The main objectives of phase 1 trials have traditionally been to determine a dose that is appropriate for use in phase 2 and 3 trials and to determine information about the pharmacokinetics of distribution of the drug. Patients with advanced disease that is resistant to standard therapy but who have normal organ function are usually included in such trials.
Phase 1 trials are usually initiated at a low dose that is not expected to produce serious toxicity. A starting dose of one-tenth the lethal dose (expressed as milligrams per square meter of body surface area) in the most sensitive species usually is used.3 The dose is increased for subsequent patients according to a series of preplanned steps. Dose escalation for subsequent patients occurs only after sufficient time has passed to observe acute toxic effects for patients treated at lower doses. Cohorts of three to six patients are treated at each dose level. Usually, if no dose-limiting toxicity (DLT) is seen at a given dose level, the dose is escalated for the next cohort. If the incidence of DLT is 33%, then three more patients are treated at the same level. If no further cases of DLT are seen in the additional patients, then the dose level is escalated for the next cohort. Otherwise, dose escalation stops. If the incidence of DLT is >33% at a given level, then dose escalation also stops. The phase 2 recommended dose often is taken as the highest dose for which the incidence of DLT is <33%. Usually, six or more patients are treated at the recommended dose.
The dose levels themselves are commonly based on a modified Fibonacci series. The second level is twice the starting dose, the third level is 67% greater than the second, the fourth level is 50% greater than the third, the fifth is 40% greater than the fourth, and each subsequent step is 33% greater than that preceding it. Escalating doses for subsequent courses in the same patient are generally not done, except at low doses before any DLT has been encountered.
Accelerated Titration Designs
There is no compelling scientific basis for the approach just outlined, except that experience has shown it to be safe. Traditional phase 1 trials have three limitations:
1. They sometimes expose too many patients to subtherapeutic doses of the new drug.
2. The trials may take a long time to complete.
3. They provide very limited information about interpatient variability and cumulative toxicity.
New trial designs have been developed to address these problems.4 The accelerated titration designs5 permit within-patient dose escalation and use only one patient per dose level until grade 2 or greater toxicity is seen. Doses are titrated within patients to achieve grade 2 toxicity. The analysis consists of fitting a statistical model to the full set of data that includes all grades of toxicity for all courses of a patient’s treatment. The model includes parameters that represent the steepness of the dose-toxicity curve, the degree of interpatient variability in the location of the dose-toxicity curve, and the degree (if any) of cumulative toxicity. All these parameters are estimated from the data.
Several variants of the accelerated titration design were studied. Design A uses conventional 40% dose steps during the initial accelerated phase, whereas designs B and C use 100% dose steps until one patient experiences DLT or two patients experience grade 2 toxicity. At that point, acceleration ceases and standard cohorts of three to six patients with 40% dose-step increments are used. These designs were compared to a control design using cohorts of three to six patients with 40% dose-step increments and no intrapatient dose escalation.
In the 20 phase 1 trials initially evaluated, only three showed any evidence of cumulative toxicity. The average number of patients required was reduced from 39.9 for the control design to 24.4, 20.7, and 21.2 for designs A, B, and C, respectively. The average number of patients who had grade 0 to 1 toxicity as their worst toxicity grade over three cycles of treatment was 23.3 for the control but only 7.9, 3.9, and 4.8 for designs A, B, and C, respectively. The average number of patients with a worst toxicity grade of 3 increased from 5.5 for the control to 6.2, 6.8, and 6.2 for designs A, B, and C, respectively. The average number of patients with a worst toxicity grade of 4 increased from 1.9 for the control to 3.0, 4.3, and 3.2 for designs A, B, and C, respectively. Accelerated titration designs appear to be effective in reducing the number of patients necessary for finding the maximum tolerated dose, for reducing the number who are undertreated, and for providing increased information. They do not necessarily reduce the length of time necessary for completion of the trial. They increase the information yield if investigators analyze the results of the trial using the model developed by Simon et al.5 Software for fitting the model is available at http://brb.nci.nih.gov. Software for determining dose assignments and for recording the data in a spreadsheet format are also available at that website. The model of Simon et al.5 uses actual worst grade toxicity for each course of treatment of each patient, and it enables one to determine whether there is cumulative toxicity and to estimate the variability among patients in toxic effects. The use of the accelerated titration design has been reviewed.6,7
Continual Reassessment Methods
