Diabetes mellitus
Mark A. Sperling, MD, William V. Tamborlane, MD, Tadej Battelino, MD, PhD, Stuart A. Weinzimer, MD and Moshe Phillip, MD
Etiology, Pathogenesis, and Genetics
Clinical Manifestations of Diabetes Mellitus
Treatment of Diabetes Mellitus
TREATMENT OF TYPE 1 DIABETES MELLITUS (T1DM)
Multiple Daily Injection (MDI) Regimens
Indications for Pumps in Pediatrics
Residual B-Cell Function (The Honeymoon Period)
Associated Autoimmune Diseases
Associated Psychosocial Problems
THE FUTURE IS NOW: CLOSED-LOOP INSULIN DELIVERY
Non-autoimmune Type 1 Diabetes
CSII-Based Treatment Regiments
Temporary Basal Rates and Basal Insulin Suspend
GENETIC DEFECTS OF BETA CELL FUNCTION
GENETIC DEFECTS IN INSULIN ACTION
ACQUIRED DEFECTS IN INSULIN ACTION
GENETIC SYNDROMES WITH DIABETES AND INSULIN RESISTANCE OR INSULIN DEFICIENCY
Introduction
Diabetes mellitus (DM) is best defined as a syndrome characterized by inappropriate fasting or postprandial hyperglycemia, caused by absolute or relative insulin deficiency and its metabolic consequences, which include disturbed metabolism of protein and fat. This syndrome results from a combination of deficiency of insulin secretion and its action. Diabetes mellitus occurs when the normal constant of the product of insulin secretion times insulin sensitivity, a parabolic function termed the “disposition index” (Figure 19-1), is inadequate to prevent hyperglycemia and its clinical consequences of polyuria, polydipsia, and weight loss. At high degrees of insulin sensitivity, small declines in the ability to secrete insulin cause only mild, clinically imperceptible defects in glucose metabolism. However, irrespective of insulin sensitivity, a minimum amount of insulin is necessary for normal metabolism. Thus, near absolute deficiency of insulin must result in severe metabolic disturbance as occurs in type 1 diabetes mellitus (T1DM). By contrast, with decreasing sensitivity to its action, higher amounts of insulin secretion are required for a normal disposition index. At a critical point in the disposition index curve (see Figure 19-1), a further small decrement in insulin sensitivity requires a large increase in insulin secretion; those who can mount these higher rates of insulin secretion retain normal glucose metabolism, whereas those who cannot increase their insulin secretion because of genetic or acquired defects now manifest clinical diabetes as occurs in type 2 diabetes (T2DM).
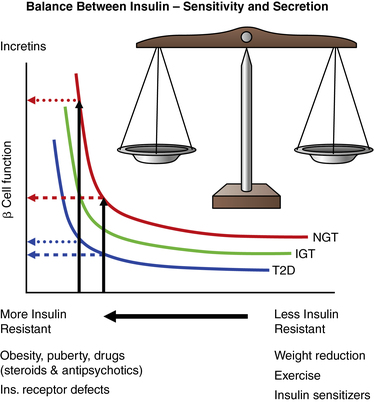
FIGURE 19-1  This figure shows the hyperbolic relationship of insulin resistance and beta cell function. On the y-axis is beta cell function as reflected in the first-phase insulin response during intravenous (IV) glucose infusion; on the x-axis is insulin sensitivity and its mirror image resistance. In a subject with normal glucose tolerance (NGT) and beta-cell reserve, an increase in insulin resistance results in increased insulin release and normal glucose tolerance. In an individual for whom the capacity to increase insulin release is compromised, increasing insulin resistance with partial or no beta-cell compensation results in progression from normal glucose tolerance, to impaired glucose tolerance (IGT), and finally to diabetes (T2D). Differences between these categories are small at high insulin sensitivity, which may be maintained by weight reduction, exercise, and certain drugs. At a critical degree of insulin resistance, due to obesity or other listed factors, only a further small increment in resistance requires a large increase in insulin output. Those that can increase insulin secretion to this extent retain normal glucose tolerance; those who cannot achieve this degree of insulin secretion (e.g., due to a mild defect in genes regulating insulin synthesis, insulin secretion, insulin action, or an ongoing immune destruction of beta cells) now unmask varying degrees of carbohydrate intolerance. The product of insulin sensitivity (the reciprocal of insulin resistance) and acute insulin response (a measurement beta-cell function) has been called the “disposition index.” This index remains constant in an individual with normal beta cell compensation in response to changes in insulin resistance. IGT, impaired glucose tolerance; NGT, normal glucose tolerance; T2D, type 2 diabetes. (From Ize-Ludlow, D., & Sperling, M. A. (2005). The classification of diabetes mellitus: a conceptual framework. Pediatr Clin North Am, 52, 1533–1552.)
This figure shows the hyperbolic relationship of insulin resistance and beta cell function. On the y-axis is beta cell function as reflected in the first-phase insulin response during intravenous (IV) glucose infusion; on the x-axis is insulin sensitivity and its mirror image resistance. In a subject with normal glucose tolerance (NGT) and beta-cell reserve, an increase in insulin resistance results in increased insulin release and normal glucose tolerance. In an individual for whom the capacity to increase insulin release is compromised, increasing insulin resistance with partial or no beta-cell compensation results in progression from normal glucose tolerance, to impaired glucose tolerance (IGT), and finally to diabetes (T2D). Differences between these categories are small at high insulin sensitivity, which may be maintained by weight reduction, exercise, and certain drugs. At a critical degree of insulin resistance, due to obesity or other listed factors, only a further small increment in resistance requires a large increase in insulin output. Those that can increase insulin secretion to this extent retain normal glucose tolerance; those who cannot achieve this degree of insulin secretion (e.g., due to a mild defect in genes regulating insulin synthesis, insulin secretion, insulin action, or an ongoing immune destruction of beta cells) now unmask varying degrees of carbohydrate intolerance. The product of insulin sensitivity (the reciprocal of insulin resistance) and acute insulin response (a measurement beta-cell function) has been called the “disposition index.” This index remains constant in an individual with normal beta cell compensation in response to changes in insulin resistance. IGT, impaired glucose tolerance; NGT, normal glucose tolerance; T2D, type 2 diabetes. (From Ize-Ludlow, D., & Sperling, M. A. (2005). The classification of diabetes mellitus: a conceptual framework. Pediatr Clin North Am, 52, 1533–1552.)
By simultaneously considering insulin secretion and insulin action in any given individual, it becomes possible to account for the natural history of diabetes in that person (e.g., remission in a patient with T1 diabetes or ketoacidosis in a person with T2DM). Thus, diabetes mellitus may be the result of absolute insulin deficiency, or of absolute insulin resistance, or a combination of milder defects in both insulin secretion and insulin action.1 Collectively, the syndromes of diabetes mellitus are the most common endocrine/metabolic disorders of childhood and adolescence. The application of molecular biologic tools continues to provide remarkable insights into the etiology, pathophysiology, and genetics of the various forms of diabetes mellitus that result from deficient secretion of insulin or its action at the cellular level.
