Diagnosis and Classification of the Acute Leukemias and Myelodysplastic Syndromes
Diagnosis and Classification of the Acute Leukemias and Myelodysplastic Syndromes
Daniel A. Arber
Attilio Orazi
INTRODUCTION
The diagnosis and classification of acute leukemias and myelodysplastic syndromes (MDSs) has grown increasingly complex.1,2 Cases can no longer be fully classified by the use of only morphologic evaluation and cytochemical studies. Historic information, such as the presence of Down syndrome, prior therapy, or prior MDS all impact the final diagnosis. Additionally, immunophenotypic studies are needed for many cases; and cytogenetic, with possible molecular genetic, studies are required for essentially all cases.
This increase in complexity in the evaluation of these neoplasms has resulted in more precise diagnostic categories and recognition that the broad categories of acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and MDS actually represent heterogeneous groups of diseases. The newer disease categories are more predictive of outcome than older classification systems, in part because of their ability to separate disease groups within each category.3,4,5 and 6 Unfortunately, many physicians continue to use older terminology for these diseases, relying on terminology from the French-American-British Cooperative Group (FAB) classification7,8,9,10 of these neoplasms. While the FAB classification provided firm diagnostic criteria and useful terminology for communication of findings using the methods available at the time, its use is no longer appropriate. The third (2001) and fourth (2008) editions of the World Health Organization (WHO) classification of hematopoietic tumors have dramatically changed the approach to diagnosis of many of these neoplasms, and the WHO system should be considered the current standard of care. Modifications from the LeukemiaNet group and others will certainly continue to aid in the refinement of our classification systems.11,12,13 This evolution from the FAB to the WHO is reminiscent of changes in lymphoma classification with evolution from the Rappaport14 and Kiel15 classifications to the Working Formulation16 to the REAL17 classification and finally to the WHO classification.2,18 Perhaps because the changes in lymphoma classification were more stepwise with shorter time intervals between the changes than the leukemia classification changes, they have been more widely adopted. We no longer refer to diffuse large B-cell lymphoma as histiocytic lymphoma and should no longer refer to AML with t(8;21)q22;q22) as M2, or to B-lymphoblastic leukemia with BCR-ABL1 as L2.
Several discoveries have impacted the classification of these neoplasms and many of them are covered in great detail in other chapters, but genetic discoveries associated with the acute leukemias and MDSs are probably the most significant. The finding of recurrent cytogenetic abnormalities with prognostic significance impacts all of these diseases.19,20,21,22,23 and 24 While balanced translocations are more common in the acute leukemias, the presence of single and complex abnormalities in the MDS has helped define disease prognosis as well as defining a specific disease category of MDS with isolated del(5q).5,6 The more recent discoveries of specific gene mutations have further impacted both acute leukemia and MDS diagnosis.13,25,26,27,28,29,30 and 31 While many of these mutations have their greatest frequency in AMLs with a normal karyotype, others offer prognostic significance that complements other morphologic and karyotypic features.
The classification of AML and MDS has also been impacted greatly by the understanding of similarities between the two. The so-called myelodysplasia-related AML and de novo AML described by Head32 helped lead to a new way of thinking about this disease; especially AML occurring in older patients.
This chapter will highlight key classification issues in the acute leukemias and MDSs, which are discussed in detail in the chapters that follow.
DIAGNOSTIC EVALUATION
The diagnostic approach to acute leukemia and MDSs still begins with a morphologic evaluation, but requires careful integration of the morphologic findings with clinical information and relevant laboratory data, including cytogenetic and molecular results.33 Laboratory data, particularly results of a recent complete blood count (CBC) must be reviewed with the samples. Morphologic evaluation requires a well-stained (usually Wright-stained) peripheral blood (PB) smear prepared from a recent sample (less than 2 hours from procurement). A 200-cell manual differential count is required for PB smears. The bone marrow (BM) aspirate smears are best prepared at the bedside immediately after procurement and promptly stained. The review of BM aspirate smear includes a 500-cell differential count. In patients with inaspirable marrow, touch preparation of the BM biopsy can be used in lieu of an aspirate smear. The BM biopsy is usually stained with hematoxylin and eosin (H&E) or Giemsa and is useful in many settings.34 The morphologic assessment allows for the appropriate use of ancillary tests in these disorders. Details of the ancillary tests used for the workup of the various diseases are provided in Chapters 2, 3 and 4 and will not be repeated here.