O’Quigley, Pepe, and Fisher8 used a dose-toxicity model to guide the dose escalation, as well as to determine the maximum tolerated dose. A Bayesian prior distribution is established for the steepness of the dose-toxicity curve and the distribution is updated after each patient is treated. The model is based on using only first-course treatment data and whether the patient experiences DLT. This approach is called the continual reassessment method. For each new patient, the model is used to determine the dose predicted to cause DLT to a specified percentage of the patients. That dose is assigned to the next patient. Many modifications of the original continual reassessment method have been subsequently proposed.9–11
For some tumor vaccines and molecularly targeted drugs, toxicity may not be dose limiting,12 and the dose selected may be based on preclinical findings or on practical considerations. For some molecularly targeted drugs, preclinical studies provide a target serum concentration of the active moiety necessary to maximally inhibit the target, and drug administration can be titrated for each patient to the targeted serum concentration. This approach can be complex because it involves developing a population pharmacokinetic model relating dose to concentration as the study progresses. A simpler approach is to have separate cohorts of patients who are treated at each of several dose levels without intrapatient dose titration. A population pharmacokinetic model relating dose to concentration is fit to the data.
Ideally, a trial design should provide the smallest dose that gives maximum biologic effect. For molecularly targeted therapeutics, the biologic effect might be a measure of the degree of inhibition of the target. Because it can be very difficult to obtain tumor samples before and after treatment, biologic effect is sometimes measured in an accessible surrogate tissue, such as peripheral blood lymphocytes or skin, or by using functional imaging.13 For therapeutic vaccines, the biologic effect might be a measure of stimulation of tumor reactive T cells.
Finding the dose that provides maximum biological effect is often not practical in a phase 1 trial, as it may require a large number of patients. For example, to have 90% power for detecting a one standard error difference in mean response between two dose levels at a one-sided 10% significance level requires 14 patients per dose level. A more limited objective is to identify a dose that is biologically active. Korn et al.14 developed a sequential procedure for finding such a dose when the measure of biologic response is binary. During an initial accelerated phase, they treat one patient per dose level until a biologic response is seen. Then, they treat cohorts of three to six patients per dose level. With zero to one biologic responses among three patients at a dose level, they escalate to the next level. With two to three responses among three patients, they expand the cohort to six patients. With five to six biologic responses from the six patients, they declare that dose to be the biologically active level and terminate the trial. With four or fewer biologic responses at a level, they continue to escalate.
Designs have also been developed for phase zero proof of concept trials.15,16 Patients are treated with single doses of a new drug at very low concentrations not expected to cause toxicity. This enables the investigator to obtain an early assessment of whether the molecular target of the drug is being inhibited by measuring a pharmacodynamic end point before and after drug administration. These trials require prior development of an assay for measuring the pharmacodynamic end point and an adequate database for estimating the variability of measurement for independent tissue samples of the same patient. This estimate should reflect variability of tissue sampling as well as technical variability of the assay. The approach developed depends on having a good estimate of assay variability and in having assay sufficiently reproducible to be able to reliably classify individual patients as responders or nonresponders based on the observed change in the level of the pharmacodynamic end point. The designs described by Rubinstein et al.16 utilize a small numbers of patients for establishing whether the drug causes target inhibition in a substantial proportion of patients.
Patient Selection
Phase 2 trials have traditionally been performed separately by tumor type in patients with the least amount of prior therapy for whom no effective therapy is available. With cytotoxics, full-dose chemotherapy is often impossible in patients debilitated by prior treatment, and lack of chemotherapeutic activity in previously treated patients may not indicate lack of clinical usefulness in earlier disease. The development of molecularly targeted drugs has introduced new complexities with regard to selection and evaluation of patients for phase 3 trials. When the target of the drug is clearly known, it may be more appropriate to select patients based on target expression than based on primary site of disease. Even if target expression is not used as an eligibility criterion, the drug should be evaluated in an adequate number of patients whose tumors express the target. Consequently, it is important to have an adequate assay for the target available at the time that phase 2 development begins.
