Disorders of mineral homeostasis in children and adolescents
Disorders of calcium, magnesium, and phosphate metabolism and of bone formation, accrual, and maintenance during the first two decades of life result from suboptimal ingestion, absorption, or retention of constituent nutrients, abnormal vitamin D metabolism or bioactivity, disorders of parathyroid hormone (PTH) synthesis, secretion, or action, and intrinsic aberrations in cartilage and bone cells. The origins of these illnesses may be intrinsic due to pathologic variations in the genes controlling these processes or to acquired insults (Table 18-1). For an integrated overview of calcium, mineral, and skeletal homeostasis and changes in this system through the life cycle, see Chapter 8.
Hypocalcemia
Hypocalcemia in the neonate and infant
Clinical manifestations of hypocalcemia occurring in the neonate—defined as values of total calcium < 7.5 to 8 mg/dL and ionized calcium (Ca2+) < 4.4 mg/dL (1.1 mmol/L) in newborns with birth weights > 1500 g and < 7 mg/dL and Ca2+ < 3.6 mg/dL (0.9 mmol/L) in newborns with birth weights < 1500 g—are principally those of neuromuscular hyperexcitability: irritability, hyperacusis, jitteriness, tremulousness, facial spasms, tetany, laryngospasm, and focal or generalized seizures.1,2 Nonspecific symptoms such as apnea, tachycardia, cyanosis, emesis, and feeding problems may also occur. Causes of neonatal hypocalcemia may be considered in relation to the age of onset (before or after 72 hours of life) (Table 18-2).
Early neonatal hypocalcemia
In the absence of hypoproteinemia, hypocalcemia occurring within the first 72 hours after birth is considered “early neonatal hypocalcemia.” It occurs most commonly in prematurely delivered or small-for-gestational-age, low-birth-weight, or asphyxiated neonates, or in those born to women with gestational or permanent forms of diabetes mellitus, and it is the consequence of impaired PTH secretion and delayed renal tubular phosphaturic response to PTH characteristic of the neonate, prolonged secretion of calcitonin, or hypomagnesemia.2 Total calcium and Ca2+ concentrations decline more rapidly from high intrauterine values to lower nadir levels in preterm than in term neonates. In low-birth-weight (LBW) neonates, hypocalcemia may be further attributed to the rapid accretion of skeletal calcium in the presence of relative resistance to the calcium absorptive and reabsorptive effects of calcitriol on the intestinal tract and bone, respectively. Offspring of severely vitamin D–deficient mothers may become hypocalcemic shortly after birth. Hypocalcemia develops in approximately one third of asphyxiated newborns who are products of complicated and compromised deliveries. In these infants, increased phosphate load due to cellular injury, reduced calcium intake, and hypercalcitonemia are important pathogenetic factors in the development of hypocalcemia. Neonates with severe infections (sepsis) or other critical illness are often hypocalcemic.
Fifty percent of infants of mothers with diabetes mellitus develop early neonatal hypocalcemia; the incidence may be reduced by strict maternal glycemic control.3 Its causes are multifactorial and include reduced placental transfer of calcium due to substantial maternal urinary excretion of calcium and magnesium, decreased neonatal secretion of PTH, hypercalcitonemia, hypomagnesemia (occurring in 40% of offspring of diabetic women), and limited intake and impaired absorption of ingested calcium.4 Maternal hypercalcemia due to unsuspected hyperparathyroidism leads to increased transfer of calcium to the fetus and still further increase in the in utero serum calcium concentrations that suppress fetal PTH synthesis and release and stimulate calcitonin secretion, aberrations in homeostatic mechanisms that persist postpartum and may result in hypocalcemic tetany/seizures in offspring. Suppression of PTH secretion may persist for several months and be undetected until symptomatic hypocalcemia develops after weaning of the infant from breast milk to higher phosphate containing cow milk formula. Maternal ingestion of large quantities of calcium carbonate in antacids has also led to neonatal hypocalcemia.5
Hypocalcemia has occurred in neonates with hyperbilirubinemia undergoing exchange transfusion due to complexing of calcium by citrate used to store blood and avoid clotting, and in those exposed to phototherapy. Neonates with acute rotavirus infection and severe diarrhea may present with hypocalcemic seizures. Aminoglycoside antibiotics (e.g., gentamycin) increase urinary excretion of calcium and magnesium, thereby facilitating the development of neonatal hypocalcemia. Compounds that complex with and sequester calcium such as citrate (present in transfused blood), phosphates (that alter the calcium × phosphate product), and fatty acids (given as caloric supplements) lower Ca2+ levels. Bicarbonate administered to correct acidosis increases calcium binding to albumin and thus lowers Ca2+ values. Hypocalcemia may also occur in hyperventilated infants with severe respiratory alkalosis as well as in those with other causes of metabolic alkalosis. Phytates in soy milk bind calcium and phosphate and interfere with their absorption. Neonates and infants with malignant osteopetrosis type II and impaired osteoclastogenesis may present with either early or late neonatal hypocalcemia.6–8
Late neonatal hypocalcemia
Hypocalcemia developing after 72 hours of postnatal age may be due to an increased intake of phosphate, hypomagnesemia, hypoparathyroidism, or vitamin D deficiency (see Tables 18-2A and B). Often neonatal hypocalcemia develops after 3 days of age in offspring born in the late winter-early spring of the year to multiparous women of lower socioeconomic status with an inadequate intake of vitamin D or exposure to sunlight. Ingestion of excessive phosphate in evaporated milk or modified cow milk formulas forms poorly soluble calcium salts, limiting the intestinal absorption of calcium while raising serum phosphate values. Premature introduction of fiber-containing cereals into the infant’s diet also decreases calcium absorption. Affected infants may have an associated defect in renal phosphate excretion or coexisting vitamin D deficiency. Hyperphosphatemia and hypocalcemia may initially suggest hypoparathyroidism, but serum PTH concentrations are high in infants with excessive phosphate loading in response to a reciprocal reduction in serum calcium values. Newborns and infants with chronic renal insufficiency due to renal hypoplasia or obstructive nephropathies often are hypocalcemic and hyperphosphatemic with elevated serum PTH levels as well, but they are also azotemic. Hypomagnesemia leads to impaired secretion of PTH and decreased peripheral responsiveness to PTH and may be transient or related to congenital abnormalities of intestinal absorption or renal tubular reabsorption of magnesium.3 Hypermagnesemia may occasionally be associated with neonatal hypocalcemia.