Morbidity and mortality stem from the metabolic derangements and from the long-term complications that affect small and large vessels, resulting in retinopathy, nephropathy, neuropathy, ischemic heart disease, and arterial obstruction with gangrene of extremities.2 The acute clinical manifestations can be fully understood in the context of current knowledge of the secretion and action of insulin.3 Genetic and other etiologic considerations implicate autoimmune mechanisms in the evolution of the most common form of childhood diabetes, known as type 1a diabetes.4,5 Genetic defects in insulin secretion are increasingly recognized and understood as defining the causes of monogenic forms of diabetes such as maturity-onset diabetes of youth (MODY) and neonatal DM and contributing to the spectrum of T2DM.6
There is strong evidence that the long-term complications are related to the degree and duration of metabolic disturbances.2 These considerations form the basis of standard and innovative therapeutic approaches to this disease that include newer pharmacologic formulations of insulin, delivery by traditional and more physiologic means, and evolving methods to continuously monitor blood glucose to maintain it within desired limits by linking these features to algorithm-driven insulin delivery pumps for an “artificial pancreas.”
Classification
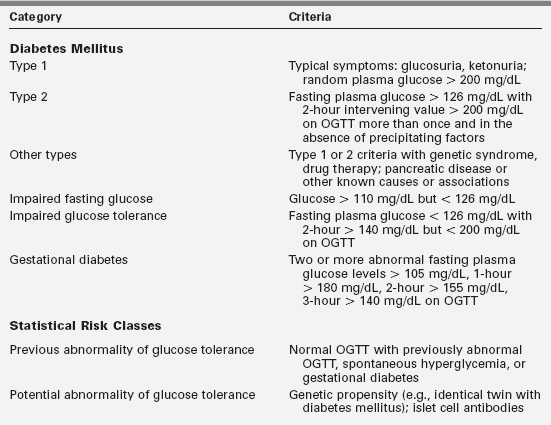
Diabetes mellitus is not a single entity but a heterogeneous group of disorders in which there are distinct genetic patterns as well as other etiologic and pathophysiologic mechanisms that lead to impairment of glucose tolerance.1,7 Box 19-1 outlines an etiologic classification of diabetes mellitus in children, based on the “Report of the Expert Committee on the Classification and Diagnosis of Diabetes Mellitus,” published by the American Diabetes Association in January of 2013.7
Our classification is modified to reflect more accurately the major categories in childhood, including the emergence of type 2 diabetes mellitus, cystic fibrosis-related diabetes, and drug-induced diabetes—largely from the antirejection agents cyclosporine, sirolimus, and tacrolimus (formerly FK-506). Table 19-1 presents a summary of the classification originally proposed in 1979 but incorporates the newer criteria for blood glucose values used to diagnose diabetes, impaired glucose tolerance, and gestational diabetes.
Among the insulin-dependent forms, severe lack of insulin secretion results most commonly from presumed autoimmune destruction of islets in genetically predisposed hosts. This form is synonymous with type 1a diabetes, formerly called juvenile-onset diabetes.4,5,8,9 Severe insulin-dependent diabetes mellitus, clinically indistinguishable from the autoimmune form, may, however, not have any evidence of autoimmunity and can result from mitochondrial or other gene defects that interfere with normal insulin secretion or rarely from pancreatic agenesis.10–13
The more severe forms of the MODY syndromes, subsequently detailed, also may require insulin.12,13 Clinically similar forms of diabetes may occur secondary to cystic fibrosis14,15 from toxic drugs such as the immunosuppressive agents cyclosporine, sirolimus and tacrolimus,16,17 the rodenticide Vacor,18 or streptozotocin as used for certain pancreatic islet cell tumors19; with the hemolytic uremic syndrome20; or after pancreatectomy, such as for persistent hyperinsulinemic hypoglycemia in infancy.21 Childhood insulin-dependent diabetes is generally type 1a diabetes mellitus.
Type 1 diabetes mellitus
This condition is characterized by severe insulinopenia and dependence on exogenous insulin to prevent ketosis and to preserve life. Thus, it was termed insulin-dependent diabetes mellitus (IDDM). The natural history of this disease indicates that there are preketotic non–insulin-dependent phases before and after the initial diagnosis. Although the onset is predominantly in childhood, the disease may occur at any age.1 Therefore, such names as “juvenile diabetes,” “ketosis-prone diabetes,” and “brittle diabetes” were abandoned in favor of the term type 1 diabetes.
Type 1a diabetes is generally distinct by virtue of its association with certain histocompatibility locus antigens (HLAs) and other genetic markers the majority of which determine the response to self (or exogenous) antigens; by the presence of circulating antibodies to cytoplasmic and cell-surface components of islet cells; of antibodies to insulin in the absence of previous exposure to exogenous injection of insulin, of antibodies to glutamic acid decarboxylase (GAD, the enzyme that converts glutamic acid to γ-aminobutyric acid found abundantly in the innervation of pancreatic islets), of antibodies to IA-2 (an islet cell-associated phosphatase and antibodies to the zinc transporter molecule ZnT8); by lymphocytic infiltration of islets early in the disease; and by coexistence with other autoimmune diseases.4,5 Occasionally, markers of autoimmunity are not found and yet there is profound insulinopenia and dependence on insulin without evidence of a mitochondrial or other genetic defect. In these cases, type 1 diabetes is considered idiopathic (type 1b). With the exceptions noted, diabetes in children is usually insulin dependent and fits the type 1a category.1
Type 2 diabetes
Persons with this subclass of diabetes (formerly known as “adult-onset diabetes,” “maturity-onset diabetes” [MOD], or “stable diabetes”) may not be permanently insulin dependent and only occasionally develop ketosis. Some may, however, need insulin to correct symptomatic hyperglycemia—and ketosis may develop in some during severe infections or other stress. Therefore, this was previously called non–insulin-dependent diabetes mellitus (NIDDM).1 This form of diabetes is becoming an increasing problem in overweight adolescents, especially those from vulnerable groups such as Africans, Mexicans, Native Indians, and other susceptible ethnic groups.22,23
Type 2 diabetes (T2DM) is not a single entity.1 T2DM may be a primary disorder, with inadequate insulin secretion caused by mutations in one of several genes encoding enzymes or transcription factors important to islet cell development and insulin secretion. Several of these defects are now part of the spectrum of the syndromes commonly associated with maturity onset diabetes of youth (MODY), which has a dominant mode of inheritance.12,13,24 However, some patients with MODY defects, which we term monogenic diabetes of youth, may require insulin from the outset or as they grow older and become insulin resistant, exceeding their ability to compensate by increasing insulin secretion (see Figure 19-1). A defect in the gene regulating glucose transport into the pancreatic beta cell, the GLUT2 transporter, may be responsible for another form of type 2 diabetes.24
