Disorders of growth hormone/insulin-like growth factor secretion and action
Measurement
Correct assessment of statural growth requires optimizing accuracy in height determinations.1–3 When feasible, measurement of supine length is employed in children younger than 2 years of age, and that of standing height is done in older children. Between 2 and 3 years of age, measurement of both length and height can be helpful to assess growth velocity more accurately whereas taking into account the measurement technique. In this age range the supine length averages about 1 cm greater than the standing height. However, the inherent inaccuracies involved in measuring length in infants are often obscured by their rapid skeletal growth characteristic of this age. For measurement of supine length, it is best to employ a firm box with an inflexible board (against which the head lies) and a movable footboard on which the feet are placed perpendicular to the plane of the supine infant. Alternatively, in many offices a counter-mounted or portable infantometer is used to assess infants’ and toddlers’ growth. To achieve reliable and accurate measurements, the technique used is of great importance. Optimally, the child needs to be relaxed—with the legs fully extended and the head positioned in the “Frankfurt plane” (meaning the line connecting the outer canthus of the eye and the external auditory meatus is perpendicular to the long axis of the trunk). Repeating the length measurement three times and using the mean is another way to improve reliability.
As with length measurements in infants, positioning of the child is critical. The patient should be fully erect, with the head in the Frankfurt plane. The back of the head, thoracic spine, and buttocks should be touching the vertical axis of the stadiometer; the heels are put together with the toes slightly separated. Every effort should be made to correct discrepancies related to lordosis (neck and lower back) or scoliosis. Ideally, serial measurements should be made at the same time of day because diurnal variation in standing height has been observed, leading to measurements in children and adolescents being up to 0.8 cm shorter at the end of the day compared with measurements obtained in the morning; this phenomenon is likely due to development of fatigue of the spine musculature throughout the day.4
It is critical that height determinations be performed by an individual with proper training, rather than (as is often the case) by an inexperienced member of the staff. We recommend that heights also be measured in triplicate, that variation be no more than 0.3 cm, and that the mean height be recorded. For determination of height velocity, it is obviously best to have the same individual performing the determinations. Even when every effort is made to maximize the precision of height determinations, a minimum interval of 3 months is necessary for accurate height velocity computation. Six months worth of data are preferable, although it is of note that seasonal variation in height velocity has been reported.5
Growth charts
Evaluation of a child’s height must be done in the context of normal standards. Such standards can be either cross-sectional or longitudinal. Most American pediatric endocrine clinics continue to use the cross-sectional data provided by the National Center for Health Statistics (NCHS), which were originally introduced in 1977 and have been updated.6–8 Epidemiologic limitations in these growth charts have been noted, however. The data included in the original infant charts, for example, were derived from a private study of a group of subjects who were primarily white, formula-fed, middle-class infants from southwestern Ohio. Data employed for older children came from national health examination surveys conducted from 1963 to 1974.
The NCHS (now a part of the Centers for Disease Control and Prevention) has provided a set of new growth charts, representing revisions of the previous charts, and has introduced new charts for body mass index9 (Figures 10-1 through 10-8). Interestingly, the new charts show little change in average height since the late 1980s, despite the common perception that today’s children are taller than peers from 10 to 25 years ago. This is in contrast to other countries, in particular, The Netherlands, where mean height continued to rise in spite of the population being the tallest in the world. In many current and formerly developing countries, the population, which was typically shorter than in the Western world, is becoming taller.
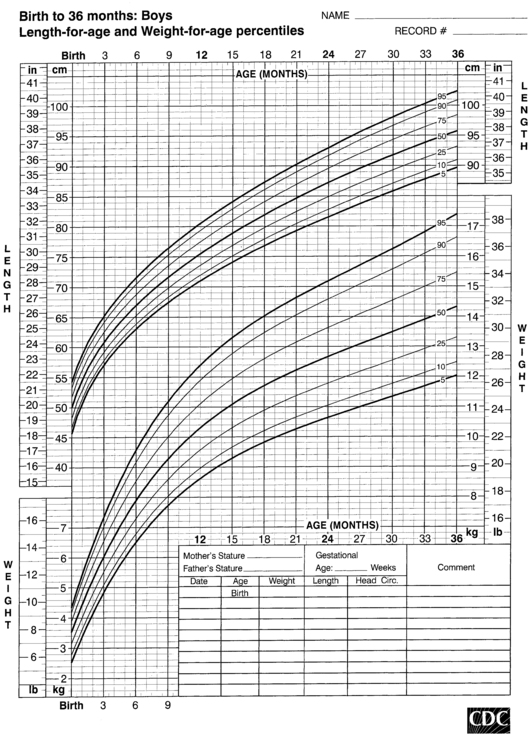
FIGURE 10-1  Length-for-age and weight-for-age percentiles for boys (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Length-for-age and weight-for-age percentiles for boys (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