TABLE 18-2B
Gene Mutations Associated with Hypoparathyroidism
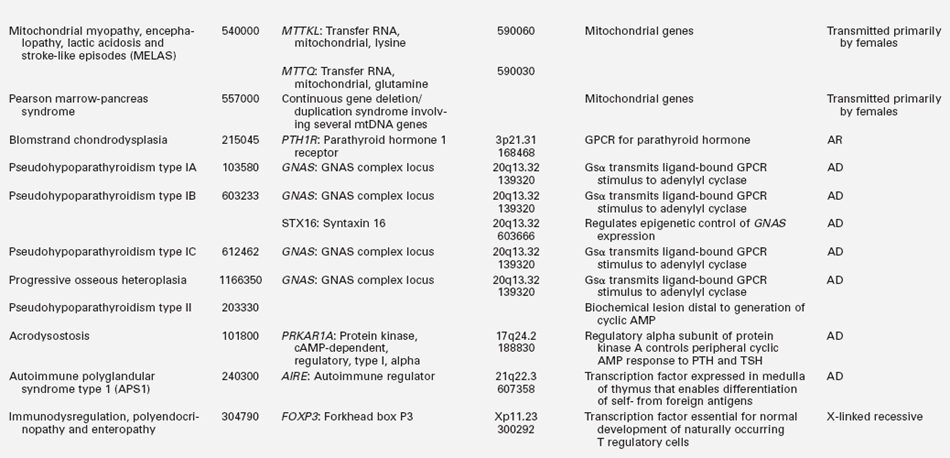
GPCR, G protein–coupled receptor; AD, autosomal dominant; AR, autosomal recessive.
Hypocalcemia due to fetal/neonatal deficiency of vitamin D occurs in offspring of mothers deprived of vitamin D (either for cultural or socioeconomic reasons); impaired renal 25-hydroxyvitamin D-1α hydroxylase activity or loss-of-function mutations of the vitamin D receptor also lead to hypocalcemia (and hypophosphatemia). Hypovitaminosis D may develop in the breastfed infant of a vegetarian mother who shields herself from sunlight and ingests a diet low in vitamin D. Marginal deficiency of vitamin D in neonates and infants is much more common than has been recognized heretofore.9–12 “Late-late” neonatal hypocalcemia occurs in premature infants with osteopenia at 3 to 4 months of age in whom the intake of calcium, phosphate, and vitamin D has been marginal; it is perhaps due to an avid deposition of available calcium into bone.3 Hypocalcemia due to vitamin D deficiency may develop rather acutely and in the absence of clinical or radiographic signs of rickets in the older infant or young child who has ingested an elimination diet low in vitamin D because of severe allergies or who has been maintained indoors with limited exposure to sunlight.
Hypoparathyroidism
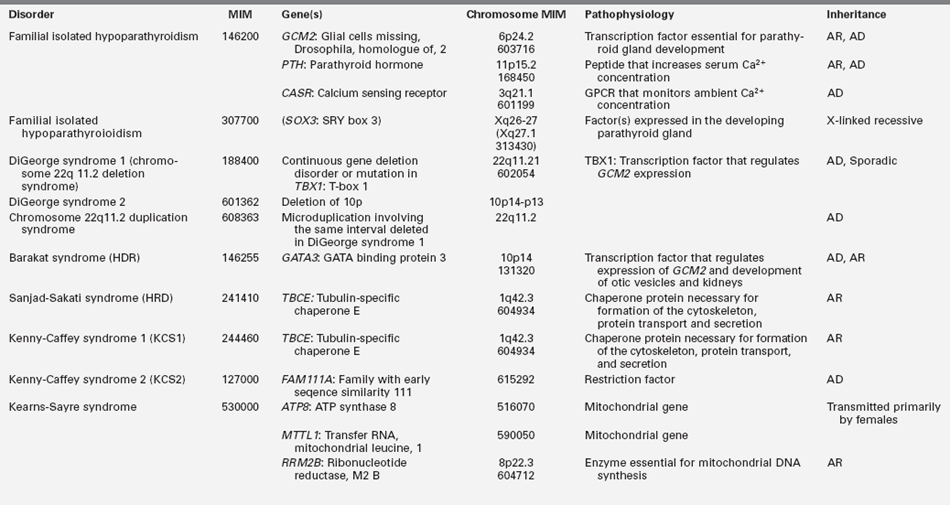
Hypoparathyroidism presenting in infancy is often transient and related to delayed developmental maturation of parathyroid gland function; it frequently resolves within the first several weeks of life (see Tables 18-2A and B). When prolonged, hypoparathyroidism is often due to an error in the embryogenesis of the parathyroid glands or in the synthesis or secretion of parathyroid hormone (PTH) or to peripheral unresponsiveness to PTH. Familial isolated congenital hypoparathyroidism may be transmitted as an autosomal recessive, autosomal dominant, or X-linked recessive trait that has been associated with loss-of-function mutations of GCM2 and PTH and possibly SOX3, respectively, and gain-of-function mutations in CASR. GCM2 is a gene with five exons that encodes a 506-amino-acid DNA-binding transcription factor whose expression is restricted to the parathyroid glands. “Knockout” of Gcm2 in mice leads to agenesis of the parathyroid glands and hypoparathyroidism. Intragenic deletions or homozygous missense mutations in exons 2, 3, and 5 of GCM2 result in hypoparathyroidism in humans.13–17 Mutations in GCM2 exons 2 and 3 (encoding DNA binding and transactivation domain 1) lead to impaired protein synthesis and stability and autosomal recessive transmission of congenital hypoparathyroidism, whereas those in exon 5 (encoding transactivation domain 2) lead to mutations with a dominant negative effect and autosomal dominant transmission of this disorder.17 (Expression of GCM2 is dependent on normal transcriptional function of GATA3, the gene mutated in patients with the Barakat syndrome of hypoparathyroidism-deafness-renal dysplasia [HDR].18) Inactivating mutations in PTH that interfere with the processing of preproPTH to active PTH result in functional hypoparathyroidism that may be transmitted as an autosomal dominant or recessive characteristic.15 Depending on the specificity of the immunoassay for PTH, serum levels of PTH may be low, normal, or even high in these patients. Hypoparathyroidism due to heterozygous activating mutations in CASR have been found in hypocalcemic neonates.19 X-linked hypoparathyroidism is associated with agenesis of the parathyroid glands; the disorder has been mapped to Xq26-q27 and may involve a deletion-insertion mutation that adversely affects the position of SOX3.20 Because Sox3 is expressed in embryonic mouse parathyroid glands, it is likely an important transcription factor for normal embryologic development of these structures.