Defects in glycogen synthase have also been implicated.25,26, A primary defect in insulin receptors—often associated with acanthosis nigricans,27 postreceptor defects (including Rad [Ras associated with diabetes]),28 and milder mitochondrial gene defects10—also may result in type 2 diabetes. Secondary causes of type 2 diabetes mellitus include excessive counterregulatory hormones, especially pharmacologic doses of glucocorticoids, antibodies to the insulin receptor, and obesity with impaired insulin secretion.29–40
In type 2 diabetes mellitus, the serum concentration of insulin may be increased, normal, or moderately depressed depending on whether the defect is one of insulin action or secretion.27–40 The onset of type 2 diabetes mellitus occurs in children generally around the time of puberty or shortly thereafter, but it is recognized, that it may occur at any age and is becoming increasingly frequent in childhood and adolescence.22,23 In some instances, there appears to be adequate secretion of insulin but resistance to its actions, and in some individuals it may represent slowly evolving type 1 diabetes mellitus.38 As an initial approach, weight reduction is indicated in children who are obese. In type 2 diabetes, there is no association with specific HLA antigens, autoimmunity, or various islet cell antibodies (ICAs).40 However, several genetic abnormalities regulating insulin secretion are increasingly implicated in T2DM.40
Type 1 diabetes mellitus
Epidemiology
The prevalence of diabetes mellitus is highly correlated with increasing age. Available data indicate a range of 1 case per 1430 children at 5 years of age to 1 case in 360 children at 16 years.41–45 Data on incidence in relation to racial or ethnic backgrounds indicate a range of more than 50 new cases annually per 100,000 population in Finland and Sardinia to about 1 per 100,000 in China and parts of South America42–46 (Figure 19-2). In all examined areas there appears to be an increasing incidence of T1DM of about 2% to 3% per year. However, the incidence of T1DM in Finland was reported to have peaked and slightly decined.41
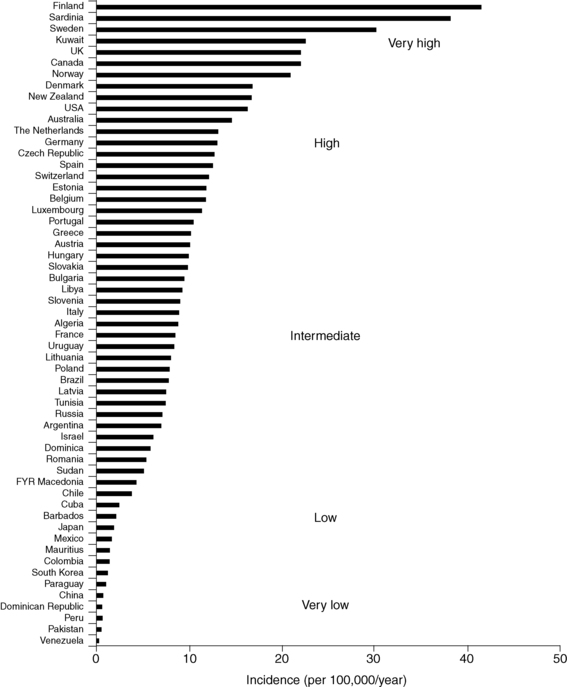
FIGURE 19-2  Age-standardized incidence (per 100,000/year) of type 1 diabetes in children younger than 14 years of age in 100 populations. Data for boys and girls have been pooled. Countries are arranged in descending order according to the incidence. (Puerto Rico and the Virgin Islands are presented separately from other populations in the United States.) (From Diamond Project Group (2006). Incidence of trends of childhood type1 diabetes worldwide 1990-1999. Diabet Med, 23, 857–866.)
Age-standardized incidence (per 100,000/year) of type 1 diabetes in children younger than 14 years of age in 100 populations. Data for boys and girls have been pooled. Countries are arranged in descending order according to the incidence. (Puerto Rico and the Virgin Islands are presented separately from other populations in the United States.) (From Diamond Project Group (2006). Incidence of trends of childhood type1 diabetes worldwide 1990-1999. Diabet Med, 23, 857–866.)
In the United States, the occurrence of type 1 in blacks had previously been reported to be only between one third and two thirds of that in whites.46 More recent data suggest that the incidence of diabetes mellitus in African Americans is increasing.46,47 It is not clear, however, whether this increase in incidence among African Americans is exclusively type 1 or includes cases of type 2 presenting in ketoacidosis and thus misclassified.46 The annual incidence in the United States is 20 to 25 cases per 100,000 of the childhood population44 (see www.cdc.gov/diabetes/projects/diabetes/children.htm and Figure 19-2).
Males and females appear to be almost equally affected. There is no apparent correlation with socioeconomic status. Peaks of presentation occur in two age groups: at 5 to 7 years of age and at the time of puberty. The first peak corresponds to the time of increased exposure to infectious agents coincident with the beginning of school. The latter corresponds to the pubertal growth spurt induced by increased pubertal growth hormone secretion that antagonizes insulin action. The incidence of type 1 diabetes is increasing worldwide, most prominently in certain populations (e.g., Finland) and in certain age groups (especially those younger than age 5 years).44,45 As mentioned, there appears to be a plateau in incidence reported from Finland.41 In younger patients, onset appears to be more abrupt and the extent of immune markers is less apparent than in older children.48 Type 1b diabetes with abrupt onset, less evidence of autoimmunity, and indicators of viral infection (including evidence of pancreatitis) has been described in Japan.49,50
Seasonal and long-term cyclical variations have been noted in the incidence of type 1 diabetes. Newly recognized cases appear to occur with greater frequency in the autumn and winter in the northern and southern hemispheres.51 Seasonal variations are most apparent in the adolescent years.51 There is no consistent pattern linking long-term cyclicity with the incidence of viral infections; however, there is a definite increased incidence of diabetes in children with congenital rubella.52,53 These changing patterns in incidence and associations with viral infections suggest a potential role for viruses or other microbial agents or their products as direct or indirect triggering mechanisms for inducing T1DM in a susceptible host.52–57
Etiology, pathogenesis, and genetics
The cause of the initial clinical findings in this predominant form of diabetes in childhood is the sharply diminished secretion of insulin.58 Although basal insulin concentrations in plasma may be normal in newly diagnosed patients, insulin production in response to a variety of potent secretagogues is blunted and usually disappears over a period of months to years. In certain individuals considered at high risk for the development of type 1 diabetes, such as the nonaffected identical twin of a diabetic, a progressive decline in insulin-secreting capacity has been noted for months to years before the clinical appearance of symptomatic diabetes that usually manifests when insulin-secreting reserve is 20% or less than normal for that individual (Figure 19-3).4,5,58
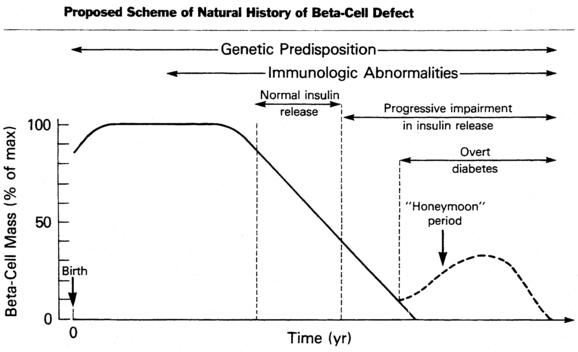
FIGURE 19-3  Proposed scheme of natural history of the evolution of insulin-dependent diabetes mellitus with progressive beta-cell failure. (From Sperling, M. A. (Ed.) (1988). Physician’s guide to insulin-dependent (type 1) diabetes mellitus: diagnosis and treatment. Alexandria, VA: American Diabetes Association.)
