Disorders of calcium and phosphorus homeostasis in the newborn and infant
Calcium (Ca), phosphorus (as phosphate [HPO42−], as phosphorus does not exist in the free state in living tissues), and magnesium (Mg) are essential nutrients that are indispensable for the structural integrity of the body and for the function of each of its cells.1,2 The genetic and physiologic mechanisms that regulate normal mineral homeostasis and bone development, composition, and strength from the prenatal period through adolescence are complex. Table 8-2 lists some of the many genes that direct these processes. Figures 8-1 and 8-2 schematically depict the factors that regulate serum concentrations of calcium and phosphate, respectively.
TABLE 8-2
Human Genes Involved in Mineral Homeostasis and Bone Metabolism
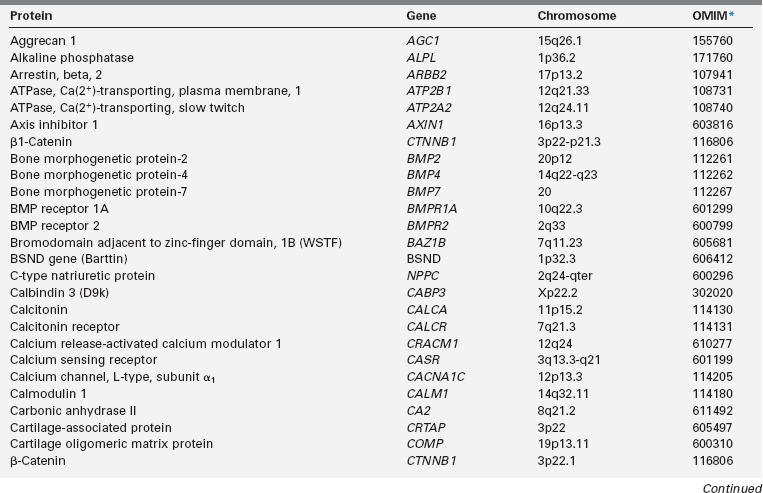
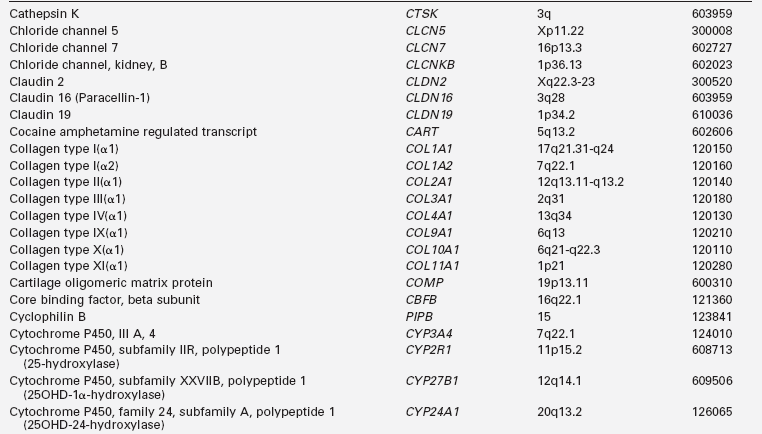

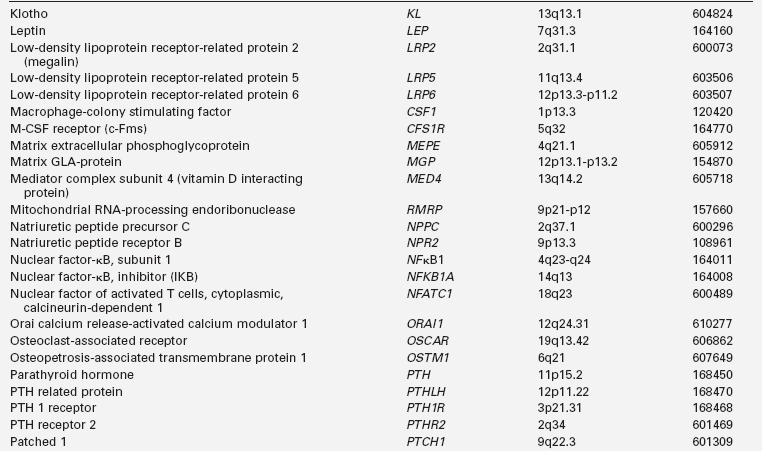
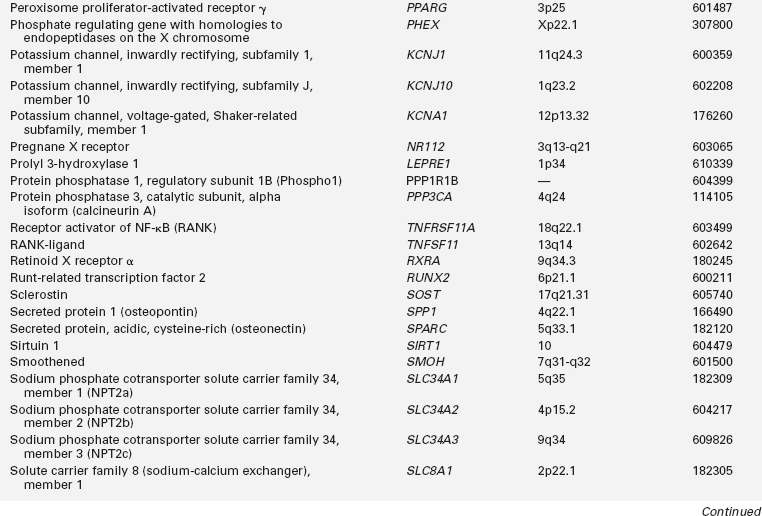
*Online Mendelian Inheritance in Man, www3.ncbi.nlm.nih.gov/htbin-post/Omim.