In many cases, the drug will have multiple targets; there may be several candidate assays available for each target. Expression of the target will often prove to be only part of the relevant genomic information. For example, the effectiveness of antiepidermal growth factor receptor antibodies cetuximab and pannitumumab turned out to depend on whether the tumor had an activating K-RAS mutation.17–19
Whereas the major objective of phase 2 trials has traditionally been to identify the primary tumor sites in which a new drug was active, a new important objective is to develop promising predictive biomarkers that identify the patients whose tumors are most (or least) likely to respond to the drug. The phase 2 development stage is also the time to select the assay(s) that will be used in the phase 3 trials of the new drug and to define the criteria that will be used to either select patients for such trials or to structure the analysis, as will be described later in this chapter.
It is often undesirable to restrict entry to phase 2 trials based on what one thinks one knows about the drug target, at least in cases where this knowledge is uncertain. It is important, however, to ensure that the activity of the drug is not missed because the phase 2 trials did not accrue enough of the right kinds of patients. The decision of whether to restrict entry based on the presumed mechanism of action will depend in part on the adverse effects of the drug.
If tumor specimens are archived for the patients entered on broad eligibility phase 2 trials, then one avoids the need to develop assays in advance for all candidate targets, but it is not possible to ensure adequate accrual for subsets of patients whose tumors are positive for the candidate markers. Pusztai, Anderson, and Hess20 described a hybrid approach that begins with conducting a standard single-arm two-stage design for evaluating whether the overall response rate for unrestricted patients is sufficiently large. If the overall response rate is sufficient in the first stage of the standard phase 2 trial, then the second stage is completed with accrual of additional unrestricted patients. If there are too few responses overall in the first stage, then one starts a two-stage phase 2 study restricting entry to patients who are marker positive. If there are multiple markers of interest, then one restricts entry to patients positive for one of the markers and ensures that each marker has sufficient number of positive patients for evaluation. LeBlanc et al.21 have described how multiple primary sites can be incorporated in a single phase 2 trial.
In some cases, the list of candidate targets can be narrowed using mRNA transcript expression profiling of the pretreatment specimens. By comparing pretreatment expression levels of responders to nonresponders, one can potentially prioritize targets for assay development. If one does not have a good list of candidate targets, genomewide expression profiling can be used to develop a classifier of the tumors likely to respond to the drug. Dobbin, Zhao, and Simon22 have provided sample size guidelines for genomewide expression profiling studies and generally recommend at least 20 responders for developing a classifier. Pusztai, Anderson, and Hess20 performed a computer simulation study to indicate that HER-2 transcript overexpression would have been missed as a predictive biomarker for treatment of advanced breast cancer with trastuzumab in whole genome expression profiling with only five responders to analyze. They recommend analysis based on candidate genes if the number of responders are very limited.
Single-Arm Phase 2 Trials
Single Agents
For most single-agent phase 2 trials, the objective is simply to determine whether the drug has activity against the tumor type in question. For this objective, response rate based on the response evaluation criteria in solid tumors guidelines may provide a satisfactory approach.23 A variety of statistical accrual plans and sample size methods have been developed for single-arm phase 2 trials. One of the most popular approaches is the optimal two-stage design.24 n1 evaluable patients are entered into study in the first stage of the trial. If no more than r1 responses are obtained among these n1 patients, then accrual terminates and the drug is rejected as being of little interest. Otherwise, accrual continues to a total of n evaluable patients. At the end of the second stage, the drug is rejected if the observed response rate is less than or equal to r/n, where r and n are determined by the design used.
Tables 36.2 and 36.3 illustrate some of these optimized designs, and a web-based interactive computer program is available at http://linus.nci.nih.gov/brb. To select a design, the investigator specifies the target activity level of interest, p1, and also a lower activity level, p0, representing inadequate activity. The first row of each triplet of optimal designs provides designs with probability 0.10 of accepting drugs worse than p0 and probability 0.10 of rejecting drugs better than p1. Subject to these two constraints, the optimal designs minimize the average sample size. The average sample size is calculated at the lower activity level p0 to optimize protection of patients from exposure to inactive drugs. The tables show for each design the optimal values of r1, n1, r, and n; the average sample size; and the probability of stopping after the first stage for a drug with activity level p0.
