secretion varies in a single individual in relation to changes in volume status and hormonal environment (e.g., pregnancy39) or glucocorticoid status.40,41 After the seventh week of gestation, osmotic thresholds for both vasopressin release and thirst are reduced by approximately 10 mOsm/kg (see Figure 11-5), such that normal blood osmolality during pregnancy is approximately 273 mOsm/kg (serum sodium 135 mEq/L).39,42 Similarly, thresholds for vasopressin release and thirst during the luteal phase of the menstrual cycle are approximately 5 mOsm/kg lower than those in the follicular phase.43,44 Human chorionic gonadotropin during pregnancy45 and luteinizing hormone during the second half of the menstrual cycle may contribute to these changes in osmotic thresholds.
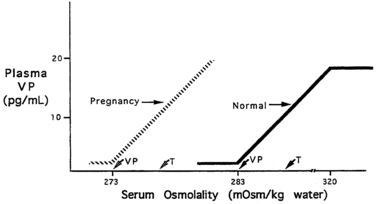
FIGURE 11-5  Osmotic thresholds for vasopressin and thirst. The threshold for vasopressin release is below that for thirst. In nonpregnant persons there is linear increase in vasopressin (VP) release up to a serum osmolality of 320 mOsm/kg, after which no further increase occurs. In pregnancy, there is a decreased threshold for vasopressin release and thirst sensation, with no change in the sensitivity (slope) of the vasopressin-osmolality relationship. Vasopressin secretion in pregnancy presumably also plateaus at some level of hyperosmolality, although this has not been studied. Normal nonpregnant persons indicated by solid line and arrows; pregnant women indicated by dashed line and arrows.
Osmotic thresholds for vasopressin and thirst. The threshold for vasopressin release is below that for thirst. In nonpregnant persons there is linear increase in vasopressin (VP) release up to a serum osmolality of 320 mOsm/kg, after which no further increase occurs. In pregnancy, there is a decreased threshold for vasopressin release and thirst sensation, with no change in the sensitivity (slope) of the vasopressin-osmolality relationship. Vasopressin secretion in pregnancy presumably also plateaus at some level of hyperosmolality, although this has not been studied. Normal nonpregnant persons indicated by solid line and arrows; pregnant women indicated by dashed line and arrows.
The sensation of thirst, a more integrated cortical activity, is determined by other anatomically distinct hypothalamic neurons, with afferents involving the ventromedial nucleus.46 The activation of the thirst mechanism is also probably mediated by angiotensin II.47 Whether the osmosensor for thirst and vasopressin release are the same is not certain, although this is suggested by lesions in the anteroventral region of the third ventricle that abolish both thirst sensation and vasopressin release.48 It makes physiologic sense that the threshold for thirst (293 mOsm/kg) is approximately 10 mOsm/kg higher than that for vasopressin release (see Figure 11-5). Otherwise, during the development of hyperosmolality, the initial activation of thirst and water ingestion would result in polyuria without activation of vasopressin release, causing a persistent diuretic state. Immediately after water ingestion, before a change in blood osmolality or volume, vasopressin concentration falls and thirst ceases.49 The degree of suppression is directly related to the coldness50 and volume51 of the ingested fluid. This effect is probably mediated by chemoreceptors present in the oropharynx, which guard against the rapid overdrinking of fluids after intense thirst during the time before the lowering of blood osmolality.
As noted earlier, water balance is regulated in two ways: (1) vasopressin secretion stimulates water reabsorption by the kidney, thereby reducing future water loss, and (2) thirst stimulates water ingestion, thereby restoring previous water loss. Ideally, these two systems work in parallel to efficiently regulate extracellular fluid tonicity (Figure 11-6); however, each system by itself can maintain plasma osmolality in the near-normal range. For example, in the absence of vasopressin secretion but with free access to water, thirst drives water ingestion up to the 5 to 10 L/m2 of urine output seen with vasopressin deficiency. Conversely, an intact vasopressin secretory system can compensate for some degree of disordered thirst regulation. When both vasopressin secretion and thirst are compromised, however, by either disease or iatrogenic means, there is great risk of the occurrence of life-threatening abnormalities in plasma osmolality.