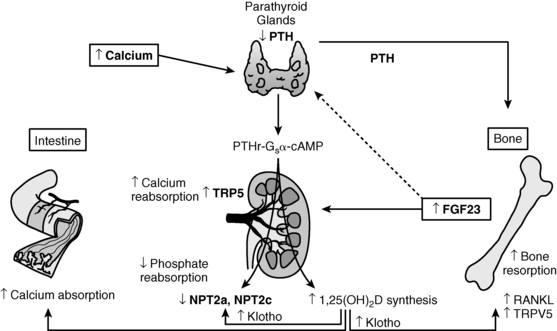
FIGURE 8-1  Regulation of calcium homeostasis. Calcium ( Ca2+) is absorbed from the intestinal tract, kidney tubule, and bone in response to calcitriol [1,25(OH)2D3] and parathyroid hormone (PTH). Calcitonin inhibits resorption of calcium from bone. The Ca2+-sensing receptor (CaSR) modulates Ca2+-mediated activity of the parathyroid glands and the renal tubules. Both hypocalcemia and hypophosphatemia enhance renal tubular generation of calcitriol and absorption of intestinal phosphate. PTH inhibits renal tubular reabsorption of phosphate. Fibroblast growth factor-23 (FGF23), a phosphatonin secreted by osteoblasts and osteocytes, inhibits renal tubular reabsorption of phosphate and synthesis of calcitriol. (Please see Figure 8-2 and the text for further details.) (Reproduced from Levine, M. A. (2010). Investigation & management of hypocalcemia. ENDO 2010: Meet the Professor, The Endocrine Society, Bethesda, MD, with permission.)
Regulation of calcium homeostasis. Calcium ( Ca2+) is absorbed from the intestinal tract, kidney tubule, and bone in response to calcitriol [1,25(OH)2D3] and parathyroid hormone (PTH). Calcitonin inhibits resorption of calcium from bone. The Ca2+-sensing receptor (CaSR) modulates Ca2+-mediated activity of the parathyroid glands and the renal tubules. Both hypocalcemia and hypophosphatemia enhance renal tubular generation of calcitriol and absorption of intestinal phosphate. PTH inhibits renal tubular reabsorption of phosphate. Fibroblast growth factor-23 (FGF23), a phosphatonin secreted by osteoblasts and osteocytes, inhibits renal tubular reabsorption of phosphate and synthesis of calcitriol. (Please see Figure 8-2 and the text for further details.) (Reproduced from Levine, M. A. (2010). Investigation & management of hypocalcemia. ENDO 2010: Meet the Professor, The Endocrine Society, Bethesda, MD, with permission.)
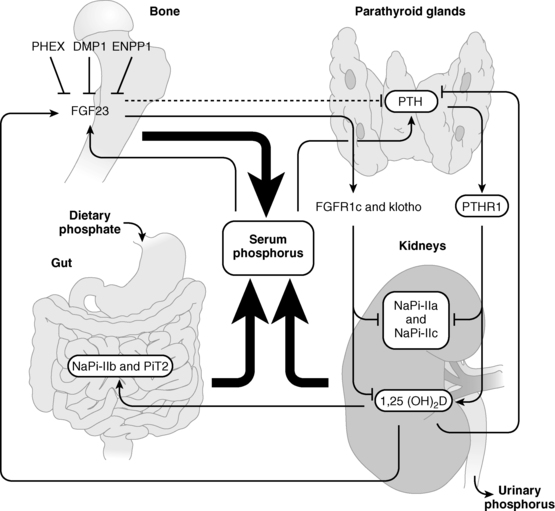
FIGURE 8-2  Regulation of phosphate homeostasis. Serum concentrations of phosphate are depressed by parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23), both of which increase urinary excretion of this cation by decreasing renal tubular expression of phosphate cotransporter proteins. Synthesis of FGF23, a product of osteoblasts and osteocytes, is depressed by low serum phosphate levels and by PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome), DMP1 (dentin matrix protein 1), and ENPP1 (ectonucleotide pyrophosphate-phosphodiesterase 1), all three of which proteins are also synthesized by bone cells. PTH increases and FGF23 depresses synthesis of calcitriol (1,25-dihydroxyvitamin D). (See text for details.) (Reproduced from Bergwitz, C., Collins, M. T., Kamath, R. S., & Rosenberg, A. E. (2011). Case 33-2011: a 56-year-old man with hypophosphatemia. N Engl J Med, 365, 1625–1635, with permission.)
Regulation of phosphate homeostasis. Serum concentrations of phosphate are depressed by parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23), both of which increase urinary excretion of this cation by decreasing renal tubular expression of phosphate cotransporter proteins. Synthesis of FGF23, a product of osteoblasts and osteocytes, is depressed by low serum phosphate levels and by PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome), DMP1 (dentin matrix protein 1), and ENPP1 (ectonucleotide pyrophosphate-phosphodiesterase 1), all three of which proteins are also synthesized by bone cells. PTH increases and FGF23 depresses synthesis of calcitriol (1,25-dihydroxyvitamin D). (See text for details.) (Reproduced from Bergwitz, C., Collins, M. T., Kamath, R. S., & Rosenberg, A. E. (2011). Case 33-2011: a 56-year-old man with hypophosphatemia. N Engl J Med, 365, 1625–1635, with permission.)