Cytochemical studies are still performed at many institutions and can often provide quick general information about the cell type of an acute leukemia (myeloid versus myeloperoxidase negative), but because more detailed information can now be obtained by flow cytometry in the same time frame, the use of these less specific cytochemical studies has decreased. Immunophenotyping, usually by multicolor flow cytometry, is now standard for acute leukemias and is absolutely required to accurately diagnose the lymphoblastic leukemias and some AMLs.7,11,12,35,36 In cases of acute leukemia with marrow fibrosis, as may occur with some acute megakaryoblastic leukemias and with acute panmyelosis with myelofibrosis (APMF) (see Fig. 73.1), paraffin section immunohistochemistry performed on a BM trephine biopsy is essential.37 The use of flow cytometry immunophenotyping in the MDSs is the subject of much study, and many centers have incorporated this technique into the evaluation of patients with potential MDS not only to help quantitate blood and marrow blast cell percentages, but also to detect abnormal maturing cell populations.11,38,39 These methods, however, do not replace morphologic evaluation, including morphologic blast cell counts on smears.
Cytogenetic studies should also be performed on all cases of suspected acute leukemia or MDS.21,40,41 While specific gene mutations and some structural abnormalities will be missed by this method, karyotype analysis currently provides the best overall assessment of chromosomal abnormalities and should not be supplanted by other studies. Fluorescence in situ hybridization (FISH) or polymerase chain reaction (PCR) studies are often helpful to detect cryptic abnormalities that may be missed by karyotype analysis and are often added in panels for suspected acute leukemia or MDS.42 Finally, molecular studies for specific gene mutations are now routinely performed on samples from patients with these disorders, but when each mutation is studied individually the specific tests performed should be ordered based on the findings of other studies. However, with the rapid growth of next-generation sequencing technology,43 more cost-effective gene mutation panels will become available that may reduce the need for selective testing.
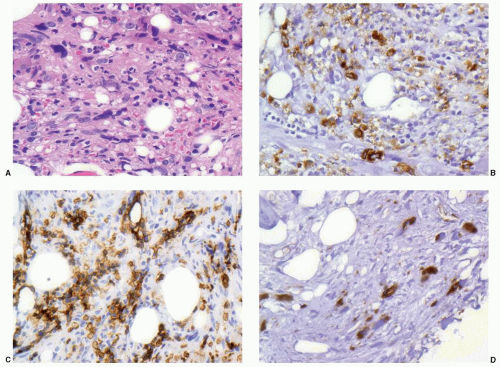
FIGURE 73.1 Acute panmyelosis with myelofibrosis illustrating the use of immunohistochemistry in diagnosis. The marrow is inaspirable due to marked marrow fibrosis (A) with a mixed cellular population that includes immature mononuclear cells. The cells show a mix of granulocyte precursors marking with myeloperoxidase (B), erythroid precursors marking with glycophorin B (C), and immature megakaryocytes marking with von Willebrand’s factor (D).
Once this broad array of studies is complete, the results should be incorporated into a single report with a final diagnosis. Because they cannot always be completed in the time interval needed to begin therapy, this approach requires the use of preliminary and amended reports. Because the WHO classification relies on use of cytogenetic studies and some prognostic risk groups are defined by mutation analysis, the diagnosis often needs to be refined, and thus amended, when these studies are complete.
MYELODYSPLASTIC SYNDROMES
Patients with MDS typically show persistent (>6 months) unexplained cytopenias. The majority of MDS patients present with anemia. Neutropenia and thrombocytopenia are less common presenting symptoms. The cytopenias are defined by a hemoglobin level of less than 10 g/dL, an absolute neutrophil count of less than 1.8 × 109/L, and a platelet count of less than 100 × 109/L.33,44 Of note, CBC values higher than those listed above are not exclusionary for a diagnosis of MDS if definitive morphologic and/or cytogenetic findings are consistent with such a diagnosis. Since the presence of cytopenia(s) is required for the diagnosis, the most current and preferably previous CBCs have to be reviewed at the time of BM exam. Other pertinent data include medications, chemical exposure history, and previous and current illnesses, since all of these can cause morphologic dysplasia indistinguishable from MDS. Comorbidities associated with morphologic dysplasia are frequent in elderly patients affected by MDS, and include liver and kidney failure, autoimmune disorders, neoplasms, and systemic infections. In particular, morphologic evaluation is best performed when the patient is off medication.