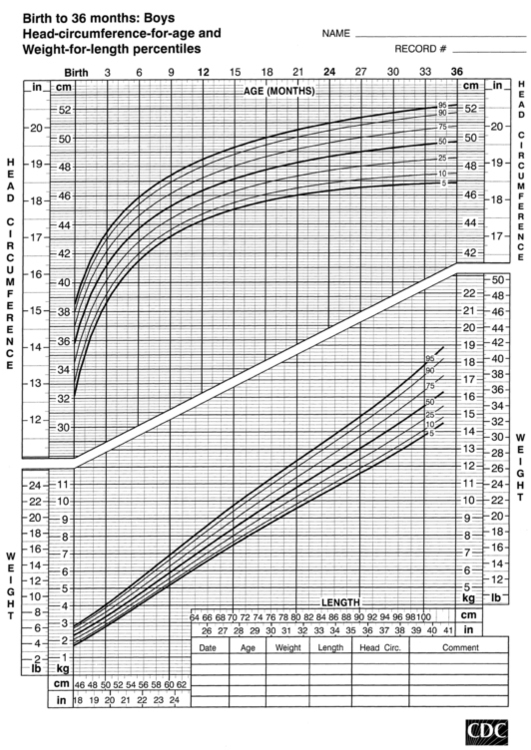
FIGURE 10-2  Head-circumference-for-age and weight-for-length percentiles for boys (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Head-circumference-for-age and weight-for-length percentiles for boys (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
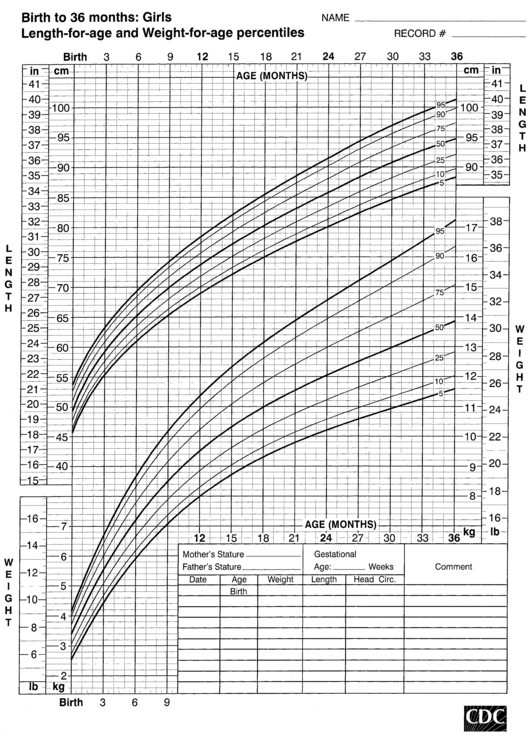
FIGURE 10-3  Length-for-age and weight-for-age percentiles for girls (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Length-for-age and weight-for-age percentiles for girls (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
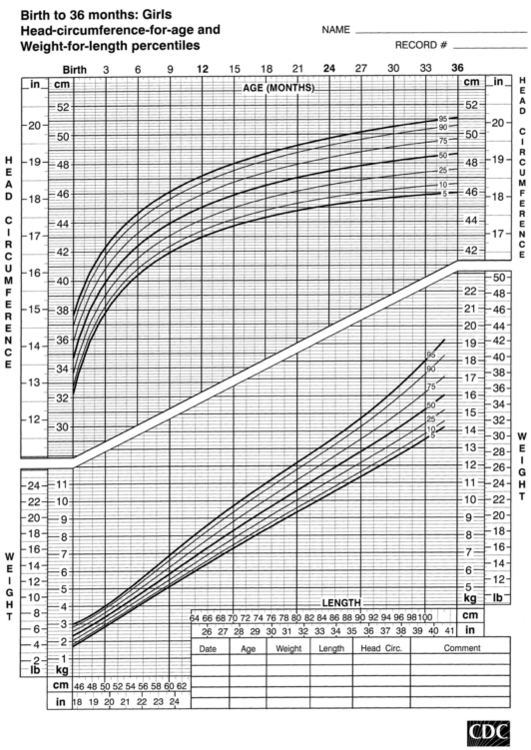
FIGURE 10-4  Head-circumference-for-age and weight-for-length percentiles for girls (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Head-circumference-for-age and weight-for-length percentiles for girls (birth to 36 months). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
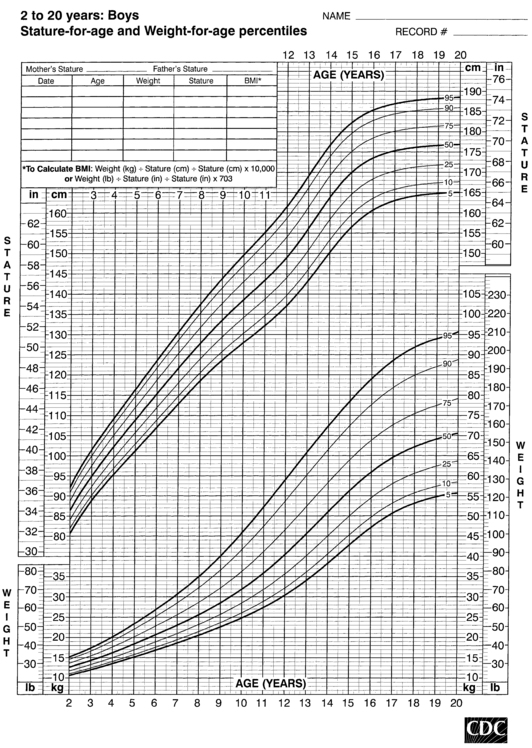
FIGURE 10-5  Stature-for-age and weight-for-age percentiles for boys (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Stature-for-age and weight-for-age percentiles for boys (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
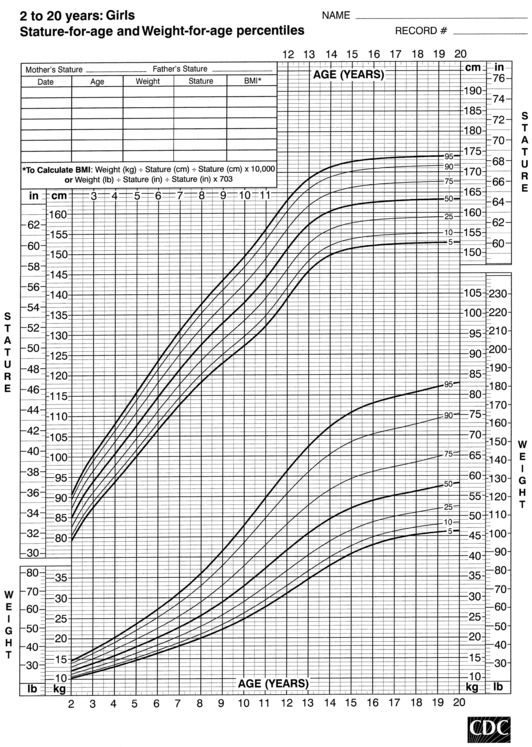
FIGURE 10-6  Stature-for-age and weight-for-age percentiles for girls (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Stature-for-age and weight-for-age percentiles for girls (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
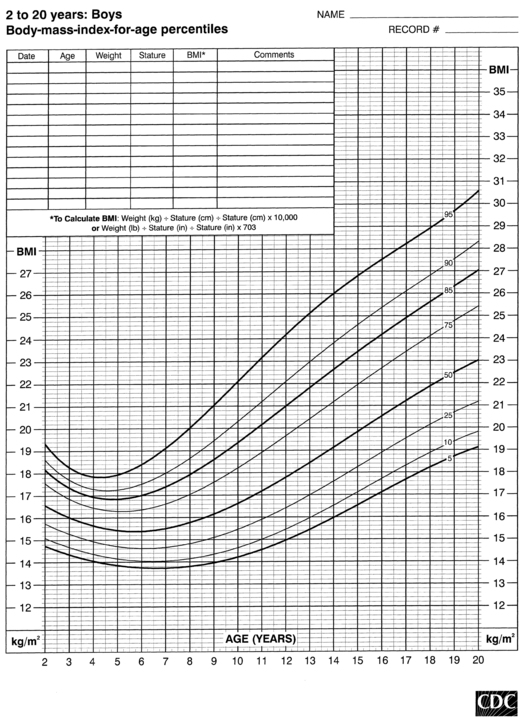
FIGURE 10-7  Body-mass-index-for-age percentiles for boys (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Body-mass-index-for-age percentiles for boys (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
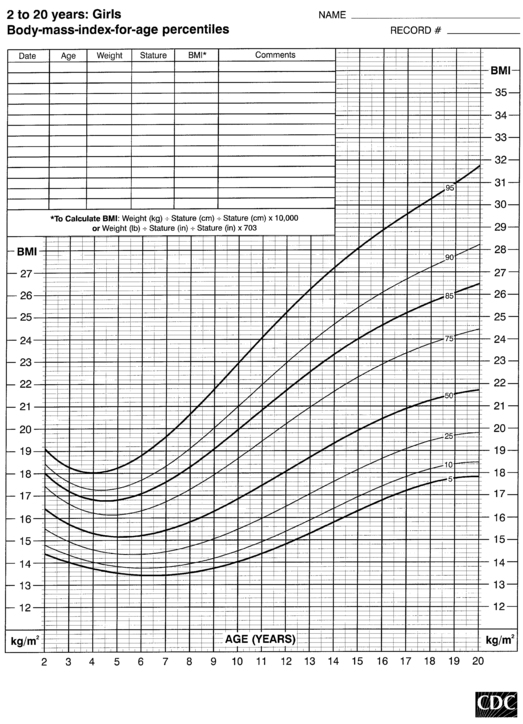
FIGURE 10-8  Body-mass-index-for-age percentiles for girls (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Body-mass-index-for-age percentiles for girls (2 to 20 years). (Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000), www.cdc.gov/growthcharts.)
Classic percentile-based growth charts are invaluable for plotting growth of children relative to the 3rd or 5th, 10th, 25th, 50th, 75th, 90th, and 95th or 97th percentiles of normal American children. There are, however, two major limitations of these charts. First, they do not satisfactorily define the growth rates of children below the 3rd or above the 97th percentile—the very children for whom it is most critical to accurately describe the degree to which their growth deviates from the normal growth percentiles. The NCHS data can be used to compute standard deviation (SD) scores (or Z scores), which are more helpful because a short child can be described as having a growth rate of (for example) 2 or 2.5 SD from normal. Because these SDs are defined by cross-sectional data, however, SD scores during childhood are not directly comparable with SD scores during adolescence—when great variation in growth rates can be normally observed. Second, cross-sectional data are of greater value during infancy and childhood than in adolescence—when differences in the timing of puberty can introduce considerable variability into normal growth rates. To address this issue, Tanner and colleagues10 have developed longitudinal growth charts that accommodate the timing of puberty. These charts are of greatest value in assessing growth during adolescence and puberty and are probably superior for plotting sequential growth data on any given child.
The data from cross-sectional and longitudinal growth studies have been employed to develop height velocity standards10 (Figures 10-9 and 10-10). It is important to emphasize that carefully documented height velocity data are invaluable in assessing the child with abnormalities of growth. Although considerable variability exists in the normal height velocity observed in children of different ages, between the age of 2 years and the onset of puberty children normally grow with remarkable fidelity relative to the normal growth curves. The physician should note any “crossing” of height percentiles during this age period, and abnormal height velocities always warrant further evaluation.
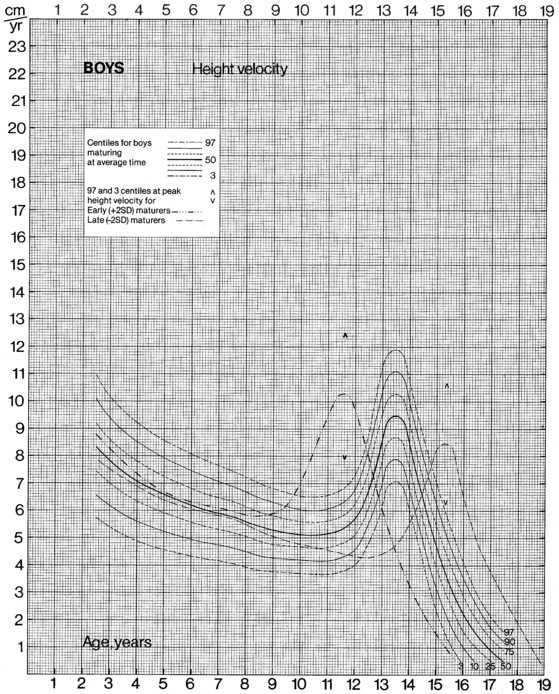
FIGURE 10-9  Height velocity chart for males from 0 to 19 years. (From Tanner, J. M., & Davies, S. W. D. (1985). Clinical longitudinal standards for height and height velocity for North American children. J Pediatr, 107, 312.)
Height velocity chart for males from 0 to 19 years. (From Tanner, J. M., & Davies, S. W. D. (1985). Clinical longitudinal standards for height and height velocity for North American children. J Pediatr, 107, 312.)