TABLE 8-1
| 1,25(OH)2D3 | 1,25-Dihydroxyvitamin D3 (calcitriol) |
| 24R,25(OH)2D3 | 24,25-Dihydroxyvitamin D3 |
| 25OHD3 | 25-Hydroxyvitamin D3 (calcidiol) |
| ADHR | Autosomal dominant hypophosphatemic rickets |
| AF | Activating function |
| AKT | see PKB |
| AMP | Adenosine monophosphate |
| APC | Adenomatous polyposis coli |
| ARHR | Autosomal recessive hypophosphatemic rickets |
| ASARM | Acidic serine aspartate-rich MEPE-associated motif |
| ATP | Adenosine triphosphate |
| ATPase | Adenosine triphosphatase |
| BAP | Bone alkaline phosphatase |
| BMAD | Bone mineral apparent density (∼volumetric BMD) |
| BMC | Bone mineral content |
| BMD | Bone mineral density |
| BMP | Bone morphogenetic protein |
| BRU | Bone remodeling unit |
| BTT | Bone transmission time |
| Ca2+e | Calcium—ionized, extracellular |
| Ca2+i | Calcium—ionized, intracellular |
| CaMK | Ca2+/calmodulin dependent protein kinase |
| CART | Cocaine amphetamine regulated transcript |
| CaSR | Calcium sensing receptor |
| Cl− | Chloride |
| CNP | Natriuretic hormone type C |
| COMP | Cartilage oligomeric protein |
| CREB | Cyclic AMP-responsive element binding protein |
| CTX | Carboxyl-terminal cross-link telopeptide α1 chain of type I collagen (see ICTP) |
| DAG | 1,2-Diacylglycerol |
| DBP | Vitamin D binding protein |
| DEXA | Dual energy x-ray absorptiometry |
| DMP | Dentin matrix protein |
| DNA | Deoxyribonucleic acid |
| DPD | Deoxypyridinoline |
| DRIP | Vitamin D interacting protein |
| ECF | Extracellular fluid |
| EGF | Epidermal growth factor |
| ER | Estrogen receptor |
| ERK | Extracellular signal-regulated kinase |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FOS | FBJ osteosarcoma oncogene |
| FRP4 | Frizzled related protein-4 |
| FRS | FGF receptor substrate |
| GABA | Gamma (γ)-aminobutyric acid |
| GDNF | Glial cell line-derived neurotrophic factor |
| Giα | Alpha subunit of inhibitory GTP-binding protein |
| Gsα | Alpha subunit of stimulatory GTP-binding protein |
| Gqα | Alpha subunit of a stimulatory GTP-binding protein |
| GH | Growth hormone |
| GMP | Guanosine monophosphate |
| GPCR | G protein–coupled receptor |
| GRB2 | GRB2-associated binding protein 2 |
| GRK | G protein–coupled receptor kinase |
| GSK3 | Glycogen synthase kinase 3 |
| GTP | Guanosine triphosphate |
| H+ | Hydrogen ion (proton) |
| HNRPD | Heterogeneous nuclear ribnucleoprotein D |
| HRpQCT | High-resolution peripheral quantitative computed tomography |
| IGF | Insulin-like growth factor |
| ICTP | Carboxyl terminal cross-link telopeptide of collagen type I generated by matrix metalloproteinase (see CTX) |
| IGF | Insulin-like growth factor (somatomedin) |
| Ihh | Indian hedgehog |
| IL | Interleukin |
| IP3 | Inositol-1,4,5-trisphosphate |
| ITAM | Immunoreceptor tyrosine-based motif |
| K+ | Potassium |
| LRP | Low-density lipoprotein receptor-related protein |
| μFE | Microfinite element |
| MAPK | Mitogen activated protein kinase |
| MARRS | Membrane-associated rapid response steroid binding protein (see TBP-2) |
| M-CSF | Macrophage colony stimulating factor |
| MEPE | Matrix extracellular phosphoglycoprotein |
| Mg2+ | Magnesium |
| MITF | Microphthalmia-associated transcription factor |
| MMP | Matrix metalloproteinase |
| MRI | Magnetic resonance imaging |
| Na+ | Sodium |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NFκB | Nuclear factor-κB |
| NFATC1 | Nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1 |
| NHERF | Sodium-hydrogen exchanger regulatory factor |
| NPT2 | Sodium/phosphate transporter 2 |
| NTX | Amino terminal cross-link telopeptide of collagen type I |
| OMIM | Online Mendelian Inheritance in Man |
| OPG | Osteoprotegerin |
| OSCAR | Osteoclast-associated receptor |
| OSTM1 | Osteopetrosis-associated transmembrane protein 1 |
| P3H1 | Prolyl 3-hydroxylase-1 |
| PHEX | Phosphate-regulating gene with homologies to endopeptidases on the X chromosome |
| PICP | Procollagen type I carboxyl-terminal propeptide |
| PINP | Procollagen type I amino-terminal propeptide |
| PIIINP | Procollagen type III amino-terminal propeptide |
| PI3K | Phosphoinositide-3-kinase |
| PKA | Protein kinase A |
| PKB | Protein kinase B (AKT) |
| PLC | Phospholipase C |
| PO4 | Phosphate (HPO42−) |
| PPARγ | Peroxisome proliferator activator receptor gamma |
| PTH | Parathyroid hormone |
| PTHrP | PTH-related protein |
| PTH1R | PTH/PTHrP-1 receptor (PTH/PTHrP-1R) |
| PYD | Pyridinoline |
| QCT | Quantitative computed tomography |
| QUS | Quantitative ultrasonography |
| RANK | Receptor activator of NFκB |
| RANKL | RANK-ligand |
| RDA | Recommended dietary allowance |
| RNA | Ribonucleic acid |
| RXR | Retinoid X receptor |
| SIBLING | Small integrin-binding ligand, N-glycosylated (proteins) |
| SOS | Speed of sound |
| SOX | SRY-Box |
| SPP1 | Secreted phosphoprotein1 (osteopontin) |
| Src | SRC oncogene |
| SRC | Steroid receptor coactivator |
| STAT | Signal transduction and transcription |
| SYK | Spleen tyrosine kinase |
| TALH | Thick ascending loop of Henle |
| TBP-2 | Thioredoxin binding protein-2 (see MARRS) |
| TCF/LEF | T cell factor/lymphoid enhancer binding factor |
| TGF | Transforming growth factor |
| TALH | Thick ascending loop of Henle |
| TIP | Tuberoinfundibular protein (hypothalamic) |
| TNF | Tumor necrosis factor |
| TNSALP | Tissue nonspecific alkaline phosphatase |
| TR | Thyroid (hormone) receptor |
| TRAF | TNF receptor associated factor |
| TRANCE | TNF-related activation induced cytokine |
| TRAP5b | Tartrate resistant acid phosphatase type 5b isoform |
| TREM2 | Triggering receptor expressed on myeloid cells-2 |
| TRP | Transient receptor potential (channel) |
| VDIR | Vitamin D–interacting repressor |
| VDR | Vitamin D receptor |
| VDRE | Vitamin D response element |
| VEGF | Vascular endothelial growth factor |
| WIF | WNT inhibitory factor |
| WINAC | WSTF including nucleosome assembly complex |
| WSTF | Williams syndrome transcription factor |
| XHR | X-linked hypophosphatemic rickets |
Calcium
Calcium is an irreplaceable component of the mineral portion of bone and is required for the function of each of the body’s cell. Together, calcium and phosphate form the hydroxyapatite crystal—Ca10(PO4)10(OH)2—of bone; hydroxyapatite accounts for 65% of bone weight and provides its mechanical and weight bearing strength and also serves as a reservoir for calcium that may be quickly mobilized for homeostatic and functional purposes. Although 99% of total body calcium is present in the slowly exchangeable, deeply deposited skeletal crystal, it is the rapidly exchangeable 1% of body calcium in recently accumulated surface bone and in the vascular, extracellular, and intracellular (soft tissues) spaces, with which it is in equilibrium, that modulates gene expression, intercellular communication and intracellular signal transduction, neural transmission, cell-to-cell adhesion, clotting, striated, smooth and cardiac muscular contraction, cardiac rhythmicity, enzyme action, synthesis and secretion of endocrine and exocrine factors, fertilization, and cellular proliferation and apoptosis.3,4 Approximately 50% of total serum calcium is bound to albumin and globulin; 5% is complexed or chelated to citrate, phosphate, lactate, bicarbonate, and sulfate; and 45% is present as biologically active and closely regulated ionized extracellular calcium (Ca2+e). Approximately 75% of the variability in total serum calcium concentrations is accounted for by genetic factors. Serum total and ionized calcium concentrations are related to levels of albumin, creatinine, parathyroid hormone (PTH), phosphate, and serum pH. For every 1 g/dL decline in the serum concentration of albumin below 4 g/dL, the total serum calcium level declines by 0.8 mg/dL. The measured Ca2+e level is dependent on the serum pH (normal adult range 1.1 to 1.3 mmol/L at pH 7.4); increase in alkalinity (higher pH) raises calcium binding to albumin thus decreasing Ca2+e, whereas acidic changes (lower pH) decrease binding thereby increasing Ca2+e. The relationship between pH and Ca2+e is best described by an inversely S-shaped third-degree function.5