Dysplastic features can be present in a single hematopoietic lineage (unilineage dysplasia) or involve all marrow populations (multilineage dysplasia) (Fig. 73.2). At least 10% of all cells in a given lineage (erythroid, myeloid, megakaryocytic) have to be dysplastic for establishing the presence of MDS-associated dysplasia. The majority of MDS subtypes show dyserythropoiesis. PB shows normocytic, normochromic, or macrocytic anemia with macroovalocytes. Microcytosis can be present in rare cases of MDS (e.g., cases associated with congenital or acquired alpha thalassemia).45 The most common dysplastic features seen in erythroid precursors include nuclear abnormalities such as nuclear budding, nuclear fragmentation, irregular nuclear outlines, karyorrhexis, internuclear bridging, multinucleation, and megaloblastoid, coarsely condensed chromatin. Cytoplasmic vacuoles with coalescing vacuoles, defective hemoglobinization and increased numbers of ring sideroblasts can also be encountered. Similarly, dysgranulopoiesis is often manifested by abnormal nuclear features including hypolobation (pseudo-Pelger-Huët or monolobated neutrophils), hypersegmented and/or enlarged nuclei, nuclear sticks or fragments, macropolycytes, and abnormally condensed chromatin, which often coexists with nuclear hypolobation. Cytoplasmic features of neutrophil dysplasia include hypogranulation, pseudo-Chediak-Higashi granules, and rarely Auer rods. Hypogranulation occurs frequently and is related to the defective formation of secondary granules. However, the evaluation of this feature is highly subjective and dependent on the staining quality. A well-stained segmented neutrophil or neutrophilic precursor with well-developed secondary granules present, preferably in the same microscopic field, can be used as an internal control. Megakaryocyte morphology can be evaluated using both aspirate smear and the histologic sections (biopsy or clot section) with careful examination of at least 30 megakaryocytes.33,44 Dysplastic features include monolobated, hypolobated, and hyperlobated nuclei, and multiple, widely separated nuclei including “pawn-ball” forms. Normal size or small megakaryocytes with a single (nonlobated) eccentrically placed nucleus are common in 5q- syndrome and in cases with abnormalities of chromosome 3. The latter also shows typically numerous bilobated forms. Megakaryocytic dysplasia is often associated with thrombocytopenia and platelets of variable size, including large forms occasionally showing hypogranulation.
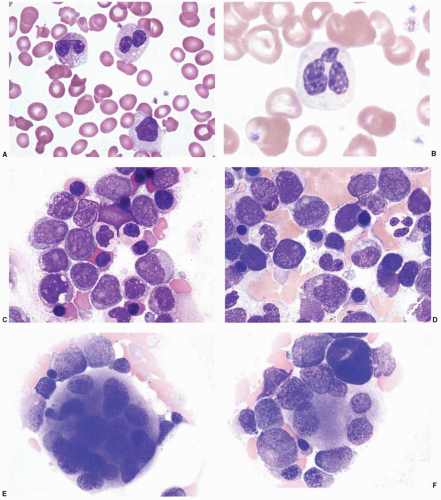
FIGURE 73.2 Dyspoietic changes in myelodysplastic syndrome and acute myeloid leukemia. Peripheral blood (A, B) and bone marrow (C, D) neutrophils may show cytoplasmic hypogranulation, clumped nuclear chromatin, and hyposegmentation, including pseudo-Pelger-Huët anomaly as shown in image A. Dyserythropoiesis may include nuclear-cytoplasmic asynchrony and irregular nuclear shapes in the marrow erythroid precursors (C, D). Megakaryocyte changes include hypersegmented cells (E) and smaller, hypolobated cells (F).
The biopsy allows for the evaluation of cellularity, architectural features, fibrosis, and the presence of previously undiagnosed focal lesions such as metastatic neoplasms or infections. In MDS marrows, the typical architectural organization is lost. Clusters of immature cells are frequently found in the center of the marrow space. This finding was originally termed abnormal localization of immature precursors and is more frequently seen in cases of high-grade MDS. The identification of blast clusters is facilitated by CD34 immunohistochemistry, which is also useful to determine the number of blasts in cases with inaspirable marrows. The presence of CD34 positive cell clusters is a prognostically significant finding that is predominantly seen in high-risk MDS.46 The BM biopsy is necessary for the assessment of BM fibrosis by the Gomori silver impregnation method. Significant fibrosis occurs predominantly in high-risk and therapy-related MDS; however, it may also occur in low-risk disease. Finally, BM biopsy is critical for the exclusion of other hematologic and also nonhematologic diseases associated with unexplained cytopenia(s) which can clinically mimic MDS.