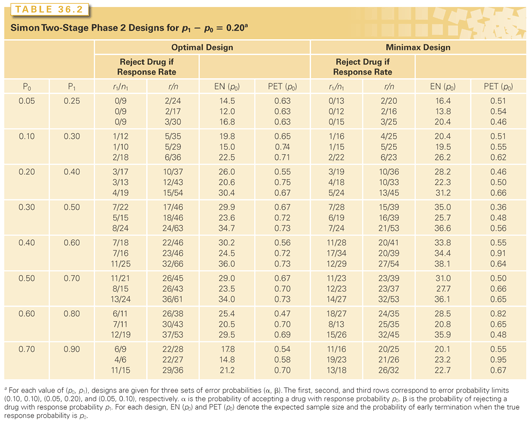
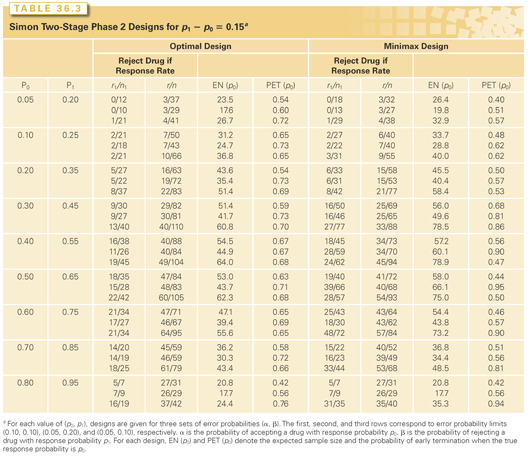
These tables also show the “minimax” designs, which provide the smallest maximum sample size n that satisfies the two constraints just described. Although minimax designs have somewhat larger average sample sizes than do optimal designs, in some instances, they are preferable because the small increase in average sample size is more than compensated for by a large reduction in maximum sample size.
The designs shown in Tables 36.2 and 36.3 are two-stage designs with the potential for early stopping for lack of activity. Optimized three-stage designs have been described by Ensign et al.25 Others have extended the design to incorporate toxicity or tumor progression information.26–28
Some authors have recommended use of progression-free survival instead of response29 for evaluating molecularly targeted drugs that may be cytostatic. Single-arm phase 2 trials can be designed using Tables 36.2 and 36.3 for testing whether the proportion of patients with stable disease at a specified landmark time like 12 months after the start of treatment is greater than a specified value p0, but that is only meaningful if the value p0 is a stable, robust, and well-characterized stable disease rate that results from multiple large studies with control regimens. Single-arm studies using stable disease are rarely planned or analyzed with that care and hence conclusions of single-arm phase 2 trials claiming that molecularly targeted agents cause disease stabilization are often dubious.30 Vidauurre et al.30 have questioned, however, whether molecularly targeted drugs are any more cytostatic than conventional chemotherapy drugs. El-Maraghi and Eisenhauer31 have also recommended that objective response is a useful end point for screening molecularly targeted agents.
Combination Regimens
Determination whether a new drug adds anticancer activity to an active regimen is inherently comparative. In using Tables 36.2 and 36.3 to design a single-arm trial, p0 should represent the level of activity of existing standard regimens. If this response probability is not well determined, however, because it varies among studies and varies based on patient prognostic factors, then a single-arm trial based on an assumed known p0 may not be appropriate.
Several approaches to single-arm study design have been developed that attempt to either account for or control the variability in p0. One approach to controlling this variability is to base the analysis of the single-arm trial on comparison to a specific set of control patients, matched for prognostic factors, and treated at the same institution as those for the new study. This can be a better approach than just using an assumed known value of p0 as described previously, but it still assumes that adjustment for known prognostic factors is sufficient to ensure comparability. Although such historic control comparisons are not considered reliable enough to eliminate the need for phase 3 trials, if done carefully, they may provide an adequate basis for decisions about which new regimens are worthy of phase 3 evaluation.