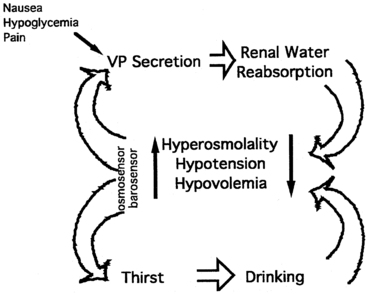
FIGURE 11-6  Regulation of vasopressin secretion and serum osmolality. Hyperosmolality, hypovolemia, or hypotension are sensed by osmosensors, volume sensors, or barosensors, respectively. These stimulate both vasopressin (VP) secretion and thirst. Vasopressin, acting on the kidney, causes increased reabsorption of water (antidiuresis). Thirst causes increased water ingestion. The results of these dual negative feedback loops cause a reduction in hyperosmolality or hypotension/hypovolemia. Additional stimuli for vasopressin secretion include nausea, hypoglycemia, and pain.
Regulation of vasopressin secretion and serum osmolality. Hyperosmolality, hypovolemia, or hypotension are sensed by osmosensors, volume sensors, or barosensors, respectively. These stimulate both vasopressin (VP) secretion and thirst. Vasopressin, acting on the kidney, causes increased reabsorption of water (antidiuresis). Thirst causes increased water ingestion. The results of these dual negative feedback loops cause a reduction in hyperosmolality or hypotension/hypovolemia. Additional stimuli for vasopressin secretion include nausea, hypoglycemia, and pain.
Nonosmotic regulation.
Separate from osmotic regulation, vasopressin has been shown to be secreted in response to alterations in intravascular volume. Afferent baroreceptor pathways arising from the right and left atria and the aortic arch (carotid sinus) are stimulated by increasing intravascular volume and stretch of vessel walls, and they send signals through the vagus and glossopharyngeal nerves, respectively, to the brainstem nucleus tractus solitarius.52,53 Noradrenergic fibers from the nucleus tractus solitarius synapse on the hypothalamic paraventricular nucleus and the supraoptic nucleus and, on stimulation, inhibit vasopressin secretion.54,55 Experimental verification of this pathway has included demonstration of increased vasopressin concentration after interruption of baroreceptor output to the brainstem and decreased plasma vasopressin concentration after mechanical stimulation of baroreceptors, an effect diminished by vagotomy.56,57
The pattern of vasopressin secretion in response to volume as opposed to osmotic stimuli is markedly different (Figure 11-7). Although minor changes in plasma osmolality above 280 mOsm/kg evoke linear increases in plasma vasopressin, substantial alteration in intravascular volume is required for alteration in vasopressin output.58–60 No change in vasopressin secretion is seen until blood volume decreases by approximately 8%. With intravascular volume deficits exceeding 8%, vasopressin concentration increases exponentially. Furthermore, osmotic and hemodynamic stimuli can interact in a mutually synergistic fashion, so that the response to either stimulus may be enhanced by the concomitant presence of the other (see Figure 11-7). When blood volume (or blood pressure61–63) decreases by approximately 25%, vasopressin concentrations are evident of 20- to 30-fold above normal and vastly exceeding those required for maximal antidiuresis. Surprisingly, the use of vasopressin antagonists has suggested that the high concentration of vasopressin observed with hypotension does not contribute to the maintenance of blood pressure in humans.64
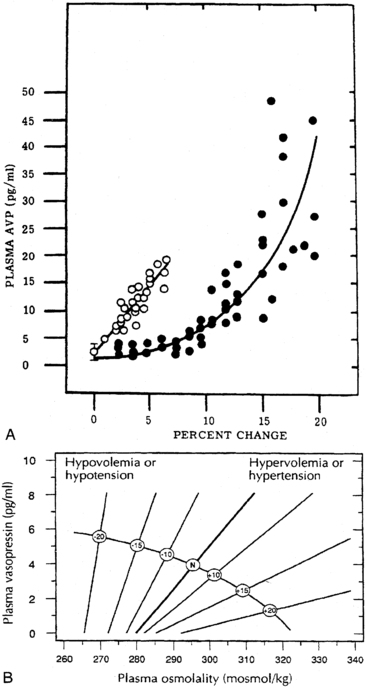
FIGURE 11-7  Relationships between osmotic and nonosmotic stimuli for vasopressin release. A, Relationship of plasma vasopressin (AVP) concentration to the percentage increase in blood osmolality (open circles) or decrease in blood volume (closed circles). B, Alteration of sensitivity of osmotic stimulation of vasopressin secretion by volume or pressure stimuli. (Reproduced with permission from referene (508) Dunn Brennan, T. J., Nelson, A. E., et al. (1973). The role of blood osmolality and volume regulating vasopressin secretion in the rat. J Clin Invest, 52, 3212; reference (509) Robertson, G. L. (1985). The kidney, physiology and pathophysiology Regulation of vasopressin secretion. In D. W. Seldin, & G. Giebisch (Eds.), New York: Raven Press.) (p. 869).