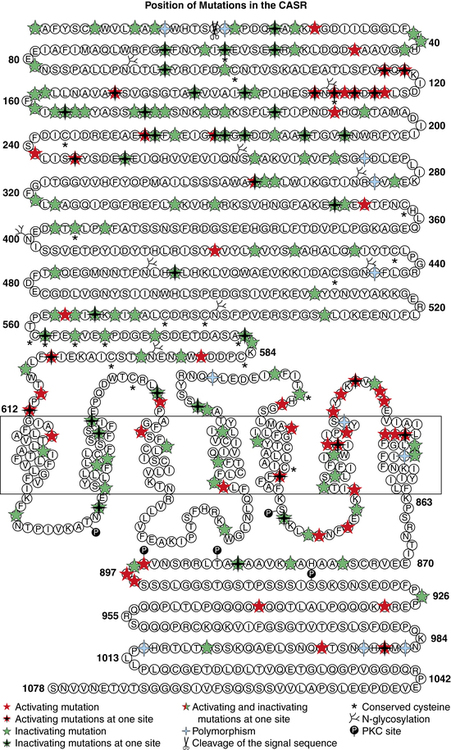
Hypercalciuric hypocalcemia is an autosomal dominantly transmitted form of hypoparathyroidism that is due to gain-of-function mutations in CASR that result in enhanced “sensitivity” to Ca2+. A lowered “set point” enables PTH secretion to be suppressed and renal tubular reabsorption of calcium to be depressed by extremely low concentrations of Ca2+; this disorder may present in the newborn period.19,21 Mutations may be scattered throughout the gene but occur predominantly in the extracellular domain of the CaSR. Activating mutations (Cys141Trp) of CASR may also inhibit function of the renal outer medullary potassium channel (KCNJ1, MIM 600359), leading to a Bartter-like syndrome with hypokalemic metabolic alkalosis, hyperreninemia, and hyperaldosteronism as well as hypercalciuric hypocalcemia; the paired metabolic defects are partially responsive to treatment with hydrochlorothiazide and low doses of calcitriol.22 Children with hypercalciuric hypocalcemia due to gain-of-function mutations in CASR are very sensitive to even low doses of calcitriol that can lead to even more marked hypercalciuria and to nephrocalcinosis. Thus, management of these patients has been quite difficult. Administration of recombinant human PTH1–34 to a 14-month-old hypocalcemic male infant with a de novo nonsense mutation in CASR (Leu727Gln) for 17 months partially restored calcium homeostasis with increased but still subnormal serum levels of calcium, whereas urinary excretion of calcium decreased into the normal range.19 During treatment the child was clinically asymptomatic, did not develop nephrocalcinosis, and tolerated the drug well. Long-term treatment of children with activating mutations of CASR may be feasible.23,24
The most common form of dysgenesis of the parathyroid glands in neonates and infants is that associated with the DiGeorge syndrome (DGS), a disorder that occurs with a frequency of 1:4000 births and is present in approximately 70% of children with isolated hypoparathyroidism.25,26 The DGS is a neurocristopathy that results from the disturbed migration of cervical neural crest cells and consequent maldevelopment of tissues of neural crest origin derived from the third and fourth pharyngeal pouches and first to fifth branchial arches. It is usually associated with microdeletions of chromosome region 22qll.2 (del22q11.2: DGCR), a segment on which more than 35 genes are sited and thus considered a contiguous gene syndrome (a disorder caused by deletion of several adjacent genes that when individually mutated may result in a distinctive clinical feature but when collectively lost lead to a group of apparently unrelated clinical findings). The chromosome 22q11.2 microdeletion is contained within regions of low copy number repeats, and it is this characteristic that results in an unequal segmental exchange between the paired 22nd chromosomes during meiosis.26 DGS may occur sporadically or be transmitted as an autosomal dominant trait. Although the clinical severity and phenotype of patients with this chromosomal anomaly are variable, characteristically subjects with DGS have the triad of hypocalcemia due to hypoplasia of the parathyroid glands often manifest in the neonatal period but which may not be detected until the child reaches an older age, defective T-lymphocyte function and impaired cell-mediated immunity due to partial or complete absence of thymic differentiation leading to increased frequency of viral and fungal infections, and conotruncal defects of the heart or aortic arch (tetralogy of Fallot, ventricular septal defect, interrupted or right aortic arch, truncus arteriosus, vascular ring).3,26,27 Immune deficiency is common as are velopharyngeal insufficiency, cleft palate, and developmental challenges.26 Other features of the DGS include gastrointestinal malformations (esophageal atresia, anal atresia), dysplastic kidneys, cervical spine instability, impaired vision, and ocular malformations.
In addition to the DGS, deletion of chromosome 22q11.2 has been associated with the conotruncal anomaly face and velocardiofacial syndromes. Collectively, these syndromes are associated with similar facial features (ocular hypertelorism, lateral displacement of inner canthi, short palpebral fissures, swollen eyelids, dysmorphic “segmented” nose, small mouth, low-set ears with abnormally folded pinnae, short philtrum, micrognathia, malar hypoplasia, velopharyngeal insufficiency with/without cleft palate), olfactory dysfunction, short stature, nonverbal learning disabilities, and various psychological maladies.25,28 Takao velocardiofacial syndrome (included in MIM 188440) consists primarily of the typical cardiac defects described earlier that may also be associated with hypocalcemia; Shprintzen velocardiofacial syndrome (MIM 192430) is characterized by craniofacial and palatal defects and cardiac anomalies; Cayler cardiofacial syndrome (MIM 125520) is associated with partial unilateral facial paresis due to hypoplasia of the depressor anguli oris muscle and anomalies of the heart and aorta. These syndromes have been grouped as the CATCH-22 syndromes of Cardiac defects, Abnormal face, Thymic hypoplasia, Cleft palate, Hypocalcemia. In addition to the anomalies and clinical findings listed, a litany of additional abnormalities may be seen in patients with del22q11.2.29 Disparate manifestations of these syndromes may be observed in different members of the same family, indicating the variable clinical expressions that accompany deletions of chromosome 22q11.2.30
A two-megabase microdeletion at chromosome 22q11.2 leads to loss of several contiguous genes within this region including HIRA (histone cell cycle regulation, MIM 600237), a transcription factor that is expressed in developing heart and upper body neural crest elements and is necessary for normal cardiac development. Also within this region is TBX1 encoding T-box 1, a transcription factor with a highly conserved DNA binding sequence (the T-box) that is essential for organogenesis and pattern formation and is expressed in the pharyngeal arches and pouches. Experimental disruption of Tbx1 impairs development of the pharyngeal arch arterial vasculature, whereas introduction of null mutations in Tbx1 results in anomalies of the cardiac outflow track and hypoplasia of the thymus and parathyroid glands. The transcription factor encoded by TBX1 is part of a network of gene products (including those encoded by ISL1, SHH, FOXA2, FOXC2) that controls development of the parathyroid glands and thymus by regulating expression of GATA3, GCM2 and PAX9.26 Evaluation of patients with clinical characteristics of the CATCH-22 syndromes but intact chromosome 22q11.2 has revealed heterozygous loss-of-function mutations (Phe148Tyr, Gly310Ser) in TBX1 in patients with the DGS and Shprintzen velocardiofacial syndrome.25,31 Thus, haploinsufficiency of TBX1 alone can account for the cardiac defects, abnormal face, thymic and parathyroid hypoplasia, and velopharyngeal insufficiency with cleft palate but not for the developmental delay characteristic of CATCH-22. Another candidate gene for the DGS sited at chromosome 22q11.2 is UFD1L (ubiquitin fusion degradation 1-like, MIM 601754), whose product is important for the posttranslational processing of proteins or their degradation by interaction with the ubiquitin fusion protein. Experimentally, the DGS and related disorders have also been linked to genes encoding endothelin-1, vascular endothelial growth factor, and fibroblast growth factor-8 (a target gene for TBX1), and to genes within the DGCR at chromosome 22q11.2—CRKL—(MIM 602007) and DGCR6 (MIM 601279). In the mouse hypomorphic for Fgf8, there are cardiovascular, craniofacial, parathyroid, and thymic defects—an experimental phenocopy of the human del22q11.2 syndrome.32 Fgf8 functions through stimulation of transcription of Crkl—its product is an adaptor protein that transduces intracellular signals from several tyrosine kinase receptors, one of which is the receptor for Fgf8; interestingly, Fgf8 interacts with Tbx1 as well. The DGS has also been associated with microdeletions of chromosomes 10p13 (DGS2), 18q21.33, and 4q21.2-q25, indicative of the cascade of genes likely involved in the generation of this phenotype. The presence of the DGS should be considered when fetal ultrasonography reveals an interrupted aortic arch or truncus arteriosus and may be confirmed by appropriate studies (microarray, fluorescent in situ hybridization [FISH]) on samples of chorionic villi or amniotic fluid.