In addition to dysplasia, the evaluation of blasts constitutes a cornerstone of morphologic diagnosis of MDS. The optimal quality of BM aspirate and PB smears cannot be overemphasized.33 The blast percentage is derived from the 500-cell differential count of marrow aspirate smear and 200-cell PB manual differential count. Both differential counts are essential for the subclassification of individual cases. The agranular and granular (type II and type III blasts), and, if present, monoblasts, promonocytes, and megakaryoblasts are included in the blast category.47 In the absence of adequate BM aspirate smears due to causes such as an inaspirable marrow, careful inspection of the histologic material aided by a CD34 immunohistochemical stain can be used. The latter is particularly helpful in cases with significant marrow fibrosis or in hypoplastic marrows, which often yield hemodilute marrow aspirates.48,49,50 Of note, blasts in MDS can be negative for CD34, therefore careful correlation with the visual blast identification is required in all cases. It is also important to emphasize that morphologic dysplasia is not equivalent to the diagnosis of MDS. Megakaryocytic and erythroid dysplasia are commonly seen in individuals in a variety of conditions. Nutritional deficiencies, heavy metal exposure, medications, and systemic diseases can produce morphologic changes resembling those seen in MDS. Therefore, as discussed previously, the correlation with clinical history is critical.
Cytogenetics is crucial and is also required for proper classification. The MDS-associated abnormalities are listed in Table 73.1. In addition, mutational analysis is becoming an important additional tool in the prognostic assessment of MDS30,51; but mutation studies are not routinely performed in the evaluation for MDS at this time.
TABLE 73.1 RECURRENT CHROMOSOMAL ABNORMALITIES AND THEIR FREQUENCY IN MYELODYSPLASTIC SYNDROME
Abnormality
Frequency (%)
Unbalanced
+8
10
-7 or del(7q)
10
-5 or del(5q)
10
del(20q)
5-8
-Y
5
i(17q) or t(17q)
3-5
-13 or del(13q)
3
del(11q)
3
del(12q) or t(12q)
3
del(9q)
1-2
idic(X)(q13)
1-2
Balanced
t(1; 3)(p36.3;q21.2)
1
t(2; 11)(p21;q23)
1
inv(3)(q21;q26.2)
1
t(6;9)(p23;q34)
1
The 2008 WHO classification further refined the criteria for diagnosis and classification of MDS, and seven categories are now recognized (Table 73.2). Cases currently recognized as refractory cytopenia with unilineage dysplasia (RCUD) were considered as refractory anemia (RA) or MDS, unclassifiable in the 2001 WHO classification. Such cases encompass patients with one isolated cytopenia or bicytopenia associated with unilineage dysplasia. An advantage of the new category of RCUD is that one can further specify the lineage: refractory anemia, refractory neutropenia, or refractory thrombocytopenia. Regardless of the lineage involved in RCUD, blasts are absent or represent less than 1% of the PB differential count. Patients with 1% blasts in PB or patients with unilineage dysplasia associated with pancytopenia are now classified as having unclassifiable MDS (MDS-U), owing to the uncertain but presumably more severe clinical significance of these findings.2 Overall, the category of MDS-U is currently better defined and also includes cytopenic patients lacking significant dysplasia, but presenting with cytogenetic abnormalities considered presumptive evidence of MDS.
The 2008 WHO classification acknowledged a subset of pediatric patients with specific MDS features different from those commonly seen in adults, and included a separate category of childhood MDS termed refractory cytopenia of childhood. This category encompasses children with MDS that have persistent cytopenia with less than 2% blasts in the PB and less than 5% blasts in the BM.52,53
Some subtypes or presentations of MDS are more challenging and will be discussed in more detail. MDS-U includes cases which do not fulfill criteria of other MDS subtypes. MDS-U can be diagnosed in patients fulfilling the following criteria: (1) patients who fit the criteria for a diagnosis of RCUD or refractory cytopenia with multilineage dysplasia (RCMD), but in whom 1% blasts in the blood are found on at least two consecutive occasions; (2) patients with MDS with pancytopenia and morphologic dysplasia limited to one hematopoietic lineage; (3) patients with persistent cytopenias, no increase in blasts, and lacking diagnostic morphologic features of MDS (less than 10% dysplastic cells in any lineage), but with clonal cytogenetic abnormalities considered as a presumptive evidence of MDS. According to the 2008 WHO classification, if characteristic features of a specific subtype of MDS develop later in the course of the disease, the case initially classified as MDS-U should be reclassified accordingly. The prognosis of MDS-U varies and close follow-up is warranted.