Relationships between osmotic and nonosmotic stimuli for vasopressin release. A, Relationship of plasma vasopressin (AVP) concentration to the percentage increase in blood osmolality (open circles) or decrease in blood volume (closed circles). B, Alteration of sensitivity of osmotic stimulation of vasopressin secretion by volume or pressure stimuli. (Reproduced with permission from referene (508) Dunn Brennan, T. J., Nelson, A. E., et al. (1973). The role of blood osmolality and volume regulating vasopressin secretion in the rat. J Clin Invest, 52, 3212; reference (509) Robertson, G. L. (1985). The kidney, physiology and pathophysiology Regulation of vasopressin secretion. In D. W. Seldin, & G. Giebisch (Eds.), New York: Raven Press.) (p. 869).
Nausea—as evoked by apomorphine,65 motion sickness,66 and vasovagal reactions—is a very potent stimulus for vasopressin secretion. This effect is likely mediated by afferents from the area postrema of the brainstem and may result in vasopressin concentrations two to three orders of magnitude above basal levels. Nicotine is also a strong stimulus for vasopressin release.67 These pathways probably do not involve osmotic or hemodynamic sensor systems, because blockade of the emetic stimulus with dopamine or opioid antagonists does not alter the vasopressin response to hypernatremia or hypovolemia.
Vasopressin secretion is inhibited by glucocorticoids; for this reason, loss of negative regulation of vasopressin secretion occurs in the setting of primary or secondary glucocorticoid insufficiency.68,69 The effects of cortisol loss of both enhancing hypothalamic vasopressin production and directly impairing free water excretion70 are important considerations in the evaluation of the patient with hyponatremia, as is subsequently discussed.
Vasopressin metabolism
Once in the circulation, vasopressin has a half-life of only 5 to 10 minutes, owing to its rapid degradation by a cysteine amino-terminal peptidase called vasopressinase. A synthetic analog of vasopressin, desmopressin, is insensitive to aminoterminal degradation and thus has a much longer half-life of 8 to 24 hours. During pregnancy, the placenta secretes increased amounts of this vasopressinase,71 resulting in a fourfold increase in the metabolic clearance rate of vasopressin.72 Normal women compensate with an increase in vasopressin secretion, but women with preexisting deficits in vasopressin secretion or action,73 or those with increased concentrations of placental vasopressinase associated with liver dysfunction74 or multiple gestations,75 may develop diabetes insipidus in the last trimester, which resolves in the immediate postpartum period.76 As expected, this form of diabetes insipidus responds to treatment with desmopressin but not with vasopressin.77,78
Sites of vasopressin action
Vasopressin receptors.