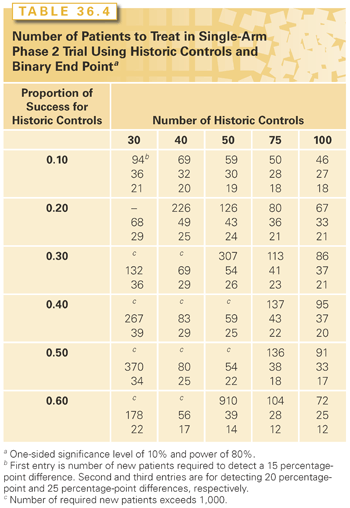
For comparative trials of response rates using specific historic controls, the sample size should be planned using the formulas appropriate for randomized clinical trials. By inserting the number of historic controls to be used, one can compute the number of patients needed to treat on the new regimen in the single-arm phase 2 trial.32 For binary end point data, the results of these calculations are presented in Table 36.4 for 80% power with a one-sided 10% significance level. The tabulated entries indicate that a 25 percentage-point difference can be detected with <40 new patients if there are at least 30 appropriate historic controls. The table entries indicate that detecting a 15 percentage-point difference is almost never feasible with this single-arm approach and that detecting a 20 percentage-point difference generally requires at least 50 appropriate historical controls and ≥60 new patients.
Thall and colleagues33,34 have developed and used Bayesian methods for planning and conducting single-institution trials comparing one or more new regimens to a specific set of historic controls who received a control treatment at the same institution. The Bayesian designs provide for continual analysis of results with either tumor response or time to event end points or for joint monitoring of efficacy and toxicity. Their methods require a substantial number of patients who have been treated on protocol with an appropriate control regimen and who have been staged comparably to the patients to be treated with the new regimen.
Korn et al.35 developed an approach for using historic control data in phase 2 multicenter trials of metastatic melanoma. They reviewed 42 previous phase 2 trials in melanoma conducted by US cancer cooperative oncology groups. They found that after adjustment for performance status, sex, presence of visceral disease, and presence of brain metastases, there was little interstudy variability in survival among the arms of the phase 2 trials. Consequently, for any single-arm phase 2 trial of metastatic melanoma, one can use their results in conjunction with the prognostic makeup of the patients in the new study to synthesize a benchmark overall survival curve or a benchmark 1-year overall survival rate for use in evaluating the new regimen. They provide an example of planning a phase 2 trial using this approach that required 72 patients to have 85% to 90% power for detecting a 15 percentage-point improvement in the 1-year overall survival rate with a one-sided type 1 error of 10%. They found that this approach was less satisfactory for use with progression-free survival because interstudy variability remained substantial after adjustment for prognostic factors.
Mick, Crowley, and Carroll36 proposed that the time to progression of a patient on a phase 2 trial be compared to the time to progression of the same patient on his/her previous trial. The ratio of these times was called a growth modulation index, and the agent was considered active if the index was >1.3 on average. In practice, however, follow-up intervals on various protocols are different, and there may be substantial variability and bias in computing the ratio of progression times. As tumors grow larger, the doubling time may increase and hence in some cases the chance of false-positive findings may be inflated.37
Randomized Phase 2 Trials
Time to tumor progression or disease-free survival has been recommended for evaluation of single-agent phase 2 trials of drugs that may be cytostatic and for trials adding a new drug to an active regimen. Even single-agent phase 2 trials of cytotoxics have been criticized on the basis that they do not provide much evidence that the drug will be able to prolong survival when incorporated into a regimen with other active drugs. Demonstrating that the regimen incorporating the new drug prolongs progression-free survival compared to the control regimen may provide a stronger basis for conducting a phase 3 trial of the new regimen.
Simon et al.12 suggested two key design differences between such randomized phase 2 designs and phase 3 designs. A randomized phase 2 design may use an end point that is a sensitive indicator of antitumor effect, although it may not be an acceptable phase 3 end point that directly reflects patient benefit. Such an endpoint does not need to be “validated.” It is not claimed to be a valid surrogate for survival; no regulatory approval or practice standard decisions should be based on the phase 2 trials using such an intermediate end point. The purpose of the phase 2 trial is merely to determine whether to conduct a phase 3 trial that will evaluate the new regimen with an accepted phase 3 end point The phase 2 trial may also serve to optimize the regimen that might be carried forward