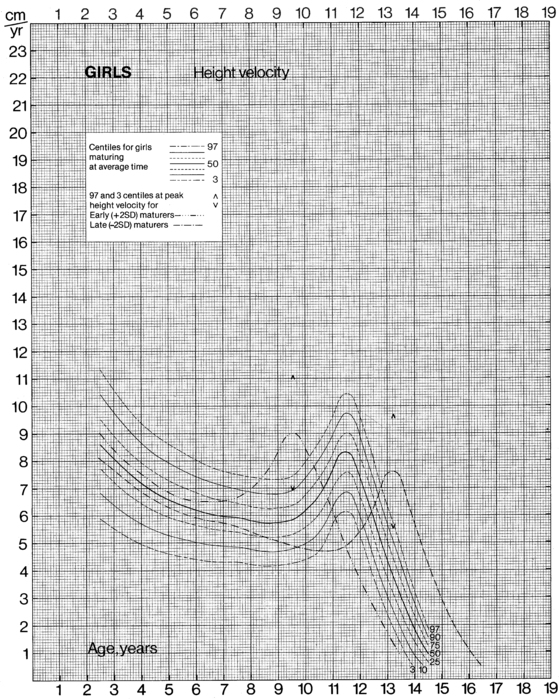
FIGURE 10-10  Height velocity chart for females from 0 to 19 years. (From Tanner, J. M., & Davies, S. W. D. (1985). Clinical longitudinal standards for height and height velocity for North American children. J Pediatr, 107, 312.)
Height velocity chart for females from 0 to 19 years. (From Tanner, J. M., & Davies, S. W. D. (1985). Clinical longitudinal standards for height and height velocity for North American children. J Pediatr, 107, 312.)
Disease-related growth curves have been developed for a number of clinical conditions associated with growth failure, such as Turner syndrome,11 achondroplasia,12 and Down syndrome.13 Such growth profiles are invaluable for tracking the growth of children with these clinical conditions. Deviation of growth from the appropriate disease-related growth curve suggests the possibility of a second underlying condition.
Body proportions
Many growth disorders, leading to either short stature or tall stature, are associated with disproportionate growth. Some of the more common disorders leading to disproportionality include the many forms of osteochondrodysplasia and rickets. Survivors of childhood cancer who received spinal irradiation are also at increased risk for disproportionate growth.14 To allow documentation of such disproportionate growth, the following measurements can be made:
• Occipitofrontal head circumference
• Lower body segment: distance from top of pubic symphysis to the floor
• Upper body segment: sitting height (height of stool should be subtracted from standing height)
• Arm span: measurement of the length from one end of a patient’s arms (measured at the fingertip) to the other with the arms raised parallel to the ground at shoulder height
Published standards exist for these body proportion measurements, which must be evaluated relative to the patient’s age.15 The upper segment-to-lower segment ratio, for example, ranges from 1.7 in the neonate to slightly below 1 in the adult. Ethnic differences also exist: black children and adolescents have shorter sitting height and longer length of the lower extremities (about one inch for each, corrected for age and sex).16
Skeletal maturation
Studies in patients with mutations of the gene for the estrogen receptor17 or for aromatase enzyme18 have demonstrated that it is estrogen that is primarily responsible for ultimate epiphyseal fusion, although it seems unlikely that estrogen is alone responsible for all skeletal maturation. Beyond the neonatal period, and especially after age 2 years, a radiograph of the left hand and wrist is commonly used to determine “bone age”—which can be related to the published standards of Greulich and Pyle.19 Although the authors recommend a bone-by-bone assessment approach to arrive at a mean bone age for the patient, most endocrinologists determine the bone age by finding the standard radiograph that best matches their patient’s using a subjective comparative approach. This technique creates significant variability in bone age reading between different interpreters. An alternative method for assessing bone age from radiographs of the left hand, involving a scoring system for each individual bone, has been developed by Tanner and Whitehouse and their colleagues.20 The left hand obviously represents a compromise, because radiographs of the entire skeleton would be tedious, expensive, and necessitate excessive radiation exposure. It is important to note, however, that the hand obviously does not contribute to the height of an individual and that accurate evaluation of growth potential might necessitate radiographs of the legs and spine.
A number of important caveats concerning bone age determination must be considered. Experience in reading bone age films is essential, and clinical studies involving bone age generally benefit from having a single reader who does all interpretations. Second, the normal rate of skeletal maturation differs between males and females (and ethnic variability exists). The standards of Greulich and Pyle are divided by sex, but they were developed in American white children. Finally, the Greulich-Pyle and the Tanner-Whitehouse standards were developed using normal children.21 They are not necessarily applicable to children with skeletal dysplasia, endocrine abnormalities, or a variety of other causes of growth retardation.
Prediction of adult height
The extent of skeletal maturation observed in a patient can be used to predict the patient’s height potential. The estimated adult height potential may be interpreted in function of the parental target height. Predictions of adult height are based on the observation that the more delayed the bone age (relative to the chronologic age), the greater the length of time before epiphyseal fusion prevents further growth. The classic method for height prediction, based on Greulich and Pyle’s Radiographic Atlas of Skeletal Development,19 was developed by Bayley and Pinneau22 and relies on the patient’s bone age and height (Table 10-1).
TABLE 10-1
Fraction of Adult Height Attained at Each Bone Age
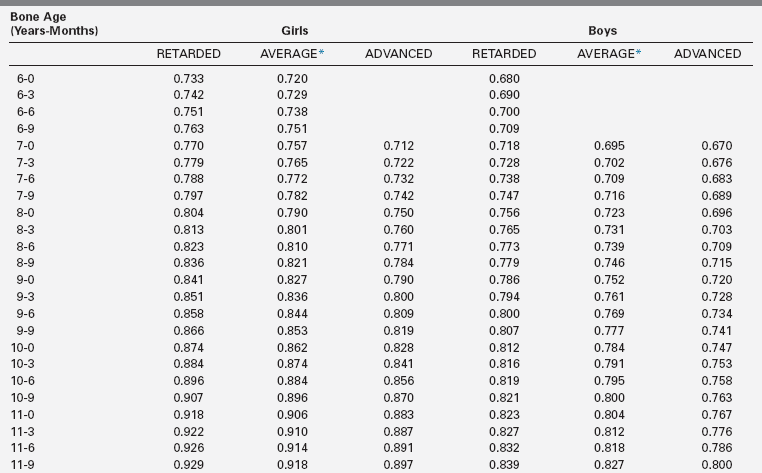
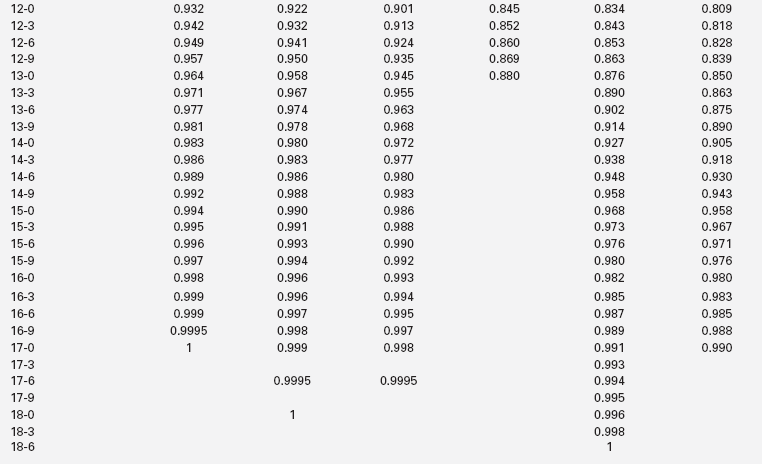
*Average: Bone age within 1 year of chronologic age.
Data from Post, E. M., & Richman, R. A. (1981). A condensed table for predicting adult stature. J Pediatr, 98, 440, based on the data of Bayley and Pinneau.22 These tables have been organized in an easy-to-use slide rule format (“Adult Height Predictor,” copyright 1987, Ron G. Rosenfeld).
Additional refinements were introduced by Tanner and colleagues20,23 with a system that employs height, bone age, and chronologic age; and by Roche and associates,24 who employ the combination of height, bone age, chronologic age, midparental height, and weight. All of these systems are by nature empirical and should never be used as absolute predictors. The more advanced the bone age, the greater the accuracy of the adult height prediction, but this is natural because a more advanced bone age places a patient closer to final height.
Endocrine regulation of growth
The pituitary
The pituitary gland lies in the sella turcica, the hypophyseal fossa of the sphenoid bone, which is located in the center of the cranial base. The concept of the pituitary as a “master gland” controlling the endocrine activities of the body has become outdated and has been replaced by an appreciation of the importance of the brain, particularly of the hypothalamus, in regulating hormonal production and secretion. Nevertheless, the pituitary gland remains central to our understanding of the regulation of growth, metabolism and homeostasis, response to stress, lactation, and reproduction.
Embryologically, the pituitary gland is formed from two distinct sources.25,26 Rathke’s pouch, a diverticulum of the primitive oral cavity (stomodeal ectoderm), gives rise to the adenohypophysis. The neurohypophysis originates in the neural ectoderm of the floor of the forebrain, which also develops into the third ventricle. The adenohypophysis normally constitutes 80% of the weight of the pituitary and consists of the pars distalis (also known as the pars anterior or anterior lobe), the pars intermedia (also known as the intermediate lobe), and the pars tuberalis (also known as the pars infundibularis or pars proximalis).
Much of our knowledge of normal hypothalamo-pituitary development is derived from animal, particularly rodent, models. In the mouse, a thickening of the ectoderm in the midline of the anterior neural ridge, forming the hypophyseal placode, heralds the onset of pituitary development at 7.5-days postcoitum (dpc). The formation of a rudimentary Rathke’s pouch follows at 9 dpc, with formation of a definitive pouch by 12 dpc and, subsequently, the anterior pituitary consisting of five different cell types secreting six different hormones (Figure 10-11). The developing Rathke’s pouch is initially associated with the presumptive hypothalamic territories, and later with the developing diencephalon.