Vasopressin released from the posterior pituitary and the median eminence affects the function of several tissue types by binding to members of a family of G protein–coupled cell surface receptors, which subsequently transduce ligand binding into alterations of intracellular second messenger pathways.79 Biochemical and cell biologic studies have defined at least three receptor types, designated V1, V2, and V3 (or V1b). The major sites of V1 receptor expression are on vascular smooth muscle80 and hepatocytes,81–84 where receptor activation results in vasoconstriction85,86 and glycogenolysis,87 respectively. The latter activity may be augmented by stimulation of glucagon secretion from the pancreas.87 The V1 receptor on platelets also stimulates platelet aggregation.88 V1 receptor activation mobilizes intracellular calcium stores through phosphatidylinositol hydrolysis.86,89 Despite its initial characterization as a powerful pressor agent, the concentration of vasopressin needed to significantly increase blood pressure is several-fold higher than that required for maximal antidiuresis,90 although substantial vasoconstriction in renal and splanchnic vasculature can occur at physiologic concentrations.91 The cloning of the V1 receptor80,81,83 has greatly elucidated the relationship of the vasopressin (and oxytocin92,93) receptors and, through sensitive in situ hybridization analysis, has further localized V1 expression to the liver and the vasculature of the renal medulla, as well as to many sites within the brain, including the hippocampus, the amygdala, the hypothalamus, and the brainstem.82,84 Compared to their normal counterparts, mice genetically modified to be deficient in the V1 receptor (V1a KO) have been found to manifest insulin resistance, increased hepatic glucose production, decreased hepatic glycogen content, decreased aldosterone secretion despite a lower plasma volume, lower basal blood pressure, a greater degree of lipolysis, and impaired nuclear transport of the renal tubular mineralocorticoid receptor.94 The V3 (or V1b) receptor is present on corticotrophs in the anterior pituitary95 and acts through the phosphatidylinositol pathway96 to increase adrenocorticotropic hormone secretion. Its binding profile for vasopressin analogs resembles more closely that of the V1 than the V2 receptor. The structure of this receptor has been determined in humans by cloning of its complementary DNA.96,97 Its structure is similar to that of the V1 and oxytocin receptors, and it is expressed in the kidney as well as in the pituitary. Mice with deletion of the V1b (V3) receptor gene (V1bKO) have been created and studied.97–99 As expected, they have defective activation of the pituitary-adrenal axis following some acute and chronic stressors. Male V1bKO mice were also found to have decreased aggression and social motivation. 100
Modulation of water balance occurs through the action of vasopressin on V2 receptors located primarily in the renal collecting tubule, along with other sites in the kidney, including the thick ascending limb of the loop of Henle and periglomerular tubules.82,84,101 It is also present on vascular endothelial cells in some systemic vascular beds, where vasopressin stimulates vasodilation,102 possibly through the activation of nitric oxide synthase.103 Vasopressin also stimulates von Willebrand factor, factor VIIIa, and tissue plasminogen activator through V2-mediated actions. For this reason, desmopressin is used to improve the prolonged bleeding times characteristic of uremia, type I von Willebrand disease, and hemophilia.104 The V2 receptor consists of 370 amino acids encoding seven transmembrane domains characteristic of the G protein–coupled receptors.101,105 These transmembrane domains share approximately 60% sequence identity with the V1 receptor but substantially less with other members of this family (Figure 11-8). Unlike the V1 and V3 receptors, the V2 receptor acts through adenylate cyclase to increase intracellular cyclic adenosine monophosphate (AMP) concentration. The human V2 receptor gene is located on the long arm of the X chromosome (Xq28),106,107 at the locus associated with congenital, X-linked vasopressin-resistant diabetes insipidus. Mice in which V2R has been deleted have a similar phenotype. 108
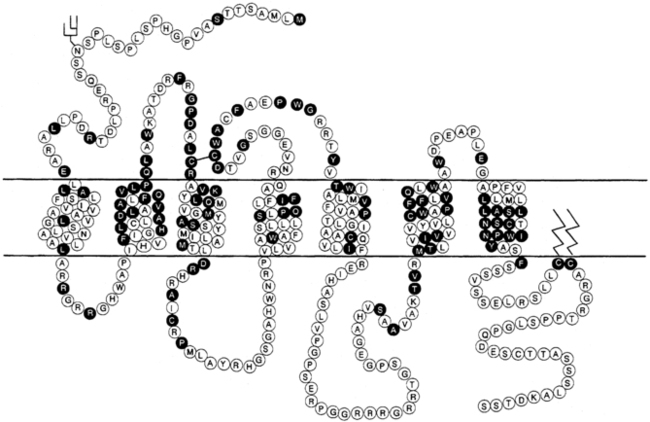
FIGURE 11-8  Structure of the V1 and V2 vasopressin receptors and the oxytocin receptor. Depicted are predicted membrane topology, with the extracellular domain at the top of the figure and amino acids in the one-letter code. Amino acids in open circles encode the V1 receptor, whereas those in black circles are common to all three receptors. (Reproduced with permission from reference (510) Bichet, D. G. (1995). The posterior pituitary. In S. Melmed (Ed.), The pituitary (p. 277). Cambridge: Blackwell Science.)