The serum concentration of Ca2+e is maintained within narrow limits by an integrated system involving the plasma membrane Ca2+e sensing receptor (CaSR), PTH and its receptor (PTH/PTH related protein [PTHrP]-1R), the thyroidal parafollicular C cell product calcitonin and its receptor, and the vitamin D hormone system acting on the intestinal tract, bone, and kidney (see Figure 8-1). With increase in serum Ca2+e concentration, the CaSR on the chief cell of the parathyroid gland is activated; the intracellular signal transduction pathway stimulated by the CaSR depresses PTH secretion instantly and decreases expression of PTH. Activation of the CaSR in the distal renal tubule decreases reabsorption of calcium filtered through the glomerulus and increases urinary calcium excretion. When the serum Ca2+e concentration falls, signaling through the CaSR also declines, thereby increasing PTH secretion and renal tubular reabsorption of filtered calcium, activating osteoclastic bone reabsorption, and somewhat later increasing synthesis of 1,25-dihydroxyvitamin D3 (calcitriol) and intestinal absorption of ingested calcium.1
The intracellular concentration of cytosolic free calcium (Ca2+i) (approximately 100 nM) is 10,000-fold less than that in serum and extracellular fluid, a gradient maintained by exchange of Ca2+ across the cell’s plasma membrane and across the membranes of intracellular structures. Within the cell, Ca2+i is primarily (99%) stored within the endoplasmic reticulum (sarcoplasmic reticulum in muscle cells) and mitochondria as well as within endosomes, lysosomes, the Golgi apparatus and secretory granules and bound to the inner plasma membrane from which sites it can be released by chemical signals—that is, inositol-1,4,5-trisphosphate (IP3).6 IP3 acts on IP3 receptors (encoded by ITPR1; see Table 8-2) located in the membrane of the endoplasmic reticulum to effect rapid egress of Ca2+ from storage, thereby quickly increasing Ca2+i levels.7 Ca2+i serves as a second messenger and signal transducer that controls many cellular activities including gene transcription, cell division and growth, cell movement, and secretion of synthesized products. Ca2+ enters the cell through transmembrane protein pores such as voltage-gated and store-operated calcium channels. Voltage-gated Ca2+ channels are active in cells that are electrically excitable such as cardiac, skeletal, and smooth muscle cells, neurons, gastric mucosa, and pancreatic β cells and that open in response to depolarization of the plasma membrane permitting rapid influx of Ca2+e into the cell cytoplasm leading to further depolarization of the membrane and activation of cell function.8 They may be activated by high or low electrical voltage. The skeletal muscle high-voltage-activated calcium channel is composed of five subunits: α1, α2, β, γ, and δ. The voltage-sensing, pore-forming Ca2+-binding α1 subunit has four repeated domains, each with 6-transmembrane spanning regions (or helices) and intracytoplasmic amino and carboxyl terminals; transmembrane helix 4 serves as the voltage sensor; the α1 subunit also has a sequence of amino acids between transmembrane helices 5 and 6 that is partially inserted into the membrane to serve as a selectivity filter.8 The assembly, intracellular movement, interaction with other proteins, activation, and kinetic properties of the α1 subunit are modified by the extracellular, glycosylated α2 subunit, the β subunit (a cytoplasmic globular protein), a small membrane spanning δ subunit that is disulfide-linked to form dimeric α2δ, and a second transmembrane subunit γ. The voltage-gated calcium channels are designated in accord with their cloned specific α1 subunits and have been termed Cav 1.1, Cav 1.2, Cav 1.3, . . . Cav 3.3; formerly, they were designated in accord with the high- or low-voltage strength required for activation and for their sensitivity to specific inhibitors (e.g., L, N, P, Q, R, T subtypes). The high-voltage–dependent L type calcium channels (Cav1.1 through Cav 1.4) are present in skeletal, cardiac, and vascular smooth muscle cells; endocrine cells; neurons; and fibroblasts. L-type calcium channels are activated by the guanine triphosphate (GTP)-binding αq subunit of Gq-proteins through stimulation of phospholipase C (PLC) leading to phosphorylation of the channel protein. L-type calcium channels modulate the growth and proliferation of fibroblasts and smooth muscle cells, the synthesis of extracellular matrix collagen proteins, and the activation of specific transcription factors.9
Ca2+e also enters the cytosol through the plasma membrane store-operated Ca2+ channel, ORAI1 (also termed CRACM1 or TMEM142A [transmembrane protein 142A]).7,10 Store-operated Ca2+ channels are active in cells that are electrically nonexcitable such as lymphocytes and other immune cells.4 Nevertheless, both voltage-gated and store-operated Ca2+ channels often coexist in the same cell. Which type of Ca2+ channel is utilized in a specific cell is determined by the expression of STIM1 (see Table 8-2) encoding a protein that transverses the membrane of the endoplasmic reticulum. When the Ca2+ content of the endoplasmic reticulum is depleted, STIM1 unfolds and bridges the cytosolic distance between the endoplasmic reticulum and the cell’s plasma membrane where it binds to and opens the ORAI1 pore, thereby permitting Ca2+ influx into the cytoplasm and the restoration of Ca2+ stores within cytoplasmic organelles. At the same time that it activates the ORAI1 Ca2+ channel, STIM1 inhibits the voltage-gated Ca2+ channel.4,11,12 Loss-of-function mutations in ORAI1 have been identified in patients with severe combined immune deficiency and Kaposi sarcoma.13,14