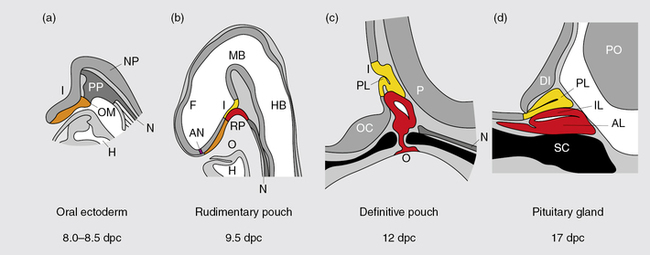
FIGURE 10-11  Mouse pituitary development. Illustration showing mouse pituitary development in sagittal section: stages of development are indicated in days post coitum (dpc). AL, anterior lobe; AN anterior neural pore; DI diencephalon; F, forebrain; H, heart; HB, hindbrain; I, infundibulum; IL, intermediate lobe; MB, midbrain; N, notochord; NP, neural plate; O, oral cavity; OC, optic chiasm; OM, oral membrane; P, pontine flexure; PL, posterior lobe; PO, pons; PP, prechordal plate; RP, Rathke’s pouch; SC, sphenoid cartilage. (Adapted from Sheng, H. Z., & Westphal H (1999). Early steps in pituitary organogenesis. Trends Genet, 1, 236–240.) This image can be viewed in full color online at ExpertConsult.
Mouse pituitary development. Illustration showing mouse pituitary development in sagittal section: stages of development are indicated in days post coitum (dpc). AL, anterior lobe; AN anterior neural pore; DI diencephalon; F, forebrain; H, heart; HB, hindbrain; I, infundibulum; IL, intermediate lobe; MB, midbrain; N, notochord; NP, neural plate; O, oral cavity; OC, optic chiasm; OM, oral membrane; P, pontine flexure; PL, posterior lobe; PO, pons; PP, prechordal plate; RP, Rathke’s pouch; SC, sphenoid cartilage. (Adapted from Sheng, H. Z., & Westphal H (1999). Early steps in pituitary organogenesis. Trends Genet, 1, 236–240.) This image can be viewed in full color online at ExpertConsult.
In humans, the pars distalis is the largest portion of the adenohypophysis and houses the great majority of hormone-producing cells. The pars intermedia is, typically, poorly developed and consists of several cystic cavities lined by a single layer of cuboidal epithelium. Pars distalis and intermedia are separated by a cleft, a vestigial structure of Rathke’s pouch from which it develops. This structure may often develop as a cyst (Rathke’s cleft cyst). In humans, in contrast to the mouse, pars intermedia is rudimentary as it largely disappears during embryogenesis. The pars tuberalis represents an upward extension of the pars distalis onto the pituitary stalk and may contain a limited number of gonadotropin-producing cells. The posterior pituitary (neurohypophysis) consists of the infundibular stem or hypophyseal stalk, the median eminence of the tuber cinereum, and the infundibular process (posterior lobe, neural lobe). The posterior pituitary contains the terminal axonal projections of magnocellular neurons from the paraventricular and supraoptic nuclei of the hypothalamus. These produce oxytocin, required during lactation and parturition, and vasopressin, required for osmotic regulation—as detailed in Chapter 11. It has no known function in the regulation of growth and will not be discussed further in this chapter.
Rathke’s pouch, the origin of the adenohypophysis, can be identified in the 3-mm embryo during the third week of pregnancy in humans. Rathke’s pouch then begins to develop, resulting in a complete pouch disconnected from the oral ectoderm by the end of the sixth gestational week. GH-producing cells can be identified by 9 weeks of gestation.27 It is at about this time that the vascular connections between the anterior lobe of the pituitary and the hypothalamus develop,28,29 although it has been demonstrated that hormonal production by the pituitary can occur in the absence of connections with the hypothalamus. Somatotropic cells in the pituitary are thus frequently demonstrable in the anencephalic newborn. Nevertheless, it appears likely that the initiation of development of the anterior pituitary is dependent on responsiveness of the oral ectoderm to inducing factors from the ventral diencephalon30–34 (Figure 10-12).
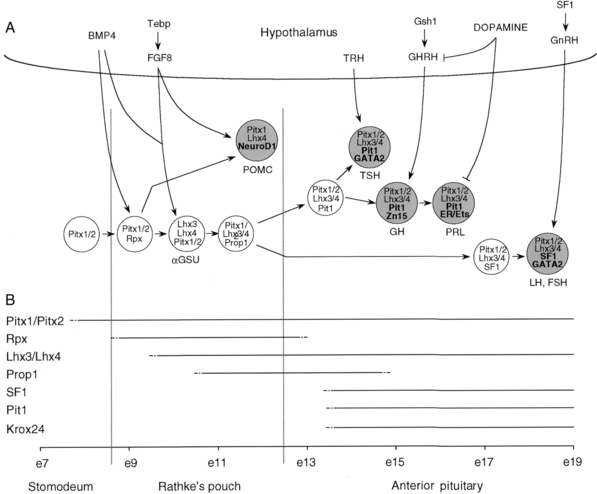
FIGURE 10-12  Development of pituitary cell lineages. A, Schematic representation of pituitary cell precursors showing the expression of prevalent transcription factors at each stage of development. Terminally differentiated cells are shown as larger and shaded circles together with the hormones produced (lineage-specific transcription factors are highlighted in bold in these cells). The interaction with transcription factors and signaling molecules in the hypothalamus is also noted. Transcription factors are represented in lowercase (except for SFI and GATA2), whereas signaling molecules appear in uppercase. B, Schema showing the timing of the appearance and disappearance of pituitary transcription factors during mouse embryogenesis. BMP4, bone morphogenic protein 4; e, embryonic day; ER, estrogen receptor; FGF8, fibroblast growth factor 8; FSH, follicle-stimulating hormone; GH, growth hormone; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin–releasing hormone; αGSU, α-glycoprotein subunit; LH, luteinizing hormone; POMC, pro-opiomelanocortin; PRL, prolactin; SFI, steroidogenic factor I; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone. (From Lopz-Bermejo, A., Buckway, C. K., & Rosenfeld, R. G. (2000). Genetic defects of the growth hormone–insulin-like growth factor axis. Trends Endocrinol, 11, 43.)
Development of pituitary cell lineages. A, Schematic representation of pituitary cell precursors showing the expression of prevalent transcription factors at each stage of development. Terminally differentiated cells are shown as larger and shaded circles together with the hormones produced (lineage-specific transcription factors are highlighted in bold in these cells). The interaction with transcription factors and signaling molecules in the hypothalamus is also noted. Transcription factors are represented in lowercase (except for SFI and GATA2), whereas signaling molecules appear in uppercase. B, Schema showing the timing of the appearance and disappearance of pituitary transcription factors during mouse embryogenesis. BMP4, bone morphogenic protein 4; e, embryonic day; ER, estrogen receptor; FGF8, fibroblast growth factor 8; FSH, follicle-stimulating hormone; GH, growth hormone; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin–releasing hormone; αGSU, α-glycoprotein subunit; LH, luteinizing hormone; POMC, pro-opiomelanocortin; PRL, prolactin; SFI, steroidogenic factor I; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone. (From Lopz-Bermejo, A., Buckway, C. K., & Rosenfeld, R. G. (2000). Genetic defects of the growth hormone–insulin-like growth factor axis. Trends Endocrinol, 11, 43.)
Maintained apposition and interactions between the oral ectoderm and neuroectoderm is critical for normal anterior pituitary development.35–38
Experimental manipulation of embryos from several species as well as Rathke’s pouch explant experiments in rodents have shown that signals from the diencephalon are essential not only for the induction and maintenance of Rathke’s pouch, but also for the regionalization within the pouch that allows the emergence of the different endocrine cell types. During gestation, proliferating progenitor cells are enriched around the pouch lumen, and they appear to delaminate as they exit the cell cycle and differentiate. During late mouse gestation and the postnatal period, anterior lobe progenitors reenter the cell cycle and expand the populations of specialized, hormone-producing cells. At birth, all cell types are present, and their localization appears stratified based on cell type. Current models of cell specification in the anterior lobe suggest that opposing gradients of FGF and BMP signaling pattern the progenitor cells within Rathke’s pouch before they move on to the anterior lobe where they differentiate. Explant studies in the mouse have demonstrated that if Rathke’s pouch is removed from the oral ectoderm on embryonic days 12 to 13 and incubated in appropriate culture medium, differentiation of each of the pituitary cell types continues, indicating that by that point organogenesis of the anterior pituitary is no longer dependent on hypothalamic signals—although such signals may remain critically involved in pituitary hormone production. These and other studies have revealed that normal pituitary development is dependent on a complex cascade of transcription factors and signaling molecules that are expressed in a spatiotemporal manner.
Signaling molecules implicated in pituitary development are either intrinsic emanating from the oral ectoderm such as sonic hedgehog (Shh) or extrinsic from the neuroectoderm such as Nkx2.1, fibroblast growth factors (Fgfs eg Fgf8), and bone morphogenetic factors (Bmps eg. Bmp4) (Figure 10-13). These molecules may activate or repress transcription factors such as Hesx1, Lhx3, and Lhx4. They may also act as morphogens creating the appropriate environment for cell differentiation, thus playing a critical role in cell fate. Such signaling molecules include members of the Shh family, Fgfs, transforming growth factors (Tgfs)/Bmps, Wingless/Wnts, and molecules in the Notch pathway, to mention a few.