Structure of the V1 and V2 vasopressin receptors and the oxytocin receptor. Depicted are predicted membrane topology, with the extracellular domain at the top of the figure and amino acids in the one-letter code. Amino acids in open circles encode the V1 receptor, whereas those in black circles are common to all three receptors. (Reproduced with permission from reference (510) Bichet, D. G. (1995). The posterior pituitary. In S. Melmed (Ed.), The pituitary (p. 277). Cambridge: Blackwell Science.)
Renal cascade of vasopressin function.
Vasopressin-induced increases in intracellular cyclic AMP as mediated by the V2 receptor triggers a complex pathway of events resulting in increased permeability of the collecting duct to water and efficient water transit across an otherwise minimally permeable epithelium (Figure 11-9).109 Activation of a cyclic AMP-dependent protein kinase imparts remodeling of cytoskeletal microtubules and microfilaments that culminate in the insertion of aggregates of water channels into the apical membrane.110 These mechanisms may involve a vesicle-associated membrane protein-2–like protein (VAMP-2), which also regulates synaptic vesicle activity in neuronal terminals111 and its associated receptor syntaxin-4.112
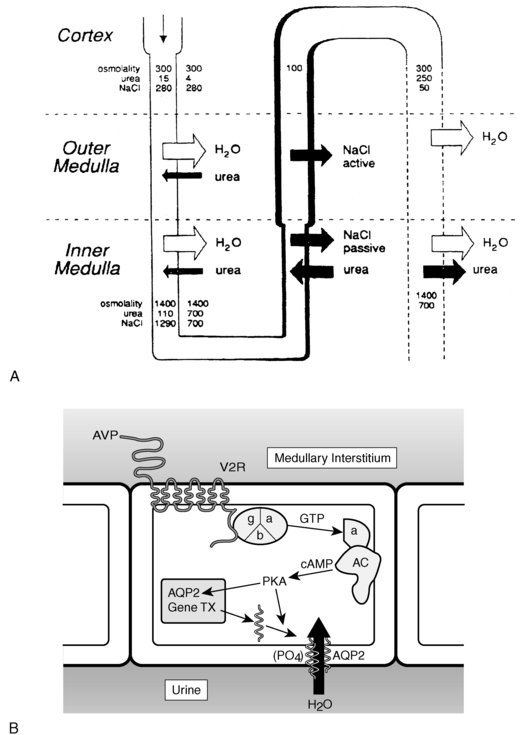
FIGURE 11-9  Vasopressin action in the kidney. A, Solute and water handling in the kidney. B, Action of vasopressin in the collecting duct cell. Vasopressin (AVP) binds to the V2 receptor (V2R), causing the binding of GTP to the stimulatory alpha G protein subunit (α). This activates adenylate cyclase (AC), resulting in an increase in cAMP and activation of protein kinase A (PKA). The catalytic subunit of PKA, via phosphorylation of serine 256 of the water channel, aquaporin-2 (AQP2), causes aggregation of AQP2 homotetramers in membrane vesicles and their fusion with the collecting duct luminal membrane, resulting in an increase in water flow from the urine into the renal medullary interstitium. Demeclocycline, lithium, high calcium, and low potassium interfere with these processes, possibly at the level of cAMP generation and AQP2 synthesis or action. (A from reference (511) Reeves, W. B., & Andreoli, T. E. (1989). Nephrogenic diabetes insipidus. In C. R. Scriver, A. L. The metabolic basis of inherited disease Beaudet, & W. S. Sly (Eds.), (6th ed.) (p. 1985) New York: McGraw-Hill.)