In addition to traversing rapidly across the cell’s plasma membrane through voltage-gated and store-operated channels, Ca2+ enters the cytosol, but at a markedly slower rate, through bifunctional membrane-associated IP3 receptors that also serve as calcium channels.7,15 Encoded by ITPR1, the tetrameric IP3 receptor has six transmembrane domains and a pore-forming loop between the fifth and sixth transmembrane segments. Whereas many IP3 receptor/channels are expressed in the membranes of the endoplasmic reticulum where IP3 activation leads to depletion of Ca2+ stored in the endoplasmic reticulum thereby triggering store-operated calcium channel activation, only one to two such channels are expressed in the plasma membrane of each cell. Ca2+ is translocated not only through specific Ca2+ channels but also through paracellular transport channels16. Ca2+i is extruded from cell cytoplasm by calcium and energy-dependent adenosine triphosphate (ATP)-driven calcium pumps and in exchange for sodium (Na+) through H+/ATPase and Na+/Ca2+ exchangers.3 The Ca2+-ATPases are encoded by four genes; ATP2B1 encodes a 1220-amino-acid plasma membrane protein with a cyclic AMP-dependent protein kinase domain and a carboxyl domain to which calbindin binds, which links hydrolysis of ATP with transport of cations across membranes. The Ca2+-ATPases have the capacity to transport Ca2+ against high concentration gradients. The Na+/Ca2+ exchangers are widely distributed plasma membrane Ca2+ export proteins.1,3 A homologue of the human Na+/Ca2+ exchanger is composed of 10 transmembrane domains consisting of two sets of five transmembrane helices in inverted reverse order with the amino and carboxyl termini within the cytoplasm.17,18 Transmembrane helices 2, 3, 7, and 8 form an ion-binding pocket with three low-affinity Na+ and one high-affinity Ca2+ binding sites symmetrically arranged in a diamond formation. Through conformational changes of the structure of the Na+/Ca2+ exchanger, the Na+ and Ca2+ binding sites are alternately exposed to either the intracellular or extracellular surfaces of the cell’s plasma membrane. As Na+ from the extracellular space occupies its three binding sites, the binding affinity for Ca2+ decreases, thereby releasing this cation into the cytosol and reversing the orientation of the Na+/Ca2+ channel; inasmuch as cytosolic levels of Na+ are low, these cations are then released into the cytosol restoring the affinity of the exchanger for Ca2+. The human cardiac Na+/Ca2+ exchanger NCX1 (encoded by SLC8A1) has Na+/Ca2+ binding configurations that are similar to those of the homologue.17 The human skeletal muscle Na+/Ca2+ exchanger NCX2 is encoded by SLC8A2. Within the cell, cytosolic Ca2+ is transported across the membrane and into the endoplasmic reticula of muscle cells through Ca2+-ATPase channels encoded by ATP2A2; linkage of small ubiquitin-like modifier type 1 (sumoylation) to this Ca2+-ATPase extends its half-life and increases its intrinsic activity.19
There are many intracellular calcium binding proteins (e.g., calcium binding proteins 1 and 4, calbindin, sorcin, calmodulin, and so forth). Calmodulin 1 (CALM1) is a widely distributed 149-amino-acid protein with an amino terminal lobe linked to a carboxyl terminal lobe that can assume more than 30 three-dimensional conformations.8 Each lobe has two globular calcium binding domains consisting of two helix-loop-helix motifs connected by an α-helical linker; calcium binding exposes hydrophobic pockets that allow calmodulin to bind to and regulate the activity of target proteins.20 When bound to a target protein, calmodulin can assume any one of an exceedingly large number of conformations. This versatility enables calmodulin to act as a calcium sensor for many different proteins subserving distinct processes within a single cell including voltage-gated calcium channels and calcium/calmodulin-dependent protein kinases.
In the gastrointestinal tract, calcium is absorbed by both active transcellular and passive paracellular processes.21 During growth in younger subjects and when calcium intake and consequently intestinal calcium concentrations are low, active transcellular absorption of calcium predominates; when calcium intake is adequate or high, passive paracellular absorption of calcium is the major process by which calcium is absorbed.21,22 Transcellular gastrointestinal absorption of calcium is an active and saturable process that is primarily stimulated by calcitriol by regulating the availability of transmembrane calcium “pumps” and channels on the luminal and basolateral surface membranes of duodenal enterocytes.1 Calcitriol acting through the vitamin D receptor (VDR) increases duodenal expression of calcium transport protein-1 (epithelial calcium channel-2 encoded by TRPV6, transient receptor potential-vanilloid 6), a luminal calcium channel with six transmembrane domains and intracytoplasmic amino and carboxyl terminals and a pore region between the fifth and sixth transmembrane domains that forms a homotetramer or heterotetramer combined with TRPV5; TRPV5 and TRPV6 specifically permit Ca2+ permeation at physiologically levels of this cation.22 Calcium is also actively absorbed in the intestinal tract by mechanisms that are independent of the VDR.23 Thus, the expression of TRPV6 can be increased by both estrogen and prolactin; prolactin can increase the transcellular absorption of calcium independently of calcitriol and the VDR.24 After entering the enterocyte, Ca2+ diffuses across its interior within the cytosol or within a lysosomal vesical bound to calbindin D9k (encoded by CALB3), a protein with two high-affinity calcium binding sites that transports calcium from the apical to the basolateral membrane of the enterocyte. After fusion of vesicle and basolateral plasma membrane, Ca2+ is extruded into the extracellular space (and circulation) through a basolateral Ca2+-Mg2+-dependent ATPase calcium channel (ATPB21), also termed plasma membrane Ca(2+) ATPase, type 1 (PMCA1b). Experimental data in mice suggest that neither TRPV6 nor calbindin D9k is essential for vitamin D–mediated intestinal absorption of calcium.25,26 However, the applicability of these observations to human physiology is uncertain.21 When calcium intake is adequate or high, the bulk of ingested calcium is passively absorbed through paracellular channels between enterocytes in the jejunum and ileum. Although calcitriol regulates synthesis of several tight junction proteins that may comprise the paracellular channels, including claudins-2 and -12, and cadherin-17, there is no known direct regulation of paracellular intestinal calcium absorption.21,27
Intestinal calcium absorption is influenced by vitamin D status, its food source (the bioavailability of calcium in cow milk formulas is 38%, that in human breast milk is 58%; leafy green vegetables are also a good source of dietary calcium), the form of the calcium salt in dietary supplements, and the presence in food of inhibitors of calcium absorption such as phytates, oxalates, or phosphates (e.g., cola beverages).28 Intestinal calcium absorption is increased during adolescence, pregnancy, and lactation, and depressed in patients with nutritional or functional vitamin D deficiency, chronic renal disease, and hypoparathyroidism. The amount of calcium ingested influences the net amount of calcium absorbed; the lower the calcium intake, the greater is the efficiency of its absorption. In the adult when the dietary calcium intake is < 200 mg/day, fecal calcium excretion exceeds intake; thus, active absorption of calcium cannot compensate for very low intake. As intake of dietary calcium increases from 200 to 1000 mg/day, active calcium absorption also increases but at a progressively decreasing rate. When dietary calcium exceeds 1000 mg/day, active calcium absorption remains relatively constant at 400 mg/day but passive absorption of calcium through paracellular channels continues to increase.1 Thus, hypercalcemia may result from dietary calcium excess as in the milk-alkali syndrome. PTH indirectly increases intestinal calcium absorption by enhancing renal 25-hydroxyvitamin D-1α-hydroxylase activity and hence calcitriol synthesis. Growth hormone (GH) and estrogens also increase intestinal absorption of calcium; glucocorticoids and thyroid hormone inhibit this process. Calcium is excreted into the intestinal tract in the ileum and in pancreatic and biliary secretions.