Hypocalcemia has been observed in some subjects with microduplication of chromosome 22q11.2, a copy number variant; clinical characteristics of individuals with this genetic anomaly vary from those who are entirely normal to patients with multiple congenital anomalies, severe developmental delay, autism, and schizophrenia.33,34 The duplication chromosome 22q11.2 syndrome appears to be transmitted as an autosomal-dominant characteristic whose expression is modified by other factors. The pathophysiology of hypocalcemia in affected subjects is uncertain. In one family in which the proband had the DGS associated with del22q11.2, the normal father had the same anomaly on one of his 22nd chromosomes and dup22q11.2 on his other 22nd chromosome; paternal quantitative expression of the genes located on chromosome 22q11.2 was normal, indicating that the adverse effects of the 22q11.2 deletion were compensated by the 22q11.2 duplication.35
Several other syndromes exhibit multisystem involvement and hypoparathyroidism. The Barakat or HDR syndrome of Hypoparathyroidism, sensorineural Deafness, and Renal disease (dysplasia, steroid-resistant nephrosis with progressive renal failure) has been attributed to haploinsufficiency of GATA3, a zinc-finger transcription factor that regulates expression of GCM2 and thus is critical for the embryonic development of the parathyroid glands as well as for the kidneys, otic vesicles, and thymus.15,18,36 The parathyroid glands of these children are dysgenetic—hypoplastic or absent. Hypocalcemia may be present in the newborn period or unrecognized until later childhood. Malformations of the female genital tract (didelphic uterus, septate vagina) may also occur in patients with HDR.37 Heterozygous deletions, insertions, missense, and nonsense mutations in GATA3 have been identified in patients and families with HDR.38,39 Patients with an isolated loss of GATA3 function do not have other features common to patients with larger terminal deletions of chromosome 10p such as growth and developmental retardation, dysmorphic facial features, or congenital heart disease.
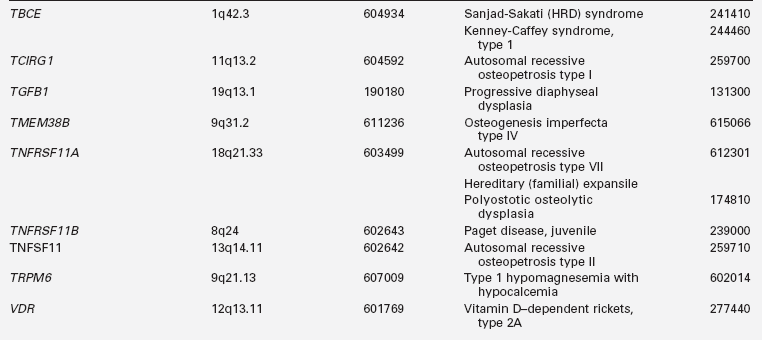
Biallelic mutations in the gene encoding tubulin-specific chaperone E (TBCE) have been identified in the Sanjad-Sakati syndrome of Hypoparathyroidism-mental Retardation-Dysmorphism (HRD) and the Kenny-Caffey syndrome type 1 (KCS1) of hypocalcemia, cortical thickening, medullary stenosis, dysmorphic face, and growth retardation. Children with HRD are short, developmentally delayed, and seizure prone; they have medullary stenosis and other skeletal anomalies; they are microcephalic with faces characterized by deeply recessed eyes or microphthalmia, depressed nasal bridge, beaked nose, long philtrum, thin upper vermillion border, micrognathia, and large earlobes. The HRD syndrome often presents in infancy with symptomatic hypocalcemia associated with low serum concentrations of PTH and normal phosphaturic responses to exogenous PTH. The cardiovascular system of these patients is intact, but as infants they are susceptible to life-threatening pneumococcal infections.40 Neonates with the closely related disorder KCS1 are often severely hypocalcemic early in the neonatal period. As children they are short, with craniofacial anomalies due to the absence of diploic space in the skull, osteosclerosis, and thickening of the cortices of the long bones with narrowing of the medullary compartment, normal or mildly delayed development, and increased susceptible to recurrent bacterial infections. TBCE is essential for formation of microtubules—cytosolic structures composed of heterodimeric α- and β-tubulin subunits that form the cytoskeleton, mitotic apparatus, cilia, and other cellular components; this chaperonin assists in the correct folding of α- and β-tubulin subunits and the formation of α-β-tubulin heterodimers. The α- and β-tubulin subunits and TBCE are necessary for normal embryogenesis of the parathyroid glands. Mutations in TBCE result in lowered microtubule formation and consequently in a decrease in subcellular components such as the Golgi apparatus and endosomal compartments required for normal intracellular movement of proteins. Interestingly, the identical mutation in TBCE—a homozygous 12 bp deletion in exon 2—may result in either the HRD or KCS1 phenotype in a specific family.41 A child with autosomal recessive HRD syndrome and intact TBCE has been identified, suggesting that this disorder is likely to be genetically heterogeneous.42 In KCS type 2, the phenotype is similar to that of KCS1 but transmission is as an autosomal dominant trait attributable to heterozygous loss-of-function mutations in FAM111A (MIM 615292).
Neonates with loss-of-function mutations in PTH1R, the gene encoding the GPCR for PTH and PTHrP, are functionally hypoparathyroid despite elevated serum concentrations of PTH.43 Because of subresponsiveness to PTHrP in utero, fetal bone formation is abnormal, resulting in Blomstrand chondrodysplasia—an osteochondrodystrophy characterized by short extremities and advanced skeletal and dental maturation—abnormalities detectable in utero by fetal ultrasonography. Histologically the proliferative zone of the cartilage growth plate is narrowed with relatively few resting and proliferating chondrocytes, whereas the hypertrophic zone is composed of irregular columns of chondrocytes. Transmitted as an autosomal recessive trait, its clinical characteristics include polyhydramnios, hydrops fetalis, short-limbed dwarfism, facial anomalies, aberrant tooth development, aplasia of the nipples and breasts, hypoplastic lungs, and preductal aortic coarctation. Although Blomstrand osteochondrodysplasia is usually lethal, skeletal malformations may be more (type I) or less severe (type II). Mutations in PTH1R that result in complete absence of normal protein (e.g., Arg104Ter) are designated type I, whereas mutations that permit some PTH1R synthesis (Pro132Leu) result in type II Blomstrand osteochondrodysplasia.44 Heterozygous inactivating mutations of PTH1R (Glu155Ter) result in autosomal dominant nonsyndromic failure of tooth eruption (MIM 125350).45 Eiken chondrodysplasia (MIM 60002) is also due to biallelic loss-of-function mutations in PTH1R but is clinically and radiographically distinct from Blomstrand osteochondrodysplasia as affected subjects have mild growth retardation, markedly delayed ossification, multiple epiphyseal dysplasia, and persistent islands of cartilage in the pelvis.46 The PTH1R mutations in patients with Eiken skeletal dysplasia occur in the carboxyl terminal (e.g., Arg485Ter).