Proposed scheme of natural history of the evolution of insulin-dependent diabetes mellitus with progressive beta-cell failure. (From Sperling, M. A. (Ed.) (1988). Physician’s guide to insulin-dependent (type 1) diabetes mellitus: diagnosis and treatment. Alexandria, VA: American Diabetes Association.)
The mechanisms that lead to failure of the function of pancreatic beta cells point to autoimmune destruction of pancreatic islets in predisposed individuals. Type 1 diabetes has long been known to have an increased prevalence among persons with such disorders as Addison disease and Hashimoto thyroiditis, in whom autoimmune mechanisms are known to be pathogenic.58 These conditions, as well as type 1 diabetes mellitus, are known to be associated with an increased frequency of genes involved in the regulation of immunity including the autoimmune regulator gene AIRE, PTPN22, CTLA4, and INS gene itself as well as certain histocompatibility loci antigens (HLAs)—in particular, DR3 and DR4.58,59 Located on chromosome 6, the HLA system is the major histocompatibility complex—consisting of a cluster of genes that code transplantation antigens and play a central role in immune responses.58–70
Increased susceptibility to a number of diseases has been related to one or more of the identified HLA antigens. The inheritance of HLA DR3 or DR4 confers a twofold to threefold increased risk for developing type 1 diabetes. When both DR3 and DR4 are inherited, the relative risk for developing diabetes is increased sevenfold to tenfold. Application of newer molecular genetic techniques has revealed further heterogeneity in the HLA D region among individuals with and without diabetes despite possessing the DR3 or DR4 markers, suggesting the participation of other susceptibility loci within these markers.58–70
Extensive genome-wide scans of markers associated with type 1 diabetes mellitus have uncovered more than a 40 loci considered to confer susceptibility (Table 19-2). Some of these loci are confirmed and replicated by at least three different data sets. Others are suggestive but as yet not definitively linked. The strongest markers are those on chromosomes 6 and 11 (IDDM1 and IDDM2), respectively, linked to the HLA DQ β chain and the insulin gene itself.
TABLE 19-2
Genome-Wide Association Study and Meta-Analysis Find More Than 40 Loci Affect Risk of Type 1 Diabetes
| Loci | Approx. Relative Risk | Effect on |
| HLA | 6.5 | Immunity |
| INS | 2.3 | Insulin production and metabolism |
| PTPN22 | 2.0 | Immunity |
| ILR2A | 1.5 | Immunity |
| SH2BE | 1.3 | Immunity |
| ERBB3 | 1.3 | Insulin production and metabolism |
| PTPN2 | 1.25 | Immunity |
| CLEC16A | 1.20 | Unknown function |
| CTLA4 | 1.20 | Immunity |
| IL18RAP | 1.20 | Immunity |
| PTPN2 | 1.20 | Immunity |
| OCR5 | 1.20 | Immunity |
| IFIHI | 1.20 | Immunity |
| CTSH | 1.20 | Unknown |
| CD226 | 1.10 | Immunity |
| IL2RA | 1.10 | Immunity |
| PRKCQ | 1.10 | Immunity |
| IL2 | 1.10 | Immunity |
| BACH2 | 1.10 | Immunity |
| UBASH3A | 1.10 | Immunity |
| RGS1 | 1.10 | Immunity |
| IL7RA | 1.10 | Immunity |
| CITNF6 | 1.10 | Unknown |
| TNFAIP3 | 1.10 | Beta-cell apoptosis protection |
| TAGAP | 1.10 | Immunity |
Adapted from Barrett, J. C., Clayton, D. G., Concannon, P., et al. for the Type 1 Diabetes Genetics Consortium (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet, 41, 703–707.
In IDDM1, the homozygous absence of aspartic acid at position 57 of the HLA DQ b chain (non-Asp/non-Asp) confers an approximately 100-fold relative risk for developing type 1 diabetes. Those who are heterozygous with a single aspartic acid at position 57 (non-Asp/Asp) are less likely to develop diabetes and are no more susceptible than individuals who contain aspartic acid on both DQ b chains (i.e., homozygous Asp/Asp; Table 19-3). Some studies suggest that type 1 diabetes mellitus is proportional to the gene frequency of non-Asp alleles in that population.69 In addition, arginine at position 52 of the DQ a chain confers marked susceptibility to type 1.63 Position 57 of the DQ b and position 52 of the DQ a chains are at critical locations of the HLA molecule that permit or prevent antigen presentation to T-cell receptors and activate the autoimmune cascade (Figure 19-4).60–63
TABLE 19-3
HLA DR and DQ Phenotype Frequencies in Patients with Type 1 Diabetes Mellitus and Healthy Control Subjects
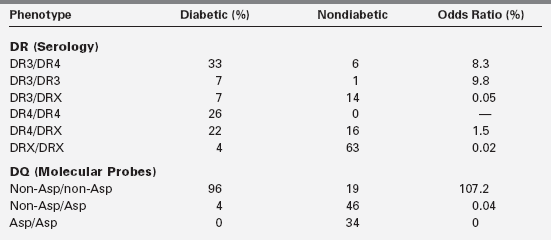
Based on Morel, P. A., Dorman, J. S., Todd, J. A., et al. (1988). Aspartic acid at position 57 of the HLA-DQ beta chain protects against type 1 diabetes: a family study. Proc Natl Acad Sci USA, 85, 8111.