Low calcium intake is associated with increased fracture rate in children and adolescents, and therefore, adequate dietary intake of calcium during infancy, childhood, and adolescence is necessary to attain a peak bone mass that may lessen the risk of fracture and the later development of osteopenia.29 In an effort to ensure optimal mineralization of the developing skeleton, age-related dietary intakes of elemental calcium for infants, children, and adolescents have been recommended (Table 8-3).30 Because calcium is an essential nutrient for bone mineralization, intake of calcium is optimal when skeletal calcium content is maximal for the age and gender of the subject.2 In normally weighted adolescents, a calcium intake above 1600 mg/day results in maximal calcium retention (approximately 35%).31 (Thus, the recommended dietary allowance [RDA] of calcium for adolescents of 1300 mg/day [see Table 8-3] may be underestimated.) Calcium retention and bone mass increase as the body mass index rises when calcium intake is sufficient. The retention of oral calcium depends not only on the level of nutrient intake but also on gender and race: white adolescent males retain more calcium than do white girls (37% versus 30%); black adolescent girls retain more calcium than do white girls (38% versus 30%). Besides dietary intake of calcium and vitamin D status, the most significant modifiable determinant of bone mineralization is weight-bearing physical activity (discussed later).32
TABLE 8-3
Recommended Dietary Intakes of Calcium and Vitamin D in Infants, Children, and Adolescents
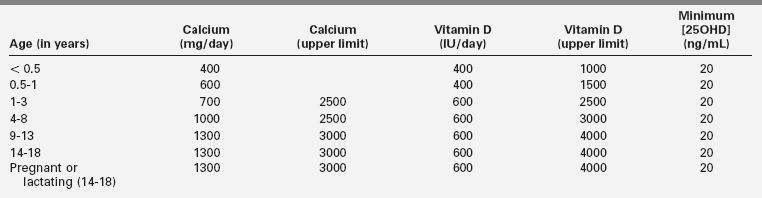
Adapted from Ross, A. C., Manson, J. E., Abrams, S. A., et al. (2011). The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab, 96, 53–58; Holick, M. F. (2011). Vitamin D: evolutionary, physiological and health perspectives. Curr Drug Targets, 12, 4–18; Holick, M. F., Binkley, N. C., Bischoff-Ferrari, H. A., et al. (2011). Evaluation, treatment, and prevention of Vitamin D deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 96, 1911–1930.
Calcium is excreted primarily by the kidney. Ultrafiltrable serum calcium (representing both the ionized Ca2+e form and calcium that is complexed or chelated) crosses the renal glomerular membrane; 98% of filtered calcium is reabsorbed in the renal tubule. Paracellular reabsorption of most (93%) of filtered calcium occurs in the proximal renal tubule and thick ascending limb of Henle (TALH). The renal distal convoluted tubule, connecting tubule, and first part of the collecting duct actively reabsorb approximately 5% of filtered calcium transcellularly through TRPV5 calcium channels expressed on the apical membrane of renal tubular epithelial cells.22 In the cytoplasm, calbindin D9k then shuttles Ca2+e across the cell for extrusion by ATPB1 channels and the Na+-Ca2+ exchanger type 1 (encoded by SLC8A1) on the basolateral membrane of the renal tubular cell. Calcitriol, PTH, low dietary calcium, estrogens and androgens, and acid/base balance increase expression of TRPV5. PTH also enhances posttranslational phosphorylation of TRPV5 and hence its movement to and insertion into the apical membrane of the renal tubular cell and inhibits its endocytosis, the paired effects thus resulting in increased numbers of TRPV5 channels on the apical surface of the renal tubular cell.22 Klotho (encoded by KL), a cofactor/coreceptor essential for normal phosphate homeostasis (discussed later) that also has intrinsic beta glucuronidase activity, increases the retention and hence the activity of the TRPV5 calcium channel by removing oligosaccharide chains during its posttranslational processing.33,34 Calmodulin, an intracellular Ca2+-sensing protein that binds to and inhibits the action of TRPV5, modulates renal tubular reabsorption of Ca2+, a process reversed by PTH-mediated phosphorylation of the TRPV5 calbindin-binding site.35 Although calbindin D9k is not expressed in the proximal renal tubule, cells lining the lumen of this segment have deep invaginations shortening the distance that reabsorbed Ca2+ must travel by diffusion between apical and basolateral membranes before extrusion.21 In the TALH, calcium and magnesium are reabsorbed through voltage-driven paracellular channels (in part through paracellin-1, a tight junction protein channel that is instrumental in renal reabsorption of filtered calcium and magnesium [discussed later]). Cells of the TALH also express the CaSR on their basolateral membrane; when activated by peritubular Ca2+, this G protein–coupled receptor (GPCR) decreases renal tubular reabsorption of calcium through the paracellular channels by inhibiting activity of the Na+-K+-2Cl– transporter and lowering lumen-positive voltage. Increased glomerular filtration or decreased renal tubular reabsorption increase renal excretion of calcium, phosphate, and magnesium. Urinary excretion of calcium is augmented by increased dietary intake, hypercalcemia of diverse pathophysiology (with the exception of that associated with familial hypocalcemic hypercalciuria [discussed later]), expansion of extracellular volume, metabolic acidosis, and loop diuretics (furosemide).1 PTH and PTHrP increase renal tubular reabsorption of Ca2+, whereas glucocorticoids, mineralocorticoids, and Ca2+ itself suppress Ca2+ reabsorption.