GNAS encodes the stimulatory alpha subunit (Gαs) of G-proteins coupled to seven transmembrane receptors. After activation by its GPCR, the guanosine diphosphate (GDP) moiety on Gαs is replaced by guanosine triphosphate (GTP), Gαs dissociates from its βγ companion subunit complex, and stimulates membrane-bound adenylyl cyclase activity generating cyclic adenosine monophosphate (AMP) and activating protein kinase A, which phosphorylates serine and tyrosine residues of further signal transduction proteins. GNAS is composed of 13 exons; it has four major transcripts that arise through splicing of unique first exons onto shared exons 2 to 13 (Figure 18-1).47 The four transcripts of GNAS encode (1) the Gαs transcript is a protein that stimulates adenylyl cyclase and generates cyclic AMP; Gαs is expressed by both maternal and paternal alleles in most tissues; in the proximal renal tubule, thyroid, gonads, and adenohypophysis, however, only the maternal allele of GNAS is expressed; (2) XLαs yields a Gαs isoform that is specifically expressed in neuroendocrine and nerve tissues and is identical to Gαs except that it has a very long amino terminal sequence of amino acids; it is expressed only by the paternally acquired allele; (3) the NESP55 (neuroendocrine secretory protein-55) transcript is a chromogranin-like protein that is expressed in neuroendocrine tissues but only by the maternal GNAS allele; (4) the alternative first exon A/B (exon 1A) transcript is expressed ubiquitously but only by the paternal GNAS allele and is not translated. The promoters of XLαs, NESP55, and exon 1A transcripts lie within the 5′ differentially methylated region (DMR) of GNAS. Methylation of the promoter usually silences expression of that GNAS transcript.
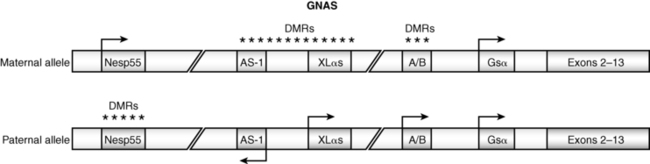
FIGURE 18-1  Schematic view of the GNAS gene complex. Three alternate exons 1—NESP55, XLαs, and AS-1—are upstream of the start codon. DMR refers to the differentially methylated region. The pattern of methylation and hence the expression of the alternate exons is dependent on the parent of origin of the allele and the specific tissue in which GNAS is expressed. (From Shore, E. M., & Kaplan, F. S. (2010). Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol, 6, 518–527.)
Schematic view of the GNAS gene complex. Three alternate exons 1—NESP55, XLαs, and AS-1—are upstream of the start codon. DMR refers to the differentially methylated region. The pattern of methylation and hence the expression of the alternate exons is dependent on the parent of origin of the allele and the specific tissue in which GNAS is expressed. (From Shore, E. M., & Kaplan, F. S. (2010). Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol, 6, 518–527.)
Loss-of-function mutations in GNAS or epigenetic aberrations that lead to failure of expression of the Gαs transcript of GNAS result in pseudohypoparathyroidism (PHP). PHP type IA is manifested by resistance to protein hormones that signal though GPCRs; resistance to PTH results in hypocalcemia, hyperphosphatemia, and elevated serum concentrations of PTH and impaired urinary excretion of cyclic AMP and phosphate after administration of exogenous PTH. (Resistance to thyroid-stimulating hormone, the gonadotropins, and growth hormone releasing hormone also occurs in patients with PHP IA. PHP IA may be suspected in the neonate with hypocalcemia in whom hyperthyrotropinemia has been detected in the neonatal screening study for congenital hypothyroidism.48) When maternally transmitted, PHP IA is coupled with the phenotype of Albright’s hereditary osteodystrophy (AHO; short stature; brachydactyly of the 3rd, 4th, and 5th metacarpals; round face; and heterotopic intramembranous subcutaneous calcifications).49 Although both maternal and paternal GNAS alleles are expressed in most tissues including bone, in renal tissue only the maternal GNAS allele is expressed. Thus, in renal tissue GNAS expression is “imprinted”—that is, there is differential gene expression depending on the parent of origin of the allele. Therefore, clinical manifestations of PHP IA in the affected patient depend on which parent donated the inactive GNAS allele. Loss-of-function mutations of the maternal GNAS allele result in PHP type IA with end-organ resistance to PTH and the skeletal defects of AHO. PHP IA patients have subnormal expression of Gsα in their erythrocytes. Inactivating mutations of paternal GNAS result in the abnormal AHO skeletal phenotype due to haploinsufficiency of GNAS expression in bone but intact proximal renal tubular responses to PTH where maternal GNAS is expressed; this clinical complex is designated pseudopseudohypoparathyroidism (PPHP). More than 80 heterozygous loss-of-function mutations in GNAS have been described. A four base pair deletion in exon 7 (codons 188 to 189) in GNAS that leads to a frameshift and premature stop codon has been found in a number of families with PHP IA and appears to be a “mutational hot spot” as it impairs DNA polymerization and replication. Other mutations alter intracellular movement of GNAS protein (Leu99Pro, Ser250Arg), increase the rate of release of GDP (Arg258Trp, Ala366Ser), or impair coupling of G-protein to PTH1R (Arg385His).