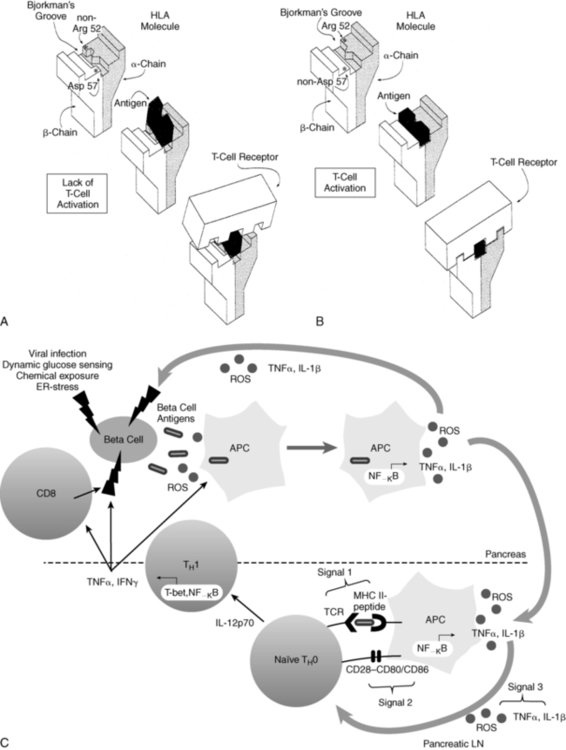
FIGURE 19-4  Representation of the interaction between antigen presentation in the context of specific HLA-DQ subtypes and the T-cell receptor. A, Presence of aspartic acid at position 57 of the DQ b chain and an amino acid other than arginine at position 52 of the a chain prevents antigen lodging in the Bjorkman groove. Therefore, antigen presentation to the T-cell receptor is impaired—and in the absence of this “fit,” T-cell activation is prevented. B, Lack of aspartic acid at position 57 of the DQ b chain and arginine at position 52 of the DQ a chain permits antigen to fit and be recognized by the T-cell receptor that is now activated. C, Role of redox in the immunopathology of type 1 diabetes. An initial genetic or environmental insult to the beta cell triggers the release of beta-cell antigens as well as the production of reactive oxygen species (ROS). Beta-cell antigens are phagocytosed, and ROS are able to stimulate redox-dependent transcription factors such as NF-κB, which leads to antigen presenting cell (APC) activation and cytokine secretion. ROS and proinflammatory cytokines secreted by APCs act as the third signal within the T-cell-APC immunologic synapse, which occurs in the pancreatic lymph node. ROS play a critical role in the progression of naïve TH0 cells to cytokine-secreting TH1 cells. Release of IFNγ by TH1 cells then works directly on the beta cells as well as activating more APCs and CD8 cells, all of which can impart deleterious effects on the islets. (A and B from Trucco M (1995). To be or not to be Asp 57, that is the question. Diabetes Care 15:705; Faas S, Trucco M (1995). The genes influencing the susceptibility to IDDM in humans. J Endocrinol Invest 17:477; C from Delmastro, M. M., & Piganelli, J. D. (2011). Oxidative stress and redox modulation potential in type 1 diabetes. Clin Dev Immunol, 2011. doi: 10.1155/2011/593863.)
Representation of the interaction between antigen presentation in the context of specific HLA-DQ subtypes and the T-cell receptor. A, Presence of aspartic acid at position 57 of the DQ b chain and an amino acid other than arginine at position 52 of the a chain prevents antigen lodging in the Bjorkman groove. Therefore, antigen presentation to the T-cell receptor is impaired—and in the absence of this “fit,” T-cell activation is prevented. B, Lack of aspartic acid at position 57 of the DQ b chain and arginine at position 52 of the DQ a chain permits antigen to fit and be recognized by the T-cell receptor that is now activated. C, Role of redox in the immunopathology of type 1 diabetes. An initial genetic or environmental insult to the beta cell triggers the release of beta-cell antigens as well as the production of reactive oxygen species (ROS). Beta-cell antigens are phagocytosed, and ROS are able to stimulate redox-dependent transcription factors such as NF-κB, which leads to antigen presenting cell (APC) activation and cytokine secretion. ROS and proinflammatory cytokines secreted by APCs act as the third signal within the T-cell-APC immunologic synapse, which occurs in the pancreatic lymph node. ROS play a critical role in the progression of naïve TH0 cells to cytokine-secreting TH1 cells. Release of IFNγ by TH1 cells then works directly on the beta cells as well as activating more APCs and CD8 cells, all of which can impart deleterious effects on the islets. (A and B from Trucco M (1995). To be or not to be Asp 57, that is the question. Diabetes Care 15:705; Faas S, Trucco M (1995). The genes influencing the susceptibility to IDDM in humans. J Endocrinol Invest 17:477; C from Delmastro, M. M., & Piganelli, J. D. (2011). Oxidative stress and redox modulation potential in type 1 diabetes. Clin Dev Immunol, 2011. doi: 10.1155/2011/593863.)
IDDM2 is a polymorphic marker near the transcription start site of the insulin gene, giving rise to variable numbers of tandem repeats (VNTR) at the promoter end of the insulin gene on chromosome 11. Each tandem repeat element consists of an approximately 14-bp DNA segment with a consensus nucleotide sequence. The number of repeats ranges from about 25 to about 200, and the three classes of alleles are based on overall size. Class I insulin VNTR consists of 26 to 63 repeats and confers susceptibility, whereas class III consists of 140 to 200 or more repeats and is protective of diabetes. Together, the gene markers on chromosomes 6 and 11 (i.e., IDDM1 and IDDM2) account for 50% to 60% of the heritability of type 1 diabetes. However, combinations of certain DQ alleles in association with certain DR alleles may confer susceptibility or protection to the development of type 1 diabetes (see Table 19-3).
In addition, other as yet undefined genetic factors play a role because the same high-risk genotypes are about sixfold more likely to develop diabetes in an individual with a positive family history than in one without a family history without type 1 diabetes (Table 19-4). Investigation of four genome-wide linkage scans in close to 1500 families with more than one affected member having T1DM identified several susceptibility loci. Of these, about 40% can still be attributed to allelic variation of HLA loci, and the influence of the VNTR in the insulin gene was confirmed.59,64
TABLE 19-4
Genetic Risk Estimates for HLA Class II in Type 1 Diabetes Mellitus
| High-Risk Genotypes | Risk in an Individual with This Genotype |
| DQB1p0302 (DQ3.2) | 1 in 60 |
| DQ3.2/DQ2 (DR3) | 1 in 25 |
| DQB1p03021 family history of IDDM | 1 in 10 |
| DQ3.2/DQ2 (DR3)1 family history of IDDM | 1 in 4 |
| Complete sharing of both HLA haplotypes | 1 in 2* |
*Individual is a sibling of patient with T1DM.
Adapted from Nepom, G. T. (1995). Class II antigens and disease susceptibility. Ann Rev Med, 46, 17; Aly, T. A., Ide, A., Jahromi, M. M., et al. (2006). Extreme genetic risk for type 1A diabetes. Proc Natl Acad Sci U S A, 103, 14, 10474–10479.