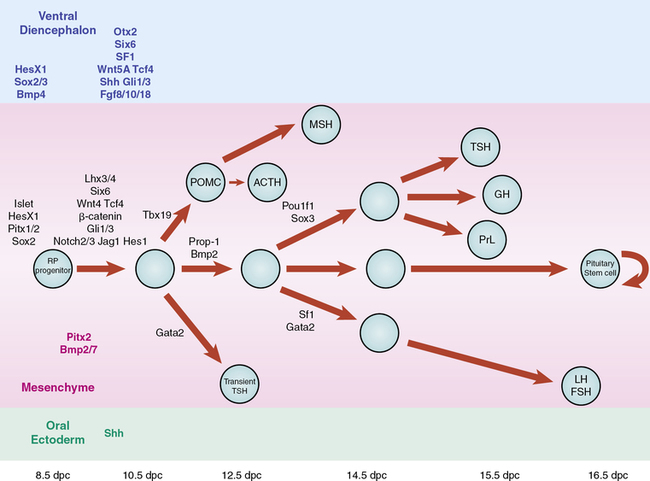
FIGURE 10-13  Schematic representation of the developmental cascade of genes implicated in human pituitary development, with particular reference to pituitary cell differentiation. (From Kelberman, D., Rizzoti, K., Lovell-Badge, R., et al. (2009). Genetic regulation of pituitary gland development in human and mouse. Endocr Rev, 30, 790–829.) This image can be viewed in full color online at ExpertConsult.
Schematic representation of the developmental cascade of genes implicated in human pituitary development, with particular reference to pituitary cell differentiation. (From Kelberman, D., Rizzoti, K., Lovell-Badge, R., et al. (2009). Genetic regulation of pituitary gland development in human and mouse. Endocr Rev, 30, 790–829.) This image can be viewed in full color online at ExpertConsult.
One study has challenged the current dogma of pituitary cell specification, using simple yet elegant experiments.40 The authors showed that, in mice, the pattern of cell specification that results in the rostral location of gonadotropes, the caudal location for somatotropes, and a more intermediate location for corticotropes and thyrotropes does not appear to be the result of an ordered cell cycle exit, as previously predicted. All anterior lobe cell types appear to begin the differentiation process concurrently (E11.5-E14.5), rather than in a temporally discrete manner.
To date, not many pituitary phenotypes have been reported in association with mutations in these signaling molecules. Importantly, the Wnt signaling pathway has recently been implicated in pituitary tumorigenesis. A number of microarray studies have identified altered expression of Wnt inhibitors in pituitary tumors, and there is clear evidence that the Wnt/β catenin pathway is involved in the pathogenesis of craniopharyngioma, a rare tumor in the hypothalamo-pituitary region.41,42
Multiple pituitary-specific transcription factors are involved in the determination of pituitary cell lineages and cell-specific expression of anterior pituitary hormones.30–34,37,38,43 To date, several homeodomain transcription factors have been shown to be involved in human anterior pituitary development and differentiation. Defects in each have now been associated with various combinations of pituitary hormone deficiencies (see Figure 10-12 and Table 10-2). Because additional gene defects have been implicated in abnormal murine hypothalamo-pituitary development, it seems likely that the number of human genetic defects will expand.
TABLE 10-2
Homeodomain Transcription Factors Involved in Human Pituitary Development and Differentiation875
| Transcription Factor | Murine Homologue |
| HESX1 (homeobox gene expression in embryonic stem cells) | Hesx1/Rpx (Rathke’s pouch, Homeobox) |
| PROP1 (prophet of Pit1) | Prop1 (Ames mouse) |
| POU1F1 (POU domain/Pit1) | Pit1/Ghf1 (Snell mouse, Jackson mouse) |
| RIEG (Rieger syndrome) | Rieg/Pitx2 |
| LHX3 (LIM homeodomain protein) | Lhx3 |
In the human adult, the mean pituitary size is 13 by 9 by 6 mm.44 Mean weight is 600 mg, with a range of 400 to 900 mg. Pituitary weight is slightly greater in women than in men and typically increases during puberty and pregnancy.45 In the newborn, pituitary weight averages about 100 mg. Infrequently, the craniopharyngeal canal (marking the embryonic migration of Rathke’s pouch) remains patent and may contain small nests of adenohypophyseal cells—giving rise to a pharyngeal hypophysis that may be capable of hormone synthesis.46
Normally, however, the pituitary resides in the sella turcica immediately above and partially surrounded by the sphenoid bone. The volume of the sella turcica provides a good measure of pituitary size, which may be reduced in the child with pituitary hypoplasia.47 It is important, however, to recognize that considerable variation in pituitary size occurs normally. The pituitary is covered superiorly by the diaphragma sellae, and the optic chiasm is directly above the diaphragma. The anatomic proximity between the optic chiasm and the pituitary is important because hypoplasia of the optic chiasm may occur together with hypothalamic/pituitary dysfunction, as in the condition of septo-optic dysplasia and because pituitary tumors may in turn impact on the optic chiasm leading to visual impairment.48 The patient with congenital blindness or nystagmus should be initially evaluated and then subsequently monitored carefully for hypopituitarism as this can evolve. In addition, suprasellar growth of a pituitary tumor may initially manifest with visual complaints or evidence of progressive impairment of peripheral vision.
The existence of a portal circulatory system within the pituitary is critical to normal pituitary function28,29 (Figure 10-14). The blood supply of the pituitary derives from the superior and inferior hypophyseal arteries, branches of the internal carotid. The anterior and posterior branches of the superior hypophyseal artery may terminate within the infundibulum and the proximal portion of the pituitary stalk. Hypothalamic peptides, produced in neurons that terminate in the infundibulum, enter the primary plexus of the hypophyseal portal circulation and are transported by means of the hypophyseal portal veins to the capillaries of the anterior pituitary. This portal system thus provides a means of communication between the neurons of the hypothalamus and the hormone-producing cells of the anterior pituitary. The blood supply of the neurohypophysis is separate, deriving from the inferior hypophyseal artery. Regulation of the posterior lobe of the pituitary does not involve the hypophyseal portal circulation but, rather, is mediated through direct neural connections.
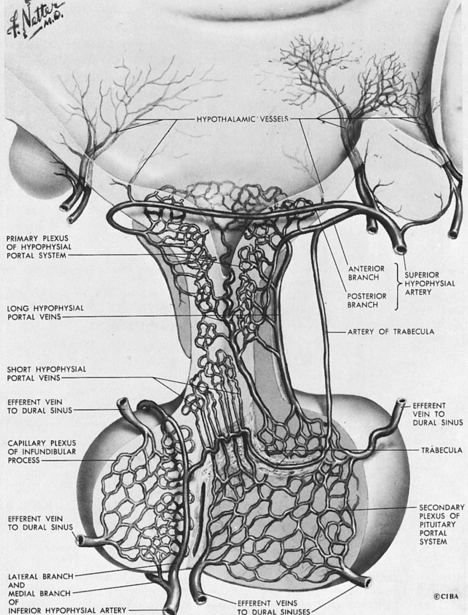
FIGURE 10-14  Illustration of the main components of the hypothalamic-pituitary portal system. (NetterImages.com #4613, © Elsevier, Inc.) See the accompanying website to view a color version of this illustration.
Illustration of the main components of the hypothalamic-pituitary portal system. (NetterImages.com #4613, © Elsevier, Inc.) See the accompanying website to view a color version of this illustration.
The definitive Rathke’s pouch comprises proliferative progenitors that will gradually relocate ventrally, away from the lumen as they differentiate. A proliferative zone containing progenitors is maintained in the embryo in a periluminal area and was found to persist in the adult. The exact nature of progenitor cells in the pituitary gland, however, remains unknown. Research has focused on the biology of these progenitor and putative stem cells within the pituitary gland.49,50 Several reports have suggested that the pituitary stem cells take the form of a SOX2+ cell population, GFRa2+ cells, a side population of cells, Nestin+ cell population, or folliculostellate cells.51 It has been hypothesized that there are two critical roles of stem cells: one is to establish the pituitary gland during development and the other is to maintain the mature pituitary gland in response to physiologic challenges and normal cell turnover. The hypothesis of two different populations of stem cells, one involved in embryogenesis and one involved in maintenance function after birth, remains highly controversial. Members of the Sox family of transcription factors are likely involved in the earliest steps of pituitary stem cell proliferation and the earliest transitions to differentiation. The transcription factor PROP1 and the NOTCH signaling pathway may then regulate the transition to differentiation. Identification of the stem cell niche is important for several reasons, and it has been proposed that the niche may be the marginal zone around the lumen of Rathke’s pouch, between the anterior and intermediate lobes of mouse pituitary, because cells in this region are able to give birth to all five pituitary hormone cell lineages.