Vasopressin action in the kidney. A, Solute and water handling in the kidney. B, Action of vasopressin in the collecting duct cell. Vasopressin (AVP) binds to the V2 receptor (V2R), causing the binding of GTP to the stimulatory alpha G protein subunit (α). This activates adenylate cyclase (AC), resulting in an increase in cAMP and activation of protein kinase A (PKA). The catalytic subunit of PKA, via phosphorylation of serine 256 of the water channel, aquaporin-2 (AQP2), causes aggregation of AQP2 homotetramers in membrane vesicles and their fusion with the collecting duct luminal membrane, resulting in an increase in water flow from the urine into the renal medullary interstitium. Demeclocycline, lithium, high calcium, and low potassium interfere with these processes, possibly at the level of cAMP generation and AQP2 synthesis or action. (A from reference (511) Reeves, W. B., & Andreoli, T. E. (1989). Nephrogenic diabetes insipidus. In C. R. Scriver, A. L. The metabolic basis of inherited disease Beaudet, & W. S. Sly (Eds.), (6th ed.) (p. 1985) New York: McGraw-Hill.)
Insertion of the water channels causes an up to 100-fold increase in water permeability of the apical membrane, allowing water movement along its osmotic gradient into the hypertonic inner medullary interstitium from the tubule lumen and excretion of a concentrated urine (see Figure 11-9). The molecular analysis of the water channels has revealed a family of related proteins, designated aquaporins, that differ in their sites of expression and pattern of regulation.113 Each protein consists of a single polypeptide chain with six membrane-spanning domains (Figure 11-10). Although functional as monomers, they are believed to form homotetramers in the plasma membrane.109
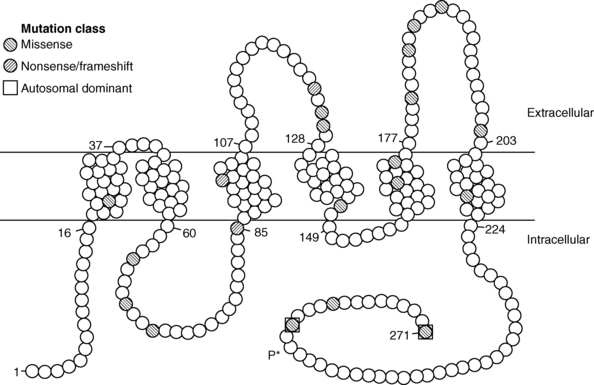
FIGURE 11-10  Structure of the aquaporin-2 protein inserted into the luminal membrane of the distal tubule. The 271-amino-acid protein consists of five transmembrane domains, four intracellular domains, and three extracellular domains. Amino acid mutations are denoted by filled circles. Most mutations are transmitted with an autosomal recessive pattern; the two dominant mutations are bounded by squares. Vasopressin-dependent, protein kinase A–mediated phosphorylation of serine at amino acid 256 (P*) is noted. (Reproduced with permission from Uyeki TM, Barry FL, Rosenthal SM, et al. Successful treatment with hydrochlorothiazide and amiloride in an infant with congenital nephrogenic diabetes insipidus. Pediatr Nephrol 1993; 7:554-6.)
Structure of the aquaporin-2 protein inserted into the luminal membrane of the distal tubule. The 271-amino-acid protein consists of five transmembrane domains, four intracellular domains, and three extracellular domains. Amino acid mutations are denoted by filled circles. Most mutations are transmitted with an autosomal recessive pattern; the two dominant mutations are bounded by squares. Vasopressin-dependent, protein kinase A–mediated phosphorylation of serine at amino acid 256 (P*) is noted. (Reproduced with permission from Uyeki TM, Barry FL, Rosenthal SM, et al. Successful treatment with hydrochlorothiazide and amiloride in an infant with congenital nephrogenic diabetes insipidus. Pediatr Nephrol 1993; 7:554-6.)
Aquaporin-2 is expressed mostly within the kidney,114 primarily within the collecting duct.115 It is also expressed in the vas deferens, at least in the rat, although it is not regulated by vasopressin in this location.116 Studies with immunoelectron microscopy have demonstrated large amounts of aquaporin-2 in the apical plasma membrane and subapical vesicles of the collecting duct, consistent with the “membrane shuttling” model of water channel aggregate insertion into the apical membrane after vasopressin stimulation.116 Studies analyzing the mechanism by which aquaporin-2 traffics to the apical plasma membrane have demonstrated that vasopressin-induced, protein kinase A–mediated serine phosphorylation at amino acid 256 is required for its exocytosis,117 a process also requiring a heterotrimeric G protein of the Gi family.118 In response to water restriction or desmopressin infusion in humans, the content of urinary aquaporin-2 in both soluble and membrane-bound forms has been found to increase.119 Mice with targeted deletion of the aquaporin-2 gene have been made.120 As expected, they have nephrogenic diabetes insipidus that is unresponsive to treatment with vasopressin.