In addition, the cytotoxic T-lymphocyte antigen 4 (CTLA4) gene on chromosome 2 and the protein tyrosine phosphatase nonreceptor 22 (PTPN22) gene on chromosome 1p13 were found to contribute significantly to predisposition to T1DM. However, the genome scan identified other potential loci conferring susceptibility on chromosomes 2q31-q33, 10p14-q11, and 16q220q24 and a locus on the long arm of chromosome 6 (6q21) distinct from the HLA region on 6p21. The precise genes in these regions that may predispose to T1DM have not been identified as yet, although some have been excluded59 and newer candidate genes—such as CBLB interacting with CTLA4,66 the decay-accelerating factor gene (DAF, a complement inhibitor),67 and the interleukin 2A receptor IL2RA—are under scrutiny.63
These considerations provide a rational framework for the long-recognized association of type 1 diabetes with genetic factors on the bases of the increased incidence in some families, of the concordance rates in monozygotic twins, and of ethnic and racial differences in prevalence.59,63,70 From multiple family pedigrees and HLA typing data, it has been estimated that if a sibling shares both HLA D haplotypes with an index case the risk for that individual is 12% to 20%; for a sibling sharing only one haplotype, the risk for IDDM is 5% to 7%; and with no haplotypes in common, the risk is only 1% to 2%.70 HLA typing is not recommended for routine practice, but for purposes of genetic counseling it can be safely assumed that in whites the overall recurrence risks to siblings is approximately 6% if the proband is younger than 10 years of age and 3% if older at the time of diagnosis. The risk to offspring is 2% to 5%, with the higher risk in the offspring of a diabetic father. 63,70
Factors other than pure inheritance must also be involved in evoking clinical diabetes. For example, DR3 or DR4 is found in approximately 50% of the general population and non-Asp/non-Asp is found in approximately 20% of white nondiabetics in the United States. However, the risk for type 1 diabetes in these subjects is only one tenth that in an HLA-identical sibling of an index case with type 1 diabetes possessing these markers.70 Even siblings sharing only one haplotype have a sixfold to tenfold greater risk of developing type 1 compared with the normal population (see Table 19-4).
Importantly, approximately 10% to 15% of patients with type 1 do not have HLA DR3 or DR4 (see Table 19-3).58 Most compelling is the fact that the concordance rate among identical twins of whom one has insulin-dependent diabetes is only about 50%, suggesting the participation of environmental triggering factors or other genetic factor such as the postnatal selection of certain autoreactive T-cell clones that bear receptors recognizing “self.” This postnatal process occurs within the thymus and implies that identical twins are not identical with respect to the T-cell receptor repertoire they possess.
Triggering factors might include viral infections.54–57 In animals, a number of viruses can cause a diabetic syndrome, the appearance and severity of which depend on the genetic strain and immune competence of the species of animal tested. In humans, epidemics of mumps, rubella, and coxsackievirus infections have been associated with subsequent increases in the incidence of type1 diabetes. The acute onset of diabetes mellitus, presumably induced by coxsackievirus B4, has been described.57 The viruses may act by directly destroying beta cells, by persisting in pancreatic beta cells as slow viral infections, or by triggering a widespread immune response to several endocrine tissues.57
A superantigen response may be involved in triggering T cells, bypassing the classic presentation by antigen- presenting cells (APCs) of the processed antigen in the context of restricted HLA molecules to T-cell receptors.71 Some viruses and certain endotoxins or exotoxins are capable of inducing a superantigen response. In addition, the virus may induce initial beta cell damage—which results in the presentation of previously masked or altered antigenic determinants. It is also possible that the virus shares some antigenic determinants with those present on or in beta cells, including GAD, such that antibodies formed in response to the virus may interact with these shared determinants of beta cells, resulting in their destruction, an example of molecular mimicry.72–79
Nitrosamines and early exposure to cow’s milk have been suggested as factors that may trigger diabetes in those genetically at risk, thus explaining the reported lower incidence of diabetes among exclusively breastfed infants. This is the basis for one ongoing primary prevention study: the Trial to Reduce Insulin-dependent Diabetes in Those Genetically at Risk (TRIGR).72–79 Antecedent stress and exposure to certain chemical toxins have been implicated in the development of type 1 diabetes. Although the rodenticide Vacor has been a cause of diabetes in individuals deliberately or inadvertently poisoned by this agent, some of these patients had islet cell antibodies (ICAs), suggesting that such antibodies are secondary to islet damage or that evolving type 1 disease preceded the drug ingestion. Nitrosamines in cured meat have also been implicated in type 1 diabetes, as have other environmental toxins.72,80
Evidence supports an autoimmune basis for the development of type 1 diabetes. but why the beta cell is the specific target remains a mystery.43,81 Is the pancreatic beta cell the sole target of immune destruction (homicide) or a contributor to its own demise (suicide)? 43,81 Histologic examination of pancreas from patients with type 1 who die of incidental causes has revealed lymphocytic infiltration around the islets of Langerhans. Later the islets become progressively hyalinized and scarred, a process suggesting an ongoing inflammatory response that is possibly autoimmune.82 However, these changes are often patchy in distribution, so that areas that appear to contain normal beta cells are interspersed with areas of beta-cell destruction, similar to the patchy distribution of depigmentation found in vitiligo.58,83 Eighty percent to 90% of newly diagnosed patients with type 1 diabetes have ICA directed at cell surface or cytoplasmic determinants in their islet cells. The prevalence of these antibodies decreases with the duration of established disease. In contrast, after pancreatic transplantation ICA may reappear in patients whose sera had become negative for ICA before transplantation. Taken together, these findings suggest that ICA disappears as the antigens intrinsic to pancreatic islets are destroyed and reappear when fresh antigen (transplanted islets) is presented.
