lymphomas (e.g., mantle cell lymphoma vs. small lymphocytic lymphoma [SLL]) (Table 86.2), can identify important nonlineagerelated markers (e.g., CD15, CD30, and CD56), and can determine the proliferative rate of lymphomas. Immunoglobulin (Ig) light chain restriction is evidence of B-cell clonality, whereas aberrant B-cell or T-cell phenotypes infer clonality.1,2 As small monotypic (light chain-restricted) B-cell or genotypically clonal B-cell or T-cell populations may be seen in reactive processes, correlation of these studies with the morphologic features is essential to prevent misdiagnosis and clinical confusion.3, 4 and 5
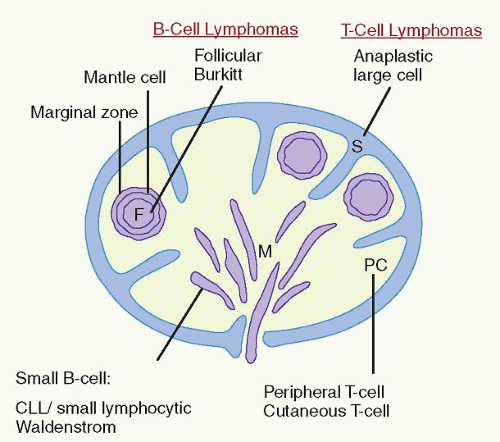 FIGURE 86.1. Sites of origin of malignant lymphomas in a lymph node according to anatomic and functional compartments of the immune system. CLL, chronic lymphocytic leukemia; F, follicles with germinal centers; M, medullary cords; PC, paracortex, or interfollicular areas; S, sinuses. (Adapted from Mann RB, et al. Malignant lymphomas: a conceptual understanding of morphologic diversity. Am J Pathol 1979;94:1.) |
TABLE 86.1 IMMUNOPHENOTYPIC MARKERS USED IN DIAGNOSIS OF MALIGNANT LYMPHOMAS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 86.2 PATHOLOGIC FEATURES IN THE DIFFERENTIAL DIAGNOSIS OF SMALL B-CELL LYMPHOMAS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
may occur at any age, the majority of these rare lymphomas have been described in children and young adults. These neoplasms present frequently in extranodal sites such as bone and soft tissue or as skin tumors of the scalp and face.31,32,33,34 They almost never present as mediastinal masses.
TABLE 86.3 WORLD HEALTH ORGANIZATION CLASSIFICATION OF MALIGNANT LYMPHOMAS | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
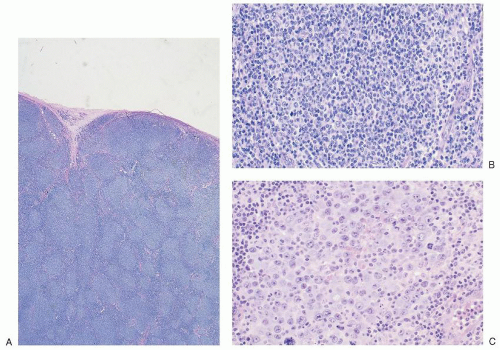 FIGURE 86.2. Lymph node: follicular lymphoma. A: Low magnification demonstrates effacement of architecture by a follicular proliferation producing a nodular pattern. B: In a higher magnification, the follicles are composed almost entirely of centrocytes (follicular lymphoma, grade 1), whereas, in (C), from another patient, the follicles are dominated by centroblasts (follicular lymphoma, grade 3). |
lymphomas of large centrocytes exhibit a predominantly diffuse growth pattern. Several studies suggest that these lymphomas have a course similar to that of low-grade follicular lymphomas composed predominantly of small centrocytes.64
and 17 in almost two-thirds of patients. Deletion of 13q is the most frequent abnormality and correlates with a stable clinical course in older patients. Trisomy 12, ATM deletions (11q), and TP53 deletions (17p) are associated with progressive disease.109,110 Molecular genetic studies divide CLL/SLL into two major groups based on the presence or absence of somatic mutation. The absence of somatic mutation is associated with a more aggressive clinical course and correlates with an increased expression of CD38 and the tyrosine kinase, ZAP-70.111,112,113,114,115 Risk stratification in CLL/SLL has therefore become an increasingly complex and controversial exercise with no clear consensus as to the relative roles of cytogenetics, molecular genetics for somatic mutation status, and use of surrogate markers such as CD38 and ZAP-70.116,117
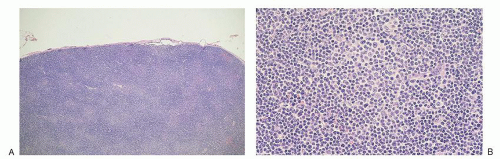 FIGURE 86.3. Lymph node: chronic lymphocytic leukemia/small lymphocytic lymphoma. A: Low magnification shows diffuse alteration of architecture, with pale areas corresponding to proliferation centers. B: A higher power of a proliferation center that is composed of intermediate-sized cells with small nucleoli that are surrounded by small round lymphocytes. |