PHP type IB occurs only in the offspring of obligate female carriers in whom loss of maternal GNAS expression is present in the renal proximal tubule, resulting in selective proximal renal tubular resistance to PTH; skeletal expression of both maternal and paternal GNAS is intact and hence bone formation is normal. In some patients, PHP type IB is due to deletions within the DMR (NESP55, XLas, or alternative first exons A/B) of GNAS or to loss of a 5′ cis-acting imprinting control center that is essential for methylation of the GNAS differentially methylated region that regulates expression of maternal GNAS in the proximal renal tubule.47,49 Within the 5′ upstream region of GNAS is STX16, a gene that is partially deleted in some patients with PHP IB; mutations of this gene are associated with loss of methylation of GNAS exons A/B. Thus, defects in STX16 and within the DMR of GNAS prevent expression of maternal GNAS in the proximal renal tubule. Paternal uniparental isodisomy of the long arm of chromosome 20 (site of GNAS) may be the cause of PHP IB in some patients. Patients with PHP IB are often resistant to thyroid-stimulating hormone (TSH) as well as to PTH.49 The phenotype of subjects with PHP type Ic is similar to that of those with PHP IA including AHO, but these patients usually express normal quantities of functional Gsα in their erythrocytes; in some PHP Ic subjects, however, inactivating mutations in GNAS have been reported.49 It must be remembered that there may be overlap in mechanisms that down-regulate expression of GNAS in various tissues and that PHP types 1 are heterogeneous in their genetic origins. Indeed, imprinting defects are present in patients with clinical manifestations of either PHP IA or PHP IB, indicating the pathogenetic overlap of these two disorders.50 Despite elevated levels of PTH and radiographic evidence of bone responsiveness to PTH in patients with PHP IA, bone mineralization is normal in these subjects.51 In patients with PHP type II, the skeletal phenotype is normal but patients are partially resistant to the renal effects of PTH; after administration of PTH, the urine excretion of cyclic AMP increases but that of phosphate does not; its pathogenesis is unknown as yet. Progressive osseous heteroplasia (MIM 166350) is an autosomal dominant disorder due to loss-of-function mutations in paternal GNAS, characterized by dermal ossifications, that begins in infancy and progresses to diffuse bone formation in skeletal muscle and deep fascia.49,52,53 This disease may lie at one end of the spectrum of PPHP. Although not as yet clinically described, an inactivating mutation in LRP6 (encoding lipoprotein receptor-related protein 6; MIM 603507) might also be associated with resistance to the biologic effect of PTH. In addition to its primary role in receptor-mediated endocytosis of lipoproteins, LRP6 is essential for the movement of Gαs to the plasma membrane and for its coupling to PTH1R.54 Experimentally, in vitro “knockdown” or inactivating mutations of Lrp6 decrease the cellular response to PTH.
Acrodysostosis is a chondrodysplasia with many features of PHP IA (short stature, obesity, brachydactyly, abnormal face with nasal and maxillary hypoplasia, advanced skeletal maturation) that is due to a mutation in PRKAR1A. This gene encodes the cyclic AMP–dependent regulatory alpha subunit of protein kinase A (PKA), the protein kinase that is downstream of Gαs and cyclic AMP and whose activation leads to cascades of intracellular signal transduction pathways that regulate cell division, differentiation, metabolism, and apoptosis. Paradoxically, prolonged activation of the regulatory alpha subunit of PKA results in a decline in the functional activity of the catalytic subunit of PKA. In a patient with acrodysostosis, a de novo germline gain-of-function mutation (Arg368Stop) in PRKAR1A was identified that led to functional resistance to PTH and TSH.55 Mutant PRKAR1A was a shortened protein whose binding avidity to the catalytic subunit of PKA was increased because it lacked one of two cyclic AMP binding domains; hence, it could only be slowly released from the catalytic subunit of PKA by cyclic AMP, thereby maintaining the catalytic subunit of PKA in the inactive state.
Deletions and mutations of the (maternal) mitochondrial genome (composed of 16,569 base pairs) have been associated with hypoparathyroidism. Patients with the Kearns-Sayre syndrome manifest ophthalmoplegia, retinal pigmentation, and cardiomyopathy; muscle biopsy may reveal ragged red fibers. Hypoparathyroidism has been observed in patients with the syndromes of Mitochondrial encephalopathy, Lactic Acidosis, and Stroke-like episodes (MELAS), Mitochondrial Trifunctional Protein Deficiency Syndrome (MTPDS, MIM 609015), and Pearson marrow-pancreas syndrome.15,43,56 Point mutations, duplications, and deletions of various length of the mitochondrial genome have been found in these patients.
Evaluation and management
Evaluation of the neonate with hypocalcemia begins with review of the maternal, gestational, peripartum, postnatal, and family histories and physical examination (Figure 18-2).1–3 Historical data include those related to maternal parity and complications of pregnancy such as maternal diabetes mellitus (gestational, types I or II), toxemia of pregnancy or ingestion of agents that may cause maternal hypercalcemia (excessive alkali), intrauterine growth restriction, abnormalities of delivery, neonatal sepsis, or other early postpartum illnesses. The family history is examined for members with abnormalities of mineral metabolism such as renal calculi, rickets, or hypocalcemia (e.g., seizure disorders). The social history provides information about the socioeconomic status of the mother and her cultural milieu that may have impacted maternal diet and exposure to sunlight during gestation. Physical examination of the neonate (abnormal face, cardiac murmur) may suggest a complex form of hypocalcemia. Determination of a complete blood count, serum concentrations of total calcium, Ca2+, magnesium, phosphate, creatinine, intact PTH, calcidiol, and calcitriol, and urinary calcium and creatinine concentrations in a spot urine should precede initial therapy of the hypocalcemic newborn whenever possible. Decreased serum concentrations of PTH are common in neonates with early onset hypocalcemia, but persistently low PTH levels suggest impaired PTH secretion. High PTH concentrations are present in patients with vitamin D deficiency or insensitivity, PTH resistance due to loss-of-function mutations in PTH1R, GNAS, or PRKAR1A or impaired renal function. Low levels of calcidiol signify decreased maternal (and hence fetal) vitamin D stores or rarely a defect in vitamin D-25 hydroxylase, whereas calcitriol concentrations are inappropriately low in subjects with severely compromised renal function, hypoparathyroidism, or those with a deficiency of 25OHD-1α-hydroxylase. Elevated calcitriol values suggest vitamin D resistance due to an abnormality in VDR, a disorder that may be associated with alopecia. Skeletal radiographs may disclose osteopenia, whereas chest x-ray may not identify a thymic shadow (although this is an unreliable sign in a severely ill or stressed neonate). Serum levels of calcium, Ca2+ phosphate, and intact PTH should be measured in the mothers of neonates with unexplained hypocalcemia.
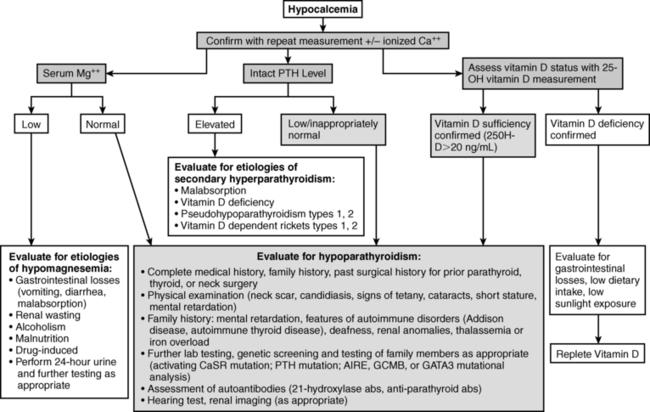
FIGURE 18-2  Evaluation of hypocalcemia. (From Bilezikian, J. P., Khan, A., Potts, J. T., Jr, et al. (2011). Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Mineral Res, 26, 2317–2337.)
Evaluation of hypocalcemia. (From Bilezikian, J. P., Khan, A., Potts, J. T., Jr, et al. (2011). Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Mineral Res, 26, 2317–2337.)