Calcium sensing receptor
Plasma Ca2+e concentrations are monitored by the CaSR, a 1078-amino-acid cell membrane GPCR encoded by CASR whose extracellular domain recognizes and binds Ca2+, Mg2+, gadolinium, aromatic amino acids, antibiotics, and other compounds.36,37 Functionally, the CaSR is a homodimer linked by disulfide bonds between extracellular domain cysteine residues 129 and 131 of each CaSR. Homodimerization of the CaSR takes place in the endoplasmic reticulum after core N-linked glycosylation; homodimeric CaSR is then packaged within the Golgi apparatus where it is further glycosylated and transported to the cell membrane.38 The amino terminal extracellular domain of the CaSR is composed of 612 amino acids with five likely Ca2+ binding sites constructed in a Venus flytrap configuration; there are 250 amino acids in the transmembrane domain composed of seven transmembrane helices and three alternating extracellular and intracellular loops, and 216 amino acids in the carboxyl terminal intracellular domain.37 Transmembrane helix 5 may also be necessary for receptor dimerization. The second extracellular loop contains sites (Asp758, Glu759, Glu767) that modulate the sensitivity of the CaSR to Ca2+. Within the intracellular domain of the CaSR are three protein kinase C (PKC) phosphorylation sites; phosphorylation of Thr888 down-regulates Ca2+-mediated CASR intracellular signal transduction.36 Because Ca2+-CaSR interaction stimulates PKC signaling, function of the CaSR can thus be autoregulated.37 Through its binding of Ca2+e, the CaSR finely regulates the extracellular concentration of this cation by modulating the secretion of PTH and the renal tubular reabsorption of filtered calcium. The CaSR is encoded by CASR and is a member of the C family of GPCRs with extremely long extracellular domains (500- to 600-amino-acid residues) and homologies with receptors that bind glutamate, γ-aminobutyric acid, and pheromones as well as with the taste receptors. The very long extracellular domain is heavily glycosylated, a posttranslational modification essential for efficient movement of the receptor to the cell surface. The CaSR is expressed on the plasma membrane of parathyroid chief cells, at the apical or basolateral membranes of most renal tubular cells—particularly the TALH and the collecting ducts, on the cell membranes of the parafollicular (C) cells of the thyroid, in cartilage and bone, lungs, adrenals, breast, intestines, skin, lens, placental cytotrophoblasts, and nervous tissue.39 Expression of CASR is regulated in part by GCM2, encoding a transcription factor essential for formation of the parathyroid glands, and enhanced by calcitriol, high Ca2+e levels, and interleukin (IL)-1β.40
The serum Ca2+e concentration is related to polymorphic variants of the CaSR; 70% of individuals are homozygous for alanine at amino acid position 986 within the intracellular domain, 3% are homozygous for serine, and the remainder are heterozygous for the two amino acids. In heterozygous Arg986Ser and homozygous Ser986 subjects, Ca2+e levels are significantly higher than in those who are homozygous for Ala986.41 Also, there are cellular Ca2+ sensors that are unrelated structurally to the CaSR as well as subtypes of the CaSR itself.39 The CaSR also serves as a Mg2+e sensor and modulates renal tubular reabsorption of this cation decreasing its reabsorption when Mg2+e levels rise. By binding to the CaSR, aromatic L-amino acids appear to “sensitize” the receptor to a given level of Ca2+e (discussed later).36
Although in the kidney the CaSR is found in greatest abundance in the medullary and cortical TALH, it is also found in glomerular cells and other segments of the renal tubule. CASR is expressed on the apical brush-border membranes of cells in the proximal renal tubule, on basolateral cell membranes in cells in the TALH, and on the apical (luminal) surface of cells in the distal renal collecting ducts where it is exposed to intratubular Ca2+.37 Increasing peritubular concentrations of Ca2+e and Mg2+e inhibit renal tubular reabsorption of filtered Ca2+ and Mg2+. In the kidney, binding of Ca2+e to the CaSR decreases not only transcellular transport of filtered Ca2+ but also its paracellular transport in the TALH. Inactivating mutations in CASR result in familial hypocalciuric hypercalcemia, whereas gain-of-function mutations in this gene are associated with autosomal dominant hypoparathyroidism (familial hypercalciuric hypocalcemia).37 Stimulation of the CaSR suppresses PTH-mediated renal tubular Ca2+ and phosphate reabsorption.37,42 In addition to effects on renal tubular cation transport, the Ca2+/CaSR complex inhibits antidiuretic hormone–induced renal tubular permeability to water by decreasing the number of apical aquaporin-2 water channels in the inner medullary collecting ducts, thus leading to polyuria. Stimulation of the CaSR expressed in cells of the renal distal collecting ducts also increases local H+-ATPase activity resulting in urine acidification.42
Acting through the CaSR, rising serum Ca2+e concentrations stimulate release of calcitonin from thyroid C cells. The CaSR is expressed throughout the intestinal tract, where it may modulate the changes in intestinal motility that accompany low (increased) and high (depressed) serum Ca2+e values.36 Additionally, in gastric cells stimulation of CaSR activity results in release of gastrin into the circulation and increased intragastric acidity.37 Expression of the CaSR in the brain suggests a mechanism whereby Ca2+ may influence neural function locally by modulating neurotransmitter and neuroreceptor function (such as the metabotropic glutamate receptor which also recognizes Ca2+). Cell membrane CaSRs are present in (mouse, rat, bovine) articular and hypertrophic chondrocytes of the epiphyseal growth plate and to a lesser extent in proliferating and maturing chondrocytes.43 Osteoblasts, osteocytes, and osteoclasts also express the CaSR, and agonist (Ca2+, neomycin, gadolinium) activation of the CaSR stimulates intracellular signal transduction in these cells. Increase in CaSR activity in osteoblasts enhances their proliferation.37 The CaSR may mediate recruitment of osteoblast precursor cells to sites of high Ca2+ levels, the residue of local osteoclast activity, one of the factors linking the bone remodeling processes of resorption and formation (discussed later).39 In mice in which expression of bone-specific Casr has been eliminated, skeletal development is impaired with diffuse decrease in bone mineralization.37 In vitro, Ca2+ inhibits bone reabsorptive activity of (rabbit) osteoclasts by causing the osteoclast to decrease secretion of acid and proteolytic enzymes and to withdraw from the site of bone resorption.44
Through its intracellular carboxyl terminal, the CaSR is linked to G proteins and their Gα subunits that couple the ligand-receptor message to several intracellular signal transduction pathways.36,37,39 After binding of Ca2+ to the extracellular domain of the CaSR, Gαq/11 dissociates from its βγ subunit complex and activates membrane bound PLC-β1; in turn, this enzyme hydrolyzes membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and IP3, the latter leading to release of Ca2+ from the endoplasmic reticulum and increased cytosolic concentrations of Ca2+i. Activation of the CaSR also stimulates activity of phospholipases A2 and D, mitogen-activated protein kinases (MAPK), and protein kinases B (Akt) and C but inhibits that of adenylyl cyclase, the latter through stimulation of adenylate cyclase-inhibitory Gαi activity.37 Increases in plasma Ca2+e and rising cytosolic levels of Ca2+i within the parathyroid gland chief cells suppress expression of PTH and increase degradation of PTH mRNA, thereby limiting synthesis and secretion of PTH; CaSR signaling also inhibits the proliferation of chief cells.37 A decline in Ca2+e leads to a decrease in CaSR activity, lower Ca2+i values, and thence to increased PTH secretion, thus enabling the CaSR to exercise minute-to-minute control over the release of PTH and hence of the concentration of Ca2+e.