Studies in identical twins and in family pedigrees demonstrate that the existence of ICA may precede by months to years the appearance of symptomatic type 1 diabetes.4 In vitro, ICA may impair insulin secretion in response to secretagogues and can be shown to be cytotoxic to islet cells—especially in the presence of complement or T cells from patients with type 1 diabetes. About 80% of patients may have antibodies to GAD, and 30% to 40% of newly diagnosed patients have spontaneous anti-insulin antibodies at initial diagnosis. These antibodies may be detected months to years before clinical diabetes becomes apparent.4,60,75,81 A more recently described antibody, the zinc transporter, ZnT8, appears to be a major marker for progressive impairment of beta-cell function.4 There is also evidence of abnormal T-cell function with an alteration in the ratio of suppressor (regulatory) to killer T cells at the onset of the disease.4,5,8 Thus, the ability of Treg to modify the activity of T effector cells in causing beta-cell destruction is an area of investigation. These findings suggest that type 1 diabetes (akin to other autoimmune diseases such as Hashimoto thyroiditis) is a disease of “autoaggression” in which autoantibodies in cooperation with complement, T cells, cytokines, FAS, and FAS’s ligand, and other factors, induce apoptosis or destruction of the insulin-producing islet cells.5–8 Thus, the inheritance of certain genes (such as those associated with the HLA system on chromosome 6 or other immunoregulatory or immunomodulatory genes) appears to confer a predisposition for autoimmune disease—including diabetes—when triggered by an appropriate stimulus such as a virus.4,5,9 Evidence of superantigen-triggered T-cell receptor activation was discussed earlier.71
Although it is understood that some insulin-dependent diabetic patients have none of the frequently associated HLA antigens, the evidence for an immune basis of islet cell destruction is sufficiently compelling to have fostered several studies of different immunosuppressive agents in the treatment of newly diagnosed diabetics (Table 19-5). None of these immunosuppressive or immunomodulatory agents has had long-term positive outcome, and some (e.g., cyclosporine) have proven toxic to beta cells. Although newer approaches are being attempted, all must be considered as experimental and not be viewed as established or recommended therapy.82–89 The Diabetes Prevention Trial for T1DM (DPT1) was a multicenter randomized but nonblinded study using daily subcutaneous insulin and an annual admission for intravenous insulin infusion in first-degree relatives with proven risk factors for developing T1DM. Whereas prediction was highly accurate in identifying those most likely to develop T1DM within 5 years of entering the study, insulin injections had no protective effect to prevent the appearance of T1DM.82–89 Figures 19-3 and 19-4 summarize current concepts of the cause of type1 diabetes as an autoimmune disease, the tendency for which is inherited and in which autoimmune destruction of beta cells is triggered by an as yet unidentified agent (possibly a virus). The slope of decline in insulin varies, and there may be periods of partial recovery such that the course of decline in insulin secretion is bumpy rather than smooth. The point at which clinical features appear corresponds to approximately 80% destruction of insulin secretory reserve. This process may take months to years in adolescent and older patients, and weeks in the very young patient in whom acute destruction by non-autoimmune mechanisms may play a significant role. Higher titers of spontaneous anti-insulin antibodies and ICA are characteristic of more active islet cell destruction, typically in the younger patient, and may prove useful in predicting evolving diabetes.82–89
TABLE 19-5
Partial List of Past and Ongoing Trials in Type 1 Diabetes*
| Intervention | Outcome | Study Status |
| Prevention Trials | ||
| Cow’s milk protein avoidance (TRIGR) | Pilot, promising | Fully powered trial enrolled, results pending |
| Gluten avoidance | Pilot, inconclusive | |
| Omega-3 fatty acid supplementation (NIP) | Pilot, did not reach primary outcome | |
| Vitamin D supplementation | Pilot, promising | |
| Parenteral insulin (DPT-1) | No effect | |
| Oral insulin (DPT-1) | Did not reach primary outcome; promising in subset of subjects | Enrolling confirmatory study |
| Intranasal insulin | Finnish study, no effect | Study enrolling in Australia (INIT II) |
| Nicotinamide (ENDIT) | No effect | |
| Anti-CD3 mAb | Currently enrolling | |
| CTLA-4 lg | Currently enrolling | |
| Preservation Trials | ||
| Anti-CD3 mAb | Series of phase 2 and 3 studies, promising | |
| CTLA-4 lg | Phase 2, promising | |
| LFA-3 lg | Phase 2, promising, under-enrolled and just missed primary endpoint | |
| ATG, cyclophosphamide, G-CSF | Three phase 1 studies, promising efficacy but safety concerns | |
| ATG | Phase 2, did not meet primary endpoint | |
| G-CSF | Small phase 2 | Fully enrolled with results pending |
| ATG + G-CSF | Small phase 2 | Fully enrolled with results pending |
| IL-2 + rapamycin | Phase 1, transient decrease in C-peptide | Study discontinued |
| IL-2, low dose | Fully enrolled with results pending | |
| Polyclonal regulatory T cells | Two phase 1 studies, promising | North American study currently enrolling |
| Umbilical cord blood | Phase 1, no effect | |
| Anti-CD20 mAb | Phase 2, promising | |
| Anti-IL – 1 mAb | Phase 2, no effect | |
| Anti-IL – 1 receptor | Phase 2, no effect | |
| Alpha – 1 antitrypsin | Pilot, promising | Phase 2, fully enrolled with results pending |
| GAD-alum vaccine | Phase 2, 3 studies, no effect | |
| Diapep277 vaccine | Phase 2, 3 studies, mixed results | |
| Sitagliptin + lansoprazole | Phase 2 | Fully enrolled with results pending |
| Intense metabolic control | Phase 2, no effect |
From Thomas, H. R., & Gitelman, S. E. (2013). Altering the course of type 1 diabetes: an update on prevention and new-onset clinical trials. Pediatr Diabetes, 14, 311–321.
Prediction and prevention
Although no presently available single marker or test can accurately predict type 1 diabetes mellitus, evidence suggests that a combination of immune and genetic markers for type1 diabetes may provide predictability.82–89 Some authorities suggest that type1 diabetes is a predictable disease, but other authorities have raised objections because predictability is not as robust in their studies. Definitive preventive therapy is not available, thereby raising ethical dilemmas, and the majority of new cases occur sporadically in the absence of a positive family history in a first-degree relative.
Most predictive studies have been performed in first-degree relatives of patients with new-onset type 1 diabetes.74 Nevertheless, there is increasing evidence that the presence of high titers of islet cell, GAD, IA2, ZnT8, and insulin autoantibodies combined with a consistently diminished first-phase response of insulin to a pulse of intravenous glucose (corresponding to the fifth percentile or less for age in insulin response) can be used to reliably predict the onset of type 1 disease.82–89
Figure 19-5 demonstrates that in a set of first-degree relatives, conversion to type1 diabetes was highly dependent on the number of antibodies detected in their sera. Of those with three antibodies, about half developed clinical diabetes within 5 years of follow-up. First-phase insulin response and genetic (HLA) markers may be used to augment the predictability. For example, Table 19-6 demonstrates that the relative risk of developing clinical diabetes within 4 years of detecting ICA is almost 230 among those who possess all four heterodimers in HLA DQ β that predispose to diabetes (i.e., Asp57 2/2 and Arg52 1/1).
TABLE 19-6
Influence of Diabetic Heterodimers (ASP57neg Arg52pos) and Islet Cell Antibody (ICA) Status on Relative Risk for Developing Diabetes after 4 Years
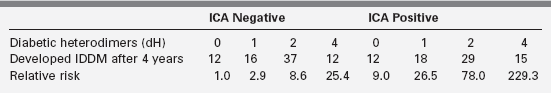
Adapted from Friday, R. P., Trucco, M., & Pietropaolo, M. (1999). Genetics of type 1 diabetes mellitus. Diabetes Nutr Metab, 12, 3.
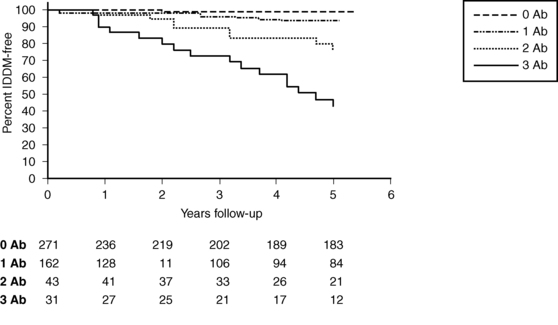
FIGURE 19-5  Survival analysis showing conversion to insulin-dependent diabetes mellitus (IDDM) in a subset of first-degree relatives according to the presence of 0, 1, 2, or 3 autoantibodies to islet antigens (ICA, GAD65, and IA-2 autoantibodies). (From Rosenbloom, A. L., Schatz, D. A., Krischer, J. P., et al. (2000). Therapeutic controversy: prevention and treatment of diabetes in children. J Clin Endocrinol Metab, 85, 494. Copyright © The Endocrine Society.)