In neonates with hypocalcemia not otherwise explained, evaluation for possible DGS should be undertaken, particularly when physical examination reveals an abnormal face, and a congenital anomaly of the outflow tract of the heart is present. The white blood and T (CD4) lymphocyte counts are low in DGS and the thymic shadow often absent. The diagnosis of the DGS is confirmed by the presence of a microdeletion of chromosome 22q11.2 as demonstrated by microarray or fluorescent in situ hybridization (FISH). Occasionally, sequence analysis of TBX1 may be needed to establish this diagnosis, if the other studies are normal. Because the DGS may be heritable, examination of the karyotype of the parents of a DGS infant is indicated as well as those of the siblings if the parent also has deletion of chromosome 22q.11.21. It should be noted that the majority of neonates and infants with DGS are recognized primarily because of cardiac anomalies and that subjects without these lesions may not be identified until mid or late childhood or adolescence.57 Neonates with PHP IA may present with elevated serum levels of TSH in the neonatal metabolic screening survey but usually do not have the characteristic skeletal phenotype (brachymetacarpals) of AHO; if no cause of congenital hypothyroidism is identified, the diagnosis of PHP IA should be suspected, and measurement of serum calcium levels and, if appropriate, genotyping and methylation studies of GNAS are indicated.49
Early neonatal hypocalcemia is often asymptomatic, but nevertheless treatment is indicated when the total serum calcium concentration is below 6 mg/dL in the preterm infant and less than 7 mg/dL in the term infant.3 Asymptomatic neonates are most easily managed by increasing the oral intake of calcium and establishing an overall ratio of calcium:phosphate intake of 4:1 (including that in feedings with a low phosphate formula such as Similac PM 60/40R—calcium:phosphate ratio 1.6:1) with calcium glubionate or calcium carbonate administered in divided doses every 4 to 6 hours (Table 18-3). Eucalcemia is almost always restored in these subjects within 3 weeks after birth and often earlier. In hypocalcemic infants with tetany or frank seizures, 10% calcium gluconate (elemental calcium 9.3 mg/mL) at a dose of 1 to 3 mg/kg and at a rate of less than 1 mL/minute with the total dose not to exceed 20 mg of elemental calcium/kg may be administered by intravenous infusion over 15 minutes; often seizures will cease after 1 to 3 mL of 10% calcium gluconate have been administered.3,58 Cardiac rate and rhythm must be carefully monitored to prevent bradycardia and asystole. Further intravenous bolus doses of calcium (∼10 mg/kg at 6-hour intervals) should be used sparingly, as they result in wide excursions in serum calcium values. After initial treatment of neonatal hypocalcemia, 500 mg of calcium gluconate/kg/24 hours may be administered by continuous intravenous infusion taking care to prevent extravasation or infiltration as extracellular calcium will precipitate in soft tissues.59 These infants may receive supplemental oral calcium if necessary (as discussed previously). Depending on the cause of the hypocalcemia, supplemental vitamin D or calcitriol may also be needed. In a neonate with hypocalcemia due to hypoparathyroidism, administration of synthetic PTH1–34 may occasionally be required to restore eucalcemia.60 Serum and urine calcium and creatinine levels should be determined frequently and treatment modified to maintain eucalcemia and the urine calcium/creatinine ratio < 0.2 – < 0.5 in an effort to avoid iatrogenic hypercalcemia, hypercalciuria, nephrocalcinosis, and renal insufficiency. In neonates who require parenteral alimentation, 50 mg of elemental calcium/kg/24 hours should be incorporated into the infused solution; elemental phosphate must also be administered as permitted and indicated.
TABLE 18-3
Preparations of Vitamin D, Calcium, Magnesium, and Phosphate
| Content | Elemental Mineral | |
| Vitamin D | ||
| Vitamin D | ||
| Calciferol | 8000 IU/mL | |
| 50,000 IU/capsule | ||
| Calcidiol | 20 or 50 μg/tablet | |
| Calcitriol | ||
| Rocaltrol | 1 μg/mL (oral solution) | |
| 0.25 μg or 0.5 μg/capsule | ||
| Vectical | 3 μg/g (ointment) | |
| Dihydrotachysterol | ||
| Hytacherol | 0.2 mg/5 mL 0.125, 0.2, 0.4 mg/tablet | |
| Calcium | ||
| Calcium acetate | ||
| Phoslyra | 667 mg/5 mL | 667 mg/capsule |
| Calcium gluconate (iv) | 93 mg/10 mL | 93 mg/g |
| 500, 650 mg/tablet | ||
| Calcium glubionate (solution) | 64 mg/g | 115 mg/5 mL |
| Calcium carbonate | 400 mg/g | 500, 600, 750, 1250 mg/tablet |
| Titralac | 420 mg tablet | 168 mg/tablet |
| 780 mg tablet | 300 mg/tablet | |
| Oscal | 650 mg tablet | 260 mg/tablet (vitamin D) |
| 1250 mg tablet | 500 mg/tablet (vitamin D) | |
| Tums | 500 mg tablet | 200 mg/tablet |
| 750 mg tablet | 300 mg/tablet | |
| 1000 mg tablet | 400 mg/tablet | |
| 1250 mg tablet | 280 mg/tablet | |
| Caltrate | 1500 mg tablet | 600 mg/tablet (vitamin D) |
| Calcium carbonate (suspension) | 1250 mg/5 mL | 500 mg/5 mL |
| Calcium chloride | 100 mg/mL | |
| Calcium citrate | 200 mg/g | 210 mg/tablet |
| Citracal | 950 mg tablet | 200 mg/tablet |
| Calcium lactate | 84 mg/g | 130 mg/tablet |
| Magnesium | ||
| Magnesium sulfate | 49 mg/mL (50% intramuscular solution) | |
| 0.32, 0.64, 4 mEq/mL (intravenous solution) | ||
| Magnesium oxide | 603 mg/g | 241 mg/tablet |
| Magnesium gluconate | 54 mg/g | 27 mg/tablet |
| Magnesium chloride | 120 mg/g | 64 mg/tablet |
| Phosphorus | ||
| Sodium phosphate (Phospha-Soda) | 127 mg/mL | |
| Sodium/potassium phosphate (Phos-NaK) | 250 mg/packet (powder) | |
| Potassium phosphate (Neutraphos-K) | 250 mg/packet (powder) | |
| Potassium phosphate (K-Phos Original) | 114 mg/tablet | |
| Sodium/Potassium phosphate (K-Phos #2) | 250 mg/coated tablet | |
| Sodium/Potassium phosphate (K-Phos Neutral) | 250 mg/tablet |
Compiled from Alon US (2006). Hypophosphatemic vitamin D–resistant rickets. In M. J. Favus (Ed.), Primer on the metabolic bone diseases and disorders of mineral metabolism, (6th ed.) Washington, DC: American Society for Bone and Mineral Research, 342-345; Shoback, D. (2008). Hypoparathyroidism. N Engl J Med 359:391-403.