Calcimimetics are agonist drugs that activate the CaSR; calcilytics are antagonists of the CaSR.45 As noted, the CaSR binds and responds to not only Ca2+e but also Mg2+e, selected L-amino acids, and some antibiotics; the latter are designated type I calcimimetics. Synthetic compounds (e.g., phenylalkylamines) that bind to the second and third extracellular loops of the transmembrane domain of the CaSR and allosterically modulate (increase or decrease) the sensitivity of the CaSR to ambient Ca2+e are designated type II calcimimetic agonists or calcilytic antagonists, respectively.36,37 The most widely employed of the clinical calcimimetics is cinacalcet [N-[1-(R)-(-)-(1-naphthyl)ethyl] -3-[3-(trifluoromethyl)phenyl]-1-aminopropane hydrochloride]; this agent has been effective in decreasing secretion of PTH in patients with either primary or secondary hyperparathyroidism.46 Calcilytics decrease the sensitivity of the CaSR to Ca2+e and thus increase the secretion of PTH and depress renal tubular reabsorption of Ca2+. Mutations in CASR may prevent its normal biosynthesis (class I), disturb movement of the CaSR protein to the plasma membrane (class II), interfere with binding of ligand to the CaSR (class III), impair activation of the CaSR (class IV), or disrupt receptor function by an unknown mechanism (class V).
Phosphate
Eighty-five percent of body phosphate is deposited in bone as hydroxyapatite. Fourteen percent of phosphate is intracellular (in the cytosol or mitochondria in the form of inorganic phosphate esters or salts, cell and organelle membrane phospholipids, and phosphorylated metabolic intermediate compounds involved in energy metabolism and formation of ATP and signal transduction and in the cytosol and nucleus as an essential component of RNA and DNA) or in interstitial fluid or serum (approximately 1%). There it circulates as free orthophosphate anions HPO42− and H2PO4− (55%), bound to proteins (10%), or complexed to calcium, magnesium or sodium (35%).1,47 At pH 7.4, serum HPO42− and H2PO4− are present in a molar ratio of 4:1; in alkalotic states the ratio increases, with acidosis it declines. (At pH 7.4, 1 mmol/L of orthophosphate = 1.8 mEq/L = 3.1 mg/dL.) The serum concentrations of calcium and phosphate are reciprocally related under normal circumstances, and the calcium × phosphate product approximates 30. Intracellularly, cytosolic-free phosphate concentrations approximate those in serum—3 to 6 mg/dL.
The serum phosphate concentration is regulated by oral intake, intestinal absorption and excretion, renal tubular reabsorption and excretion of filtered phosphate, and release from bone by dissolution of hydroxyapatite by PTH and calcitriol, and it fluctuates with age, gender, growth rate, diet, and serum calcium levels (see Figure 8-2).1,48,49 Inasmuch as phosphate is found in all cells and foods, dietary deficiency is unusual. Dietary phosphate is absorbed across the intestinal brush border as HPO42− in direct proportion to its intake, principally in the duodenum and jejunum. Intestinal phosphate is absorbed by both passive paracellular diffusion related to the luminal concentration of this anion and by an active transcellular mechanism stimulated by calcitriol. The latter is an energy-requiring transport process with sodium through the type IIb Na+-HPO42− cotransporter protein (NPT2b; encoded by SLC34A2) with low affinity for phosphate whose activities are maintained by calcitriol-dependent Na+,K+-ATPase. Decreased intestinal absorption of phosphate is compensated by increased renal tubular reabsorption of this anion.47 Phosphate is also secreted into the intestinal tract. When dietary phosphate intake falls below 310 mg/day in the adult, net phosphate absorption is negative. At low phosphate intakes, absorption is active in the duodenum, jejunum, and distal ileum, whereas at high intakes 60% to 80% of ingested phosphate is absorbed primarily by diffusion. Phosphate absorption can be impaired by its intraluminal precipitation as an aluminum or calcium salt and by intestinal malabsorption disorders.
The kidney regulates moment-to-moment phosphate homeostasis. Phosphate is filtered in the renal glomerulus and reabsorbed primarily in the proximal renal tubules. It is actively transported across the luminal membrane against an electrochemical gradient through renal specific type II Na+-HPO42− cotransporter proteins expressed on the apical (tubular) surface of cells in the proximal renal tubule—NPT2a encoded by SLC34A1 and NPT2c encoded by SLC34A3—with the aid of a Na+,K+-ATPase pump.1,47,50 Expression of SLC34A1 is regulated by serum phosphate levels (hypophosphatemia increases expression), PTH, PTHrP, and fibroblast growth factor (FGF)-23 (discussed later). NPT2a actively transports approximately 70% and NPT2c 30% of proximal renal tubular reabsorbed phosphate in the mouse; in the human, NPT2c may be the major renal tubular transporter of phosphate. The Na+-H+ exchanger regulatory factor 1 (NHERF1; encoded by SLC9A3R1) is a scaffolding protein that is essential for trafficking of NPT2a and NPT2c to the luminal membrane of cells in the proximal renal tubule; NHERF1 also interacts with PTH1R.50 Another family of phosphate transporters (PiT2, encoded by SLC20A2, OMIM ID: 158378

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