Survival analysis showing conversion to insulin-dependent diabetes mellitus (IDDM) in a subset of first-degree relatives according to the presence of 0, 1, 2, or 3 autoantibodies to islet antigens (ICA, GAD65, and IA-2 autoantibodies). (From Rosenbloom, A. L., Schatz, D. A., Krischer, J. P., et al. (2000). Therapeutic controversy: prevention and treatment of diabetes in children. J Clin Endocrinol Metab, 85, 494. Copyright © The Endocrine Society.)
As technologic improvements continue, it is likely that population-wide screening for antibody markers (alone or combined with specific genetic markers) will be available to identify those at risk for developing type 1 diabetes. Such population-wide screening would be ethically justified if prevention could be proven effective. Presently, the data are sufficiently persuasive to have fostered national trials in Europe and the United States to predict and possibly prevent the clinical onset of type 1 diabetes through immune intervention strategies (see Table 19-5).
The European Nicotinamide Diabetes Intervention Trial (ENDIT) was a multicenter trial that screened approximately 22,000 first-degree relatives of patients with type 1 diabetes to identify 500 considered to be at high risk for developing this disease.86–89 These at-risk individuals were treated with nicotinamide or a placebo in a double-blind fashion. The purported advantage of nicotinamide was that it was not known to be toxic or harmful in humans at the recommended doses; its major disadvantage was that the proposed protective effects in delaying diabetes were based on a small sample cohort. The results of ENDIT were disappointing, with positive protection not apparent.
The U.S. Diabetes Prevention Trial for T1DM (DPT1) was based on promising pilot data that suggested preservation of insulin secretion and prevention of progression to diabetes mellitus in at-risk individuals treated with insulin.85 Daily subcutaneous insulin, coupled with intensive intravenous insulin every 9 months, prevented diabetes for at least 3 years in five subjects considered to be at risk because of genetic markers, islet cell and insulin autoantibodies, and diminished first-phase insulin response. Among seven similar at-risk subjects who chose not to be treated, six developed insulin-dependent diabetes within 3 years. DPT-1 was concluded in 2001, and there was no difference in the rates of developing diabetes among the placebo and insulin-treated groups. However, the ENDIT and DPT1 studies proved that large-scale multicenter studies could be successfully undertaken and that the prediction of progression to clinical diabetes was remarkably accurate. Thus, in those at highest risk (such as first-degree relatives of patients with T1DM) prediction is feasible and the discovery of a successful means of arresting or reversing progression to clinical diabetes is the subject of intense research.
Another study (TRIGR) involves 3000 families in whom half will avoid cow’s milk for the first 9 months of life to test the hypothesis that the ingestion of breast milk and avoidance of cow’s milk formula (with its bovine serum albumin [BSA]) may protect participants from the appearance of diabetes.78 Several other studies are examining the utility of antibodies to the IL2 receptor, CD3 antibodies, immune suppressors such as mycophenolate mofetil, and immune modulators in preventing diabetes, including oral insulin. These studies are conducted by worldwide consortia of participating institutions (see www2.diabetestrialnet.org).
In animal models, oral insulin or oral GAD has been successfully used to prevent diabetes.90 It is postulated that ingestion of T lymphocyte–dependent antigens can establish immunologic tolerance. Such oral strategies have been proposed and oral insulin is being tested in humans.90 The subjects of primary prevention trials and secondary intervention trials to preserve residual insulin secretion at initial diagnosis are of major interest to investigators and clinicians alike. Progress is likely, but at present all of these strategies must be viewed as experimental and not currently in the domain of daily clinical practice.
Insulin biosynthesis
Insulin is synthesized on the ribosomes of pancreatic islet beta cells and released into the circulation as a molecule composed of two separate straight polypeptide chains linked by disulfide bridges between and within these chains.91–98 The two chains are not synthesized separately but are derived from a larger precursor, proinsulin, a single coiled chain in which the NH2 terminus of the A chain is linked to the COOH terminus of the B chain by a connecting peptide, known as C-peptide (Figure 19-6). An even larger precursor (preproinsulin, containing an additional peptide chain on the NH2 terminus of the A chain) is first synthesized, but this additional piece (important to the initiation of synthesis) is rapidly excised. Further processing of proinsulin within the beta cell cleaves the C-peptide, consisting of 31 amino acids, from the insulin molecule at the sites indicated in the figure.
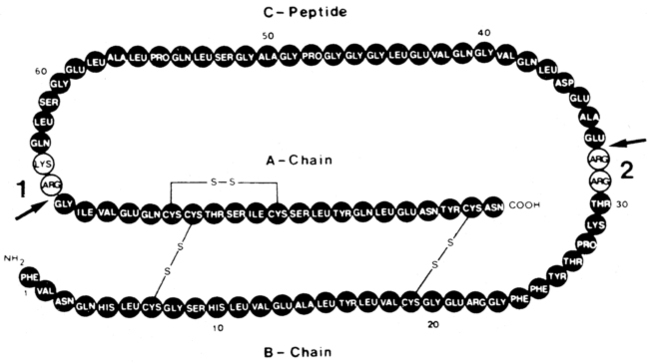
FIGURE 19-6  Structure of proinsulin. Arrows 1 and 2 indicate the two sites of normal cleavage that yield insulin and C-peptide when the amino acid residues indicated in the open circles are removed. These cleavage points are known mutation sites, are inherited in an autosomal-dominant manner, and can yield two types of familial hyperproinsulinemia. During insulin secretion, equimolar amounts of insulin and C-peptide are released.
Structure of proinsulin. Arrows 1 and 2 indicate the two sites of normal cleavage that yield insulin and C-peptide when the amino acid residues indicated in the open circles are removed. These cleavage points are known mutation sites, are inherited in an autosomal-dominant manner, and can yield two types of familial hyperproinsulinemia. During insulin secretion, equimolar amounts of insulin and C-peptide are released.
Defects in these cleavage sites are inherited in an autosomal-dominant manner and result in insulin molecules with less-than-normal biologic activity that can give rise to two types of familial hyperproinsulinemia. One defect yields B-C proinsulin, cleaved at site 1 but not at site 2 (see Figure 19-6). This intermediate has 50% of the biologic activity of insulin, which is sufficient to prevent any abnormality in carbohydrate metabolism. The defect at site 1 yields A-C proinsulin, cleaved at site 2 but not at site 1, which has inadequate biologic activity to prevent carbohydrate intolerance. A structural mutation in the proinsulin molecule, between the C-peptide and insulin, has been confirmed.90–96 In addition, a defect also occurs in the enzymatic conversion of a normal proinsulin molecule to insulin—yielding hyperproinsulinemia and mild carbohydrate intolerance.91
The proconvertases responsible for correct conversion of proinsulin to insulin are also involved in the processing of other hormones. Thus, impaired prohormone processing may lead to obesity and secondary hypocortisolism owing to defective processing of pro-opiomelanocortin (POMC) and to hypogonadotropic hypogonadism.97–98
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