In addition to aquaporin-2, different aquaporins appear to be involved in other aspects of renal water handling. In contrast to the apical localization of aquaporin-2, aquaporin-3 and aquaporin-4 are expressed on the basolateral membrane of the collecting duct epithelium. They appear to be involved in the flow of water and urea from the inside of the collecting duct cell into the extracellular renal medullary space. Mice made genetically deficient in aquaporin-4 demonstrate a mild urinary concentrating defect,121 whereas those with deficiency of aquaporin-3 alone, or together with aquaporin-4, demonstrate more severely impaired urinary concentrating ability.122 Mice made genetically deficient in aquaporin-1 demonstrate a urinary concentrating defect caused by decreased water permeability in the proximal tubule.123
Volume sensor and effector pathways
Renin-angiotensin-aldosterone system
In contrast to the vasopressin system, the classic, or peripheral, renin-angiotensin system primarily affects maintenance of intravascular volume as opposed to plasma tonicity. In addition to the well-established endocrine regulatory system, several local renin-angiotensin systems have emerged, with both autocrine and paracrine effects in their tissue of synthesis, whose regulation is independent of the classic system. Finally, brain and pituitary angiotensin systems involved in blood pressure, autonomic function, and fluid balance have been characterized with extensive interaction with the vasopressin system, and vasopressin has been found to play a role in the normal action of aldosterone on the renal tubular epithelium.124
Endocrine renin-angiotensin-aldosterone system
Anatomy and biochemistry.
Renin, which is synthesized by the renal juxtaglomerular apparatus, is a proteolytic enzyme that catalyzes the cleavage of angiotensinogen, synthesized by hepatocytes, into the decapeptide angiotensin I.125,126 Angiotensin I possesses no intrinsic vasoreactive or mineralocorticoid secretagogue activity but is efficiently cleaved by angiotensin-converting enzyme in the lungs, as well as other peripheral sites, to generate the octapeptide angiotensin II. Angiotensin II is further metabolized to the heptapeptide angiotensin III by removal of one aminoterminal amino acid. Angiotensin II possesses greater vasopressor activity and is present in approximately a fourfold greater amount than angiotensin III. Angiotensins II and III possess equivalent mineralocorticoid secretory activity on the adrenal glomerulosa cells.
Angiotensin II and III act through cell surface receptors (AT1) on the adrenal glomerulosa cells to activate the phospholipase C/protein kinase C pathway.127–131 This activation results in increased production of pregnenolone from cholesterol by side-chain cleavage enzyme (20,22-desmolase) and of aldosterone from corticosterone by the glomerulosa-specific corticosterone methyloxidase I and II activities (18-hydroxylation and dehydrogenation, respectively).132–135 A distinct receptor subtype for angiotensin II, the AT2 receptor, is not G protein coupled and is of unclear physiologic significance in the periphery.136–138 Aldosterone, the primary and most potent endogenous mineralocorticoid released by the zona glomerulosa, acts on target tissues expressing the nuclear mineralocorticoid (or type I glucocorticoid) receptor to promote sodium absorption and potassium excretion. For control of intravascular volume, the primary target of action of aldosterone is the distal nephron. Here, aldosterone increases synthesis of apical membrane sodium channels, mitochondrial enzymes involved in adenosine triphosphate production, and components of Na+,K+ adenosine triphosphatase to increase sodium reabsorption and potassium excretion.139
Regulation of secretion.
Decreased intravascular volume as sensed by the renal juxtaglomerular apparatus results in release of renin.125,140 Increased plasma renin activity then allows increased conversion of angiotensinogen to angiotensin I, which in turn is converted peripherally to angiotensins II and III. Increased angiotensin II activity causes vasoconstriction and blood pressure elevation, whereas both angiotensins II and III stimulate aldosterone release from the zona glomerulosa and subsequent salt and water retention and potassium excretion by the distal tubule of the kidney. Conversely, expanded intravascular volume causes decreased renin output and less sodium and water resorption in the kidney, serving to decrease intravascular volume and restore homeostasis.