Stem cells have been shown to play a role in tumorigenesis in some tissues, and their role in pituitary hyperplasia, pituitary adenomas, and tumors is an important area for future investigation. The ability to cultivate and grow stem cells in a pituitary predifferentiation state might also be helpful for the long-term treatment of pituitary deficiencies. Indeed, a seminal study has resulted in the efficient self-formation of a three-dimensional anterior pituitary tissue in an aggregate culture of mouse embryonic stem (ES) cells.52 ES cells were stimulated to differentiate into non-neural head ectoderm and hypothalamic neuroectoderm in adjacent layers within the aggregate and treated with agonists of sonic hedgehog signaling. This resulted in increased expression of the Rathke’s pouch marker Lhx3 followed by self-organization of Rathke’s pouch–like three-dimensional structures at the interface of these two epithelia. The resulting 3D structures had a central cavity, and the resemblance to Rathke’s pouch was striking, as was the topographic location between the neuroepithelium and the rostral headlike ectoderm. The juxtaposition of the two tissues, mimicking the spatial organization in embryonic development, was indeed critical as Rathke’s pouch–like vesicles did not develop when neuroepithelium tissue was not present. Various endocrine cells were subsequently produced and these cells were able to respond to trophic hormones. For example, adrenocorticotropic hormone (ACTH)–expressing cells developed from the vesicle like structures, and activation of Wnt signaling led to expression of Pit1, growth hormone (GH), and prolactin. Luteinizing hormone (LH)/follicle-stimulating hormone (FSH)/thyroid-stimulating hormone (TSH) expression was also achieved, albeit after more intense manipulation of the culture conditions. When ES-derived cell aggregates were implanted under the kidney capsule in hypophysectomized mice, corticosterone was produced. These studies may therefore reflect the first step toward stem cell treatment.
Terminally differentiated secreting cells are not distributed randomly in a patchwork-like fashion throughout the pituitary gland. Instead, it is starting to emerge that these cells organize themselves in same-cell type networks. This was first shown by Bonnefont and colleagues when GH cells in murine pituitary slices were visualized using high-resolution imaging.53 The connectivity between the cells of this network is important to deliver coordinated secretory pulses of hormones to their target tissues. Additionally, the two least abundant pituitary cell types, corticotropes and gonadotropes, are also organized in homotypic cell networks, as are pituitary lactotropes.54,55 This distribution of cells into networks facilitates the coordinated physiologic response to stimuli.
Growth hormone
Chemistry
Human GH is produced from somatotrope cells within the anterior pituitary as a single-chain nonglycosylated 191-amino-acid 22-kd protein (Figure 10-15)56,57 that comprises a core of four helices in a parallel/antiparallel orientation with two disulfide bonds between cysteines 53 to 165 and 182 to 189.58 The 217aa GH-precursor is transported in the lumen of the endoplasmic reticulum (ER) via a mechanism that involves the recognition of the signal peptide (first 26 aa). Following its cleavage, the mature protein is transported to the Golgi apparatus and secretory vesicles; the presence of zinc ions facilitates the formation of soluble GH dimer complexes within the secretory granules, as well as the storage and secretion of GH aggregates.59GH is homologous with several other proteins produced by the pituitary or placenta, including prolactin, chorionic somatomammotropin (CS, placental lactogen), and a 22-kd GH variant (hGH-V) secreted only by the placenta.60 The latter differs from pituitary GH by 13 amino acids. The genes for these proteins have probably descended from a common ancestral gene, even though the genes are now located on different chromosomes (chromosome 6 for prolactin and chromosome 17 for GH).61
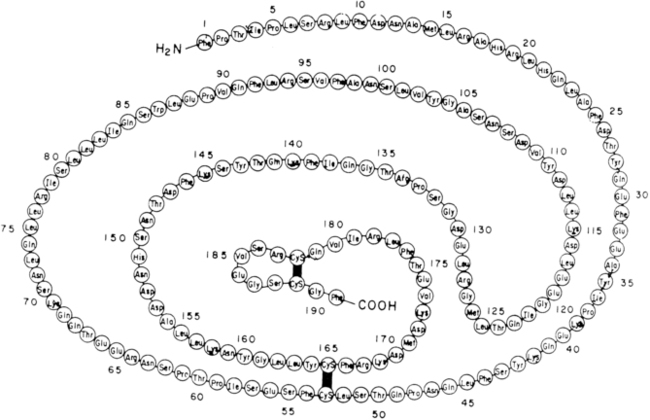
FIGURE 10-15  Covalent structure of human growth hormone. (From Chawla, R. K., Parks, J. S., & Rudman, D. (1983). Structural variants of human growth hormone: biochemical, genetic and clinical aspects. Annu Rev Med, 34, 519.)
Covalent structure of human growth hormone. (From Chawla, R. K., Parks, J. S., & Rudman, D. (1983). Structural variants of human growth hormone: biochemical, genetic and clinical aspects. Annu Rev Med, 34, 519.)
The genes for GH, prolactin, and placental lactogen share a common structural organization—with four introns separating five exons. GH1 is located on the long arm of chromosome 17 (17q22-24) within a cluster of five homologous genes encompassing a distance of about 65kb—CSHP (chorionic somatomammotropin pseudogene), CSH-1 (chorionic somatomammotropin gene), GH-2, and CSH-2.62 Expression of GH1 is regulated by the highly polymorphic proximal promoter and a locus control region (LCR) 15-32kb upstream of the gene that confers the pituitary-specific and high-level expression of GH.63 Normally, the majority of GH (75%) produced by the pituitary is of the mature 22-kd form. Alternative splicing of the second exon results in deletion of amino acids 32 through 46, yielding a 20-kd form that normally accounts for less than 10% of pituitary GH.61,64,65 The remainder of pituitary GH includes desamidated and N-acetylated forms, as well as various GH oligomers. A 17.5-kDa variant that results from complete skipping of exon 3 and lacks amino acids 32-71 is much less abundant (1% to 5%).
Secretion
The pulsatile pattern characteristic of GH secretion largely reflects the interplay of multiple regulators, including two hypothalamic regulatory peptides: GH-releasing hormone (GHRH)66,67 and somatostatin (somatotropin release–inhibiting factor [SRIF]).68 The amino terminus of the 44-amino-acid protein GHRH is required for stimulation of GH secretion. GHRH activity is species specific, presumably reflecting the specificity of binding to a G protein–coupled receptor on the pituitary somatotropes.
Regulation of GH production by GHRH is largely transcriptionally mediated and is dependent on stimulation of adenylate cyclase and increases in intracellular cyclic adenosine monophosphate (AMP) concentrations. The GHRH receptor is a member of the G protein–coupled receptor family B (also called the secretin family) and has partial sequence identity with receptors for vasoactive intestinal polypeptide, secretin, calcitonin, and parathyroid hormone.69 Solid tumors secreting GHRH are a rare cause of GH excess. GHRH has previously been approved in the United States for treatment of growth hormone deficiency but has been withdrawn from the market for therapeutic purposes. It can still be used as a diagnostic agent—if available, especially for the identification of adult growth hormone deficiency, and then it is frequently used in combination with arginine as part of a stimulation testing protocol.
The actions of the 14-amino-acid protein somatostatin appear to be related to the timing and amplitude of pulsatile GH secretion, rather than to GH synthesis. The binding of somatostatin to its specific receptor results in an inhibition of adenylate cyclase activity and a reduction in intracellular calcium concentrations.68 Treatment of cultured somatotropic cells with GHRH and somatostatin has indicated a dominant effect of somatostatin, with a reduction of intracellular calcium concentrations and an inhibition of GH secretion. The pulsatile secretion of GH observed in vivo is believed to result from a simultaneous reduction in hypothalamic somatostatin release and increase in GHRH activity.70 Conversely, a trough of GH secretion occurs when somatostatin release is increased in the presence of diminished GHRH activity. Somatostatin analogs are used therapeutically for the treatment of acromegaly, underscoring its role as a GH secretion inhibitor.
The regulation, on a neuronal basis, of this reciprocal secretion of GHRH and somatostatin is imperfectly understood. Multiple neurotransmitters and neuropeptides are involved in regulation of release of these hypothalamic factors, including serotonin, histamine, norepinephrine, dopamine, acetylcholine, gamma-aminobutyric acid (GABA), thyroid-releasing hormone, vasoactive intestinal peptide, gastrin, neurotensin, substance P, calcitonin, neuropeptide Y, vasopressin, corticotrophin releasing hormone,71 and galanin.72 These factors are clearly implicated in the alterations of GH secretion observed in a wide variety of physiologic states (such as stress, sleep, hemorrhage, fasting, hypoglycemia, and exercise) and form the basis for a number of GH-stimulatory tests employed in the evaluation of GH secretory capacity/reserve.