After restoration of eucalcemia in the infant with DGS, other components of this disorder must be addressed. Cardiac anomalies often require surgical correction, as do palatal clefts. In DGS infants who are immunocompromised and experience recurrent infections due to thymic aplasia, appropriate anti-infectious therapy is mandatory. Transplantation of fetal or cultured postnatal thymic tissue, bone marrow, or peripheral blood mononuclear cells has restored immune function in infants with the DGS. Supplemental calcitriol (20 to 60 ng/kg/day) and calcium are necessary for restoration and maintenance of eucalcemia in infants with hypoparathyroidism. Poor growth due to feeding difficulties and learning disabilities due to developmental delay must be managed on an individual basis and illustrate the need for a multidisciplinary approach to the care of DGS patients.57 Hypocalcemia due to hypomagnesemia is managed acutely by the intravenous infusion over 1 to 2 hours or by the intramuscular injection of 50% magnesium sulfate at a dose of 0.1 to 0.2 mL/kg while carefully monitoring cardiac status.
Hypocalcemia in the child and adolescent
Etiology
Causes of hypocalcemia in the child and adolescent include many that also result in neonatal hypocalcemia and are listed in Tables 18-2A and B. Hypocalcemia is defined by the norms of the analytic laboratory and its parameters dependent on the age of the subject (total calcium concentrations: 1 to 5 years 9.4 to 10.8; 6 to 12 years 9.4 to 10.2; > 20 years 8.8 to 10.2 mg/dL).58 Total calcium levels are low in the hypoalbuminemic patient; a correction for hypoalbuminemia may be calculated by adding 0.8 mg/dL to the recorded total calcium concentration for every decrease in albumin concentration of 1 g/dL. Thus, it is appropriate to measure both total and Ca2+ values when evaluating the hypocalcemic child, but reliance on Ca2+ determinations alone is discouraged given the technical difficulties with this assay. Hypocalcemia develops as a consequence of either too little inflow of calcium from the gastrointestinal tract, bone, or kidney into the extracellular and vascular spaces or an excessive loss of calcium from these spaces into urine, stool, or bone. Thus, hypocalcemia may be due to decreased intake or absorption or excessive loss of calcium, decreased production of bioactive PTH due to congenital abnormalities of parathyroid gland development or PTH synthesis or of the CaSR, destruction of parathyroid glands by autoantibodies, metal overload (copper, iron), surgical or radiation insults, granulomatous infiltration, or impaired cellular responsiveness to PTH. Restricted exposure to sunlight or reduced intake, absorption, metabolism, or activity of vitamin D leads to hypocalcemia. Hypomagnesemia impairs the secretion (but not the synthesis of PTH) and blunts tissue responsiveness to PTH. Hypocalcemia occurs in the very ill child and after exposure to a number of drugs and medications. Hypocalcemic tetany may develop after the administration of phosphate containing enemas by rectum or laxatives by mouth.61 At times, the hypocalcemic child or adolescent may be asymptomatic and identified by chemical screening for an unrelated problem, or she or he may present with intermittent muscular cramping either at rest or during exercise (when the increase in systemic pH due to hyperventilation further lowers the concentration of Ca2+); paresthesias of fingers, toes, or circumoral regions; tetany (carpopedal spasm, laryngospasm, bronchospasm); or seizures (grand mal, focal, petit mal, adynamic, or syncopal). Prolonged and severe hypocalcemia may lead to congestive heart failure.62 Physical examination often reveals a positive Chvostek or Trousseau sign (carpopedal spasm) and hyperreflexia. However, a Chvostek sign is commonly present in normal adolescents also.
Hypoparathyroidism may occur as a solitary disorder, as part of a multidimensional autoimmune polyendocrinopathy, or as one manifestation of a group of complex congenital anomalies (DGS, HDR, HRD, KCS1, Blomstrand, and other syndromes).62 There are sporadic and familial forms of hypoparathyroidism; when familial, hypoparathyroidism may be transmitted as an autosomal dominant, autosomal recessive, or X-linked recessive trait (see Tables 18-2A and B). Abnormalities in the development of the parathyroid glands, in the transcription of PTH, and in the processing of the translated product have been associated with inherited forms of hypoparathyroidism. Autosomal dominant dyshormonogenic hypoparathyroidism due to a monoallelic T d C transition in codon 18 (Cys18Arg) of the 25 amino acid signal peptide of prepro-PTH impaired efficient transport of protein from the ribosome and interaction of prepro-PTH with the signal recognition particle, movement of the precursor peptide into and exit from the rough endoplasmic reticulum, its cleavage by a signal peptidase, and its incorporation into a secretory granule.63 Autosomal recessive dyshormonogenic hypoparathyroidism has been associated with a homozygous G d C transversion in nucleotide 1 of intron 2 of PTH within the signal sequence that prevented normal cleavage of prepro-PTH and decreased secretion of PTH. However, inactivating mutations in PTH are uncommon in patients with sporadic idiopathic hypoparathyroidism.64 Isolated hypoparathyroidism may be found in patients with deletion of chromosome 22q11.2 without other signs or symptoms of the DGS or the velocardiofacial syndrome.
Autosomal dominant hypoparathyroidism (MIM 146200) due to heterozygous gain-of-function mutations in the extracellular, transmembrane, and intracellular domains of CASR that transcribe a CaSR that is not intrinsically constitutively active but is exceptionally sensitive to and easily activated by very low serum Ca2+ concentrations may be identified in infancy or in older subjects (Figure 18-3). Even at hypocalcemic levels, Ca2+ binds avidly to the CaSR and activates phospholipase C-β1, increasing cytosolic levels of inositol phosphate and Ca2+ and stimulating the mitogen-activated protein kinase (MAPK) signal transduction pathway in parathyroid chief cells—suppressing PTH synthesis and secretion—and in the kidney—decreasing renal tubular calcium and magnesium resorption leading to urinary wasting of these cations (hypercalciuric hypocalcemia); urinary concentrating ability is also depressed. In these subjects, serum levels of phosphate are increased and magnesium values decreased; PTH concentrations are low or inappropriately normal. Affected patients frequently have symptomatic hypocalcemia such as tetany and seizures. They are very sensitive to vitamin D; the dose of calcitriol must be limited to that which raises serum calcium to asymptomatic values even if not within the normal range, as larger doses lead to hypercalciuria (sometimes even when serum calcium levels remain subnormal), nephrocalcinosis, and functional renal insufficiency. Administration of recombinant human (rh)PTH1–34 restores calcium homeostasis in this disorder but does not necessarily prevent nephrocalcinosis.19,23,24 Although there has been reluctance to administer rhPTH to children because there is an increased incidence of bone tumors in young rats receiving very large amounts of this agent, primates appear to be less susceptible to PTH-induced bone tumor formation than do rodents.65 Development of stimulatory autoantibodies to the CaSR results in an acquired variant of spontaneous hypoparathyroidism that may be isolated or part of a complex autoimmune endocrinopathy.66 Indeed, in perhaps as many as one third of patients with acquired isolated idiopathic hypoparathyroidism, antibodies directed against epitopes in the extracellular domain of the CaSR may be present. This form of acquired hypoparathyroidism may be reversible, as these antibodies do not destroy the parathyroid glands. In patients with other forms of autoimmune hypoparathyroidism, the antibodies are cytotoxic and accompanied by lymphocytic infiltration, atrophy, and fatty replacement of parathyroid tissue.