TABLE 73.2 THE WHO 2008 CLASSIFICATION OF MDS
Subtype
Blood findings
Bone marrow findings
Refractory cytopenias with unilineage dysplasia (RCUD)
≥10% of the cells of the affected lineage are dysplastic
Refractory neutropenia (RN)
No or rare blasts (<1%)
<5% blasts
Refractory thrombocytopenia (RT)
<15% ring sideroblasts
Refractory anemia with ring sideroblasts (RARS)
Anemia
No blasts
Erythroid dysplasia only
<5% blasts
≥15% ringed sideroblasts
Refractory cytopenia with multilineage dysplasia (RCMD)
Cytopenia(s)
No or rare blasts (<1%)
No Auer rods
<1 × 109 L
monocytes
Dysplasia in ≥10% of cells in two or more myeloid lineages <5% blasts
No Auer rods
±15% ring sideroblasts
Refractory anemia with excess blasts-1 (RAEB-1)
Cytopenias
<5% blasts
No Auer rods
<1 × 109 L
monocytes
Unilineage or multilineage dysplasia
5-9% blasts
No Auer rods
Refractory with excess blasts-2 (RAEB-2)
Cytopenias
5-19% blasts ±Auer rods <1 × 109 L
monocytes
Unilineage or multilineage dysplasia 10-19% blasts ±Auer rodsb
Myelodysplastic syndrome, unclassified (MDS-U)
Cytopenias
No or rare blasts (≤1%)
No Auer rods
Unequivocal dysplasia in <10% of cells in one or more myeloid cell lines <5% blasts
MDS associated with isolated del(5q)
Anemia
No or rare blasts
(<1%) Platelet count usually normal or increased
Normal to increased megakaryocytes with hypolobated nuclei <5% blasts
Isolated del(5q) cytogenetic abnormality
No Auer rods
aBicytopenia may occasionally be observed. Cases with pancytopenia should be classified as MDS-U.
bIf the diagnostic criteria for MDS are fulfilled and Auer rods are present, the patient should always be categorized as RAEB-2.
MDS with fibrosis (MDS-F) is defined by the presence of significant fibrosis (at least 3+ reticulin fibrosis according to the Manoharan scoring system, or at least 2+ reticulin fibrosis according to the European consensus grading system).44,54,55 In the past, select cases with 2+ reticulin fibrosis according to Manoharan scoring may have been classified as MDS-F. Approximately 10% to 15% of MDS patients show a significant increase in reticulin fibers and/or collagen fibrosis at presentation.54 These patients are diagnosed with an MDS subtype according to the 2008 WHO classification, with the annotation to indicate the presence of fibrosis. The majority of MDS-F cases fall into the category of refractory anemia with excess blasts (RAEB). Therapy-related MDS cases frequently show significant marrow fibrosis, but are excluded from the MDS-F category in favor of placing them in a special entity of therapy-related myeloid neoplasms. The majority of MDS-F patients present with severe pancytopenia. Organomegaly is minimal or absent. The patients with MDS-F are reported to have shorter overall survival and disease-free survival times than those without fibrosis, even in the absence of excess blasts.54,56,57 BM fibrosis is also associated with a poor outcome after BM transplantation.58 BM and PB often show trilineage dysplasia with excess blasts. There is prominent dysmegakaryopoiesis, often associated with increased numbers of megakaryocytes. Cellular streaming is frequently seen on H&E biopsy section and is indicative of significant fibrosis. CD34 immunostaining may be helpful in assessing the number of blasts, since the aspirate smears are often inadequate in patients with fibrotic marrows. Differential diagnoses of MDS-F include other myelofibrotic myeloid neoplasms such as APMF, myeloproliferative, and myelodysplastic/myeloproliferative neoplasms. Similar to MDS-F, APMF, a subtype of AML-NOS (not otherwise specified), shows marked fibrosis, trilineage dysplasia, and dwarf megakaryocytes. The higher number of blasts (in the range of 20% to 25%), abrupt onset with fever and bone pain, and rapidly progressive course seen in APMF are helpful in establishing the definitive diagnosis.37 Classic myeloproliferative neoplasms, such as primary myelofibrosis, can usually be easily distinguished from MDS-F by their morphologic characteristics (e.g., large-to-giant megakaryocytes with cloud-like nuclei, lack of other myelodysplastic features), and by the presence of significant splenomegaly.33,59 Myelodysplastic/myeloproliferative neoplasms such as chronic myelomonocytic leukemia may show morphologic features similar to MDS, yet demonstrate laboratory and clinical features of a proliferative process such as elevated WBC and monocytosis.
Only gold members can continue reading. Log In or Register to continue
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