GH secretion is also impacted by a variety of nonpeptide hormones, including androgens,73,74 estrogens,75 thyroxine,76 and glucocorticoids.77,78 The precise mechanisms by which these hormones regulate GH secretion are complex, potentially involving actions at the hypothalamic and pituitary levels. Practically speaking, hypothyroidism and glucocorticoid excess may each blunt spontaneous and provocative GH secretion (and therefore should be corrected prior to GH testing). Sex steroids, at the onset of puberty or administered pharmacologically, appear to be responsible for the rise in GH secretion characteristic of puberty.79
Synthetic hexapeptides capable of stimulating GH secretion have been developed80 and termed GH-releasing peptides (GHRPs). These peptides, later recognized as analogs of the gastric hormone ghrelin, are capable of directly stimulating GH release and enhancing the GH response to GHRH.81 These agents have the potential advantage of oral administration, and in the patient with an intact pituitary they may be capable of greatly enhancing GH secretion. When these agents were administered chronically to elderly patients and to some GH-deficient children, the amplitudes of GH pulses were significantly increased. Ghrelin-mimetic ligands were used to characterize a common receptor termed the GH secretagogue receptor (GHS-R) for the GH-releasing substances. The GHS-R is distinct from the GHRH receptor.82
Subsequently, the GHS-R gene was cloned and shown to encode a unique G protein–coupled receptor with a deduced protein sequence that was 96% identical in human and rat. Three receptor isotypes were isolated from human genomic libraries. The receptor is strongly expressed in the hypothalamus. Specific binding sites for GH releasing peptides have also been identified in other regions of the central nervous system (CNS) and peripheral endocrine and nonendocrine tissues in both humans and other organisms. Ghrelin is a 28-amino-acid peptide that has been identified as the endogenous ligand for the growth hormone secretagogue receptor (GHS-R).83 It is expressed predominantly in the stomach, but smaller amounts are also produced within the bowel, pancreas, kidney, the immune system, placenta, pituitary, testis, ovary, and hypothalamus. Ghrelin is a unique gene product that requires octanoylation for normal function. Intravenous, intracerebroventricular, and intraperitoneal administration of ghrelin in animal models stimulates food intake and obesity84 and raises plasma GH concentrations85—and to a lesser extent prolactin and adrenocorticotropic hormone (ACTH) concentrations. Additionally, it influences endocrine pancreatic function and glucose metabolism, gonadal function, and behavior. It also controls gastric motility and acid secretion and has cardiovascular and antiproliferative effects. Both ghrelin and GH-releasing peptides release GH synergistically with GHRH, but the efficacy of these compounds as growth-promoting agents is poor. Mutations in the ghrelin receptor have been identified as a possible cause of idiopathic short stature (ISS) and GH deficiency.86 However, it is important to note that mouse models with targeted deletion of the receptor (ghsr -/-) have a near normal phenotype.87
This suggests that ghrelin is an important stimulus for nutrient allocation for growth and metabolism and that it may represent a key component of the GH regulatory system. A second peptide encoded by the same gene as ghrelin has been identified and termed obestatin. This appears to regulate weight but not GH secretion.88,89
In addition to the complex regulatory processes described previously, the synthesis and secretion of GH are also regulated by feedback by the insulin-like growth factor (IGF) peptides.90–95 IGF receptors have been identified in the pituitary.96–98 Inhibition by IGF-1 of GH secretion has been demonstrated in multiple systems.99 In addition, inhibition of spontaneous GH secretion has been demonstrated in humans treated with subcutaneous injections of recombinant IGF-1.100,101 GH can be identified in fetal serum by the end of the first trimester. Serum concentrations are lower in term infants than in premature infants, perhaps reflecting feedback by the higher serum levels of IGF peptides characteristic of the later stages of gestation.102 However, at birth, GH concentration in normal newborns is of the order of 40 ng/mL, declining gradually in the initial weeks of life to levels less than 10 ng/mL.
Twenty-four hour GH secretion peaks during adolescence,79 undoubtedly contributing to the very high serum concentrations of IGF-1 characteristic of puberty. GH secretion begins to decline by late adolescence and continues to fall throughout adult life. Indeed, puberty may be considered with some justification a period of “acromegaly,” whereas aging (with its characteristic decrease of GH secretion) has been termed the somatopause.75,103,104
Twenty-four-hour GH production rates for normal men range from 0.25 to 0.52 mg/m2.78,105 However, a wide variety of physiologic conditions (in addition to aging) affect GH secretion. These include stage of sleep,106,107 nutritional status,108 acute fasting, exercise,109 stress,109 and sex steroids.73,74 Ho and associates75 have reported that serum estradiol concentrations are the dominant factor affecting GH secretion. Neither age nor sex influenced the integrated serum concentrations of GH when the effects of estradiol were removed from analysis. The effects of testosterone on serum IGF-1 concentrations may be at least in part independent of GH because individuals with mutations of the GH receptor (GHR) still experience a rise in serum IGF-1 during puberty.110
The pulsatile nature of GH secretion is readily demonstrable by frequent serum sampling, especially when coupled with sensitive assays for GH.108 Such assays demonstrate that under normal conditions serum GH concentrations are less than 0.2 ng/mL between bursts of GH secretion. It is consequently impractical to assess GH secretion by random serum sampling. Maximal GH secretion occurs during the night, especially at the onset of the first slow-wave sleep (stages III and IV). Rapid-eye-movement (REM) sleep is, on the other hand, associated with low GH secretion.
Normal young men generally experience 12 GH secretory bursts per 24 hours. Obesity is characterized by decreased GH secretion, reflected by a decreased number of GH secretory bursts.111 Fasting increases the number and amplitude of GH secretory bursts, presumably reflecting decreased somatostatin secretion and possibly increased ghrelin secretion. The impact of the pulsatile secretory nature of GH secretion on its biologic actions remains uncertain.
GH receptor/GH-binding protein
The gene for the human GHR has been localized to chromosome 5p13.1-p12, where it spans more than 87 kb.112,113 In the mouse114,115 and rat,116 on the other hand, multiple transcripts for the GHR have been identified. The larger (3.4 to 4.8 kb) transcript codes for the intact receptor, whereas the 1.2- to 1.9-kb transcript codes for the soluble GHBP. The coding and 3′ untranslated regions of the human GHR are encoded by the nine exons, numbered 2 through 10.117 Exon 2 corresponds to the secretion signal peptide, whereas exons 3 through 7 encode the extracellular domain. Exon 8 encodes the transmembrane domain. Exons 9 and 10 encode, respectively, the intracellular domain and the 3′ untranslated region. Two genomic GHR isoforms that exist only in humans have arisen from ancestral homologous recombination. They differ in the retention or deletion of exon 3. Exon 3 of the GHR has been shown to be deleted in a substantial number of normal individuals. This delta-3 GHR polymorphism has been shown by some but not all investigators to determine responsiveness to GH and to be associated with birth size and postnatal growth.118–120
The GHR has been found to be highly homologous with the prolactin receptor and to share sequence homology with many of the receptors for interleukins, as well as receptors for erythropoietin, leptin, granulocyte-macrophage colony-stimulating factor, and interferon.117 The GHR is a member of the class 1 hematopoietic cytokine family. A complex of the GH and the GHBP molecule has been shown to be more effective as an agonist of GH action than GH alone—indicating a physiologic and possible therapeutic role for the GHBP.121 Examination of the crystal structure of the GH-GHR complex revealed that the complex consisted of one molecule of GH bound to two GHR molecules, indicating a GH-induced receptor dimerization—which is necessary in GH action.58 Interestingly, as noted previously, a genetically engineered fusion complex of GH and the GHR has been shown to have a significantly improved efficacy and a dramatically longer half-life compared to growth hormone alone when tested in rodent models.121
The GHR, like its family group member EPO-R, is preformed as a dimer and is transported in a nonligand bound state to the cell surface.122,123 GH then binds in a sequential manner to the GHR dimer where the first GHR binds to the stronger site 1 of the GH molecule followed by the second GHR binding to the weaker site 2. Binding of GH results in a conformational change whereby rotation of the GHRs results in repositioning of the intracellular domains and of Box1-associated Janus Kinase 2 (JAK2), a major GHR-associated tyrosine kinase. As a result, JAK2 is auto-phosphorylated and activated, leading in turn to cross phosphorylation of distal tyrosine residues of GHR that enables SH2 (Src homology 2) domain molecules to dock to these sites.124,125 The GHR itself appears to have no intrinsic kinase activity. It is likely that co-localization of two JAK2 molecules by the dimerized GHR results in transphosphorylation of one JAK2 by the other, leading to JAK2 activation. Stat5a and Stat5b molecules contain SH2 domains and bind to these phosphorylated tyrosine sites. In turn they then become phosphorylated. Phosphorylated Stat5 molecules (homo- and hetero-) dimerize and translocate to the nucleus where they bind DNA, as dimers or as tetramers, and activate target genes.126,127
GH can activate both Stat5a and Stat5b, and they have both overlapping and distinct functions.128–135 Gene inactivation mouse models have shown that deletion of Stat5b, but not of other Stat genes, even though GH also activates Stat1 and Stat3, affects growth, and that Stat5b is of greater importance for stimulation of growth than Stat5a. Stat5b null mice have severe postnatal growth retardation especially in males, although this is not as severe as in Ghr null mice. They have increased GH secretion; reduced hepatic IGF1-, IGF-binding protein (IGFBP)3-, and acid labile subunit (ALS)-expression; and increased obesity.135 Stat5a null mice have normal growth but impaired mammary gland formation and lactogenesis, reflecting impaired signaling of prolactin.130 Stat5a/b double null mice are more severely affected than the single null mice and display more severe growth retardation, although Ghr null mice are still more severely affected.136,137 Stat5a/b double null mice also have a severe combined immunodeficiency, with reduced CD8 T cell number and a failure of hematopoietic stem cells to develop lymphoid lineages.138 Transgenic Stat5 expression results in expansion of CD8 cells and lymphomagenesis139 and also in increased proliferation and differentiation in mammary cells,140 suggesting a critical role for Stat5 in cell proliferation, particularly in immune cells. Stat5b is phosphorylated upon stimulation by a pulse of GH, and after this rapid activation it becomes temporarily refractive to further or continuous stimulation.141,142 GH secretion is more continuous in females and, indeed, in female rodents, Stat5b is phosphorylated to a lesser extent although phosphorylation still occurs. This gender-specific signaling plays an important role in the regulation of gender-specific proteins, especially CYP450 enzymes,143,144 which play a role in hepatic metabolism of steroids and foreign compounds. The Stat5b null male mice are resistant to GH pulses,145and their hepatic male-specific genes are decreased to female levels, whereas female predominant genes are expressed at higher levels than in WT males. Hepatic nucleic factors (HNFs), especially HNF3, 4α, and 6, interact with Stat5b to induce these Stat5b-dependent gender-specific gene expression patterns.134,144,146
Phosphorylated Stat5a and Stat5b bind Stat5 response elements (Stat5 RE) as dimers or tetramers, but their binding is enhanced by the interaction of coactivators binding to adjacent DNA binding sites. Stat5 RE are located in the second and third intron of the human IGF1 gene and 73 kb upstream of the initiation site, although the effectiveness of the distant site is much less.147,148 Activation of JAK-STAT signaling occurs rapidly, within minutes after GH stimulation, but is transient due to the tight control of the termination of signaling. This negative regulation of signaling occurs at several levels: GHR internalization, suppressors of cytokine signaling (SOCS), protein tyrosine phosphatases (PTPs), and protein inhibitors of activated stats (PIAS). The inhibition of GH signaling by several members of the GH-inducible suppressors of the cytokine signaling (SOCS) family has been demonstrated. The SOCS family of proteins comprises cytokine inducible-Src homology 2 protein (CIS) and SOCS 1-7. GH, PRL, and many other cytokines can induce CIS, SOCS1, -2, and -3. SOCS family proteins inhibit JAK-STAT signaling by inhibiting JAK proteins, binding positive regulators of signaling or docking sites, and promoting GHR ubiquitination. The role of the several family members has become clearer from phenotypes associated with their overexpression. Mice overexpressing CIS have mild growth retardation and have also altered T-cell function and impaired mammary gland development, whereas Socs2 null mice show gigantism similar to bovine GH overexpessing mice (30% to 40% overgrowth), are hyperresponsive to GH, and have increased extrahepatic IGF-1 production.149
All of these agents can also induce SOCS proteins.150 SOCS-3 induced by IL-1b and TNFα or by endotoxin in vivo may play a role in the GH resistance induced by sepsis.151 Critically ill patients with septic shock who had been treated with GH were shown to have increased mortality, possibly related to the induction of GH resistance in specific tissues as a consequence of sepsis.146 Thus, the role of SOCS proteins as intracellular GH signaling antagonists appears to be important in a variety of pathophysiologic states.