Changes in vascular volume are not the only regulators of the renin-angiotensin-aldosterone system. Serum potassium concentration directly modulates aldosterone release by the adrenal glomerulosa by its effects on plasma membrane potential and activation of voltage-gated calcium channels.126,141 By membrane depolarization, increased serum potassium leads to increased aldosterone synthesis, which promotes renal potassium excretion, whereas low serum potassium reduces aldosterone synthesis and decreases urinary potassium losses. Pituitary adrenocorticotropin hormone and vasopressin act through their respective receptors on the glomerulosa cells to increase acute aldosterone secretion. These effects are of short duration because long-term chronic infusions do not chronically elevate aldosterone concentrations. Direct inhibitors of aldosterone secretion, and thus promoters of natriuresis, include atrial natriuretic peptide (ANP),142,143 somatostatin,144–146 and dopamine.147,148
Local renin-angiotensin systems
Anatomy and biochemistry.
In addition to the well-defined endocrine circuit, the components of the renin-angiotensin system have been found in a wide variety of tissues, including brain, pituitary, arterial wall, heart, ovary, kidney, and adrenal where paracrine and autocrine regulatory functions149–153 have been postulated, undergoing regulation independent of the systemic counterpart. From the standpoint of regulation of water and volume homeostasis, the brain renin-angiotensin system merits further description.154 It has long been known that peripherally synthesized angiotensin II could increase blood pressure by effects on the brain outside the blood-brain barrier, at sites such as the OVLT, SFO, area postrema, and median eminence as revealed by ligand-binding studies.35,154–156 Since the early 2000s, it has become clear that the complete system for generation of angiotensin II is present within the brain. Angiotensinogen has been localized to astrocytes by both immunohistochemical peptide localization and in situ hybridization analysis of messenger RNA.157 In contrast, renin has been found in high concentration in nerve terminals, with enhanced release on nerve depolarization.158 Angiotensin-converting enzyme has been found within vascular, choroid plexus, and neuronal components of the central nervous system,159–161 most notably the SFO and many hypothalamic nuclei, sites of endogenous angiotensin II receptor expression, primarily of the AT1 subtype, as well as sites not expressing the angiotensin II receptor such as the basal ganglia. The primary effector molecule, angiotensin II, has been localized specifically to neurons and subcellularly to synaptic vesicles.162 Two of the most significant sites include the circumventricular organs and the paraventricular nucleus of the hypothalamus. Within the paraventricular nucleus, angiotensin II immunoreactivity colocalizes with magnocellular vasopressin, whereas its receptors are within the parvocellular region of the paraventricular nucleus.163
Regulation of secretion.
The forebrain angiotensin II pathway, of which the paraventricular nucleus is one component, and circumventricular organ angiotensin II pathway are important control centers for maintenance of osmotic and volume homeostasis.164 Increased concentration of peripheral angiotensin II, as would be expected in intravascular volume depletion, stimulates drinking behavior.35 This action of peripheral angiotensin II can be abolished by destruction of the OVLT or SFO, regions whose destruction has long been recognized as causing adipsia.36 Further effects of central angiotensin II action include augmentation of sodium appetite and stimulation of vasopressin release, all serving, as with peripheral angiotensin II, to restore intravascular volume and maintain blood pressure.35 The signal of hypovolemia is transduced through the vagal nerve from volume sensors to the brainstem and the region of the nucleus tractus solitarius. Efferents from these brainstem centers project to the median preoptic nucleus and paraventricular nucleus, as does the forebrain angiotensin II pathway, where drinking and pressor effects as well as vasopressin release are elicited.165–170
Separate pathways for vasopressin release mediate the response to either peripheral angiotensin II or purely osmotic stimulation of the osmosensors.171 The release of vasopressin in response to osmotic stimulation is not increased by peripheral angiotensin II, and pure osmotic stimulation does not increase salt appetite. Central angiotensin II, in contrast, may function as a transmitter in the osmosensing circuit, leading to vasopressin release.172