antibodies to pan-B-cell antigens CD19, CD20, and CD22. CD23 is negative or sometimes partially expressed, and FMC7 is positive in contrast to the tumor cells of CLL/SLL.107,142 Overexpression of cyclin D1 is almost universal in mantle cell lymphoma143 (Fig. 86.4C), is not seen in follicular lymphoid hyperplasia, and is uncommon in other small B-cell malignancies, with the exception of plasmacytic neoplasms and hairy cell leukemia.144 Recent gene expression studies have identified SOX11 expression as another specific marker for mantle cell lymphoma.145 Rare “in situ” mantle cell lymphomas have been recognized in which cyclin D1-positive cells fill unexpanded mantle zones without distortion of nodal architecture.146 The clinical significance of these cases is unclear at this time. High Ki-67 expression is associated with a poor prognosis.147,148
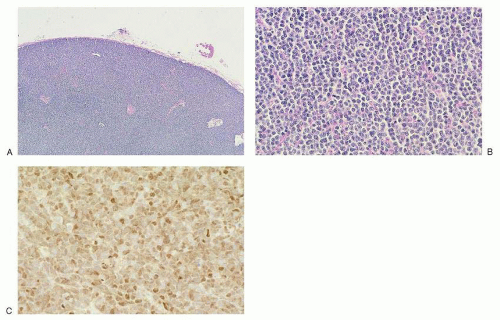 FIGURE 86.4. Lymph node: mantle cell lymphoma. A: Low magnification shows diffuse architectural effacement, which, on higher magnification in (B), is composed of sheets of small irregular lymphocytes with few large cells. C: Small lymphocytes exhibit nuclear staining for cyclin D1. |
TABLE 86.4 RISK FACTORS FOR EXTRANODAL MARGINAL ZONE B-CELL LYMPHOMAS OF MUCOSA-ASSOCIATED LYMPHOID TISSUE (MALT LYMPHOMAS) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
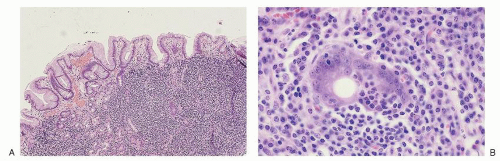 FIGURE 86.5. Stomach: extranodal marginal zone B cell lymphoma of mucosa-associated lymphoid tissue. A: The submucosa contains a diffuse infiltrate of small lymphocytes. B: Centrocyte-like cells with moderate amounts of clear cytoplasm invade gastric glands, producing lymphoepithelial lesions. |
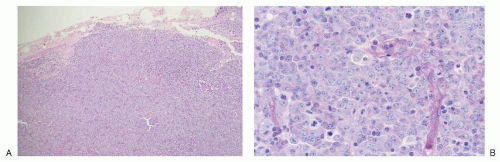 FIGURE 86.6. Lymph node: diffuse large B cell lymphoma. A: On low magnification, a neoplastic large lymphocyte population diffusely effaces lymph node architecture. B: On high magnification, round nuclei, partially clumped chromatin, small nucleoli and modest amounts of pale cytoplasm characterize the tumor cells. Note the apoptotic bodies and mitotic figures. |
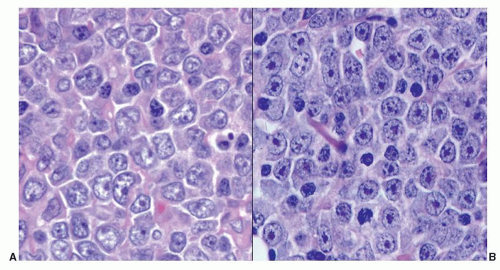 FIGURE 86.7. Lymph node: cytologic variability in diffuse large B cell lymphoma. A: Centroblastic. B: Immunoblastic. |
for practical purposes, several immunohistochemistry algorithms, Choi,204 Hans,205 Muris,206 Nyman,207 and Talley,208 have been proposed by which GCB and non-GCB are assigned. All have been shown to predict prognosis of R-CHOP-treated DLBCL patients in some, but not all studies. Therefore, phenotyping to assign a DLBCL to the GCB vs. non-GCB subtype for the purpose of selecting optimal therapy has been inconsistently applied to routine practice. Approximately 5% of DLBCL cases express CD5.200,209 These cases are distinguished from the blastoid variant of mantle cell lymphoma because they are cyclin D1–. They tend to occur in older patients, are more frequent in women than in men, and tend to be disseminated at the time of diagnosis.209 They are biologically distinct by genetic and gene expression profiling studies,210,211 but are not recognized as a separate entity in the WHO classification. One other marker of interest in DLBCL is Ki-67, a marker of cells in cycle. The fraction of Ki-67+ cells in a tumor is a general indicator of its proliferative rate. Ki-67 positivity ranges from 30% to nearly 100% in DLBCL cases, and it too, if expressed in greater than 90% of the cells, has been suggested as an adverse prognostic indicator in DLBCL.212
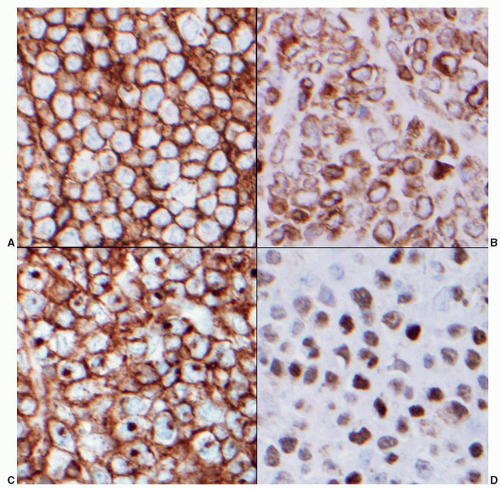 FIGURE 86.8. Lymph node: germinal center B cell lymphoma phenotype. By the criteria of the Hans algorithm this DLBCL has a germinal center phenotype. Using immunohistochemistry on paraffin sections the neoplastic cells express: A: CD20, B: bcl-2, C: CD10, and D: bcl-6. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