PTPs dephosphorylate activated phosphorylated proteins in cytokine signaling pathways but also insulin signaling pathways. Several PTPs—such as PTP-1, PTP-H1 and PTP-B1, and TC-PTP—attenuate GH signaling, and PTP-1B is involved in fasting-induced GH insensitivity.153,154 PTPN11 mainly functions in RAS-MAPK signaling, and abnormalities result not only in Noonan syndrome but also in associated mild GH resistance.155
Other GH-activated pathways include mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase (ERK)-1 and ERK2, the insulin-signaling pathway (by means of insulin receptor substrate [IRS]-1 and IRS-2), and protein kinase C (PKC).156,157 How all of these pathways interact to mediate the various anabolic and metabolic actions of GH remains to be elucidated (Figure 10-16).
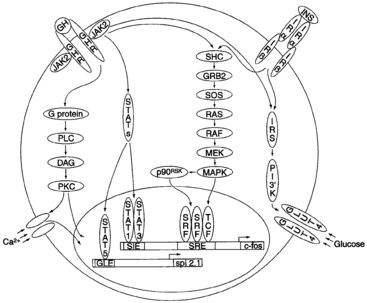
FIGURE 10-16  A model depicting intracellular signaling intermediates induced by binding of growth hormone (GH) with the GH receptor (GHR). DAG, diacylglycerol; ERK, extracellular signal-regulated kinase; GLE, interferon-γ–activated sequence (GAS)–like response element; GLUT, glucose transporter; GRB, growth factor receptor–binding protein; JAK, janus kinase; INS, insulin; Irα, insulin receptor α subunit; MAPK, mitogen-activated protein kinase; MEK, MAPK-ERK kinase; PKC, protein kinase C; PLC, phospholipase C; SHC, SrC homology complex; SIE, sis-inducible element; SOS, son of sevenless; SRE, serum response element; SRF, serum response factor; STAT, signal transducer and activator of transcription; TCF, ternary complex factor. (From Kopchick, J. J., Bellush, L. L., & Coschigano, K. T. (1999). Transgenic models of growth hormone action. Annu Rev Nutr, 19, 437. With permission, from the Annual Review of Nutrition, Volume 19 © 1999 by Annual Reviews. www.AnnualReviews.org.)
A model depicting intracellular signaling intermediates induced by binding of growth hormone (GH) with the GH receptor (GHR). DAG, diacylglycerol; ERK, extracellular signal-regulated kinase; GLE, interferon-γ–activated sequence (GAS)–like response element; GLUT, glucose transporter; GRB, growth factor receptor–binding protein; JAK, janus kinase; INS, insulin; Irα, insulin receptor α subunit; MAPK, mitogen-activated protein kinase; MEK, MAPK-ERK kinase; PKC, protein kinase C; PLC, phospholipase C; SHC, SrC homology complex; SIE, sis-inducible element; SOS, son of sevenless; SRE, serum response element; SRF, serum response factor; STAT, signal transducer and activator of transcription; TCF, ternary complex factor. (From Kopchick, J. J., Bellush, L. L., & Coschigano, K. T. (1999). Transgenic models of growth hormone action. Annu Rev Nutr, 19, 437. With permission, from the Annual Review of Nutrition, Volume 19 © 1999 by Annual Reviews. www.AnnualReviews.org.)
The main GH binding protein (GHBP) is the soluble extracellular domain of the GHR with an apparent molecular weight of approximately 55 kDa and has an identical affinity for GH as GHR.112 It binds to GH with high specificity and affinity, but with relatively low capacity.158–160 It increases its half-life in the circulation and may serve a function in the transportation of GH to target tissues and subsequent binding to the receptor. Although in rodents GHBP arises through alternative splicing, human GHBP arises from proteolytic cleavage of the cell membrane anchored GHR, for example, by TNF α converting enzyme (TACE).161 GHBP is, like GHR, present in many tissues, but GHBP in the circulation is mostly derived from the liver. Although GHR and GHBP are regulated by and are very sensitive to GH, and GHR and GHBP often change in parallel,162–164 measurement of plasma GHBP has not been shown to reflect GHR and GH responsiveness.165 Initial assays for GHBP involved incubation of serum with 125-I-GH and separation of bound from free radioligand by gel filtration, high-pressure liquid chromatography, or dextran-coated charcoal. Carlsson and colleagues166 developed a ligand-mediated immunofunctional assay (LIFA) that measures GHBP capable of binding GH. Assays of serum concentrations of GHBP have been instrumental in identifying patients with GH insensitivity (GHI) caused by genetic abnormalities of the GHR.167,168 Patients with GHI from nonreceptor abnormalities, abnormalities of the intracellular portion of the GHR, or inability of the receptor to dimerize may, however, have normal serum concentrations of GHBP.169
GH actions
According to the somatomedin hypothesis, the anabolic actions of GH are mediated through the IGF peptides.170–175 Although this hypothesis is at least in part true, it appears that GH is capable of stimulating a variety of effects that are independent of IGF activity. Indeed, the effects of GH and IGF are on occasion contradictory—as evident in the “diabetogenic” actions of GH175,176 and the glucose-lowering activity of IGFs. Green and colleagues177 attempted to resolve some of these differences in a “dual-effector” model in which GH stimulates precursor cells, such as prechondrocytes, to differentiate.
When differentiated cells or neighboring cells then secrete IGFs, these peptides act as mitogens and stimulate clonal expansion. This hypothesis is based on the ability of IGF peptides to work not only as classic endocrine factors that are transported through the blood but as paracrine or autocrine growth factors. GH also stimulates a variety of metabolic effects, some of which appear to occur independently of IGF production—such as lipolysis,178 amino acid transport in diaphragm179 and heart,180 and production of specific hepatic proteins. Thus, there are multiple sites of GH action—and frequently it is not entirely clear which of these actions are mediated through the IGF system and which might represent IGF-independent effects of GH.181 The sites of action of GH include the following:
• Epiphysis: Stimulation of epiphyseal growth.
• Bone: Stimulation of osteoclast differentiation and activity, stimulation of osteoblast activity, and increase of bone mass by endochondral bone formation. Lupu and coworkers182 have shown that mice with a complete knockout of IGF-I (making them substantially smaller than wild-type mice) become even smaller when mated into a GH receptor knockout strain, indicating a direct IGF-independent effect of GH on growth.
• Adipose tissue: Acute insulin-like effects, followed by increased lipolysis, inhibition of lipoprotein lipase, stimulation of hormone sensitive lipase, decreased glucose transport, and decreased lipogenesis.183
• Muscle: Increased amino acid transport, increased nitrogen retention, increased lean tissue, and increased energy expenditure.183
The concept of IGF-independent actions of GH is supported by in vivo studies, in which IGF-1 cannot duplicate all of the effects of GH (such as nitrogen retention and insulin resistance). The effects of GH in normal human aging184 and in catabolic conditions185 are subjects of active investigation.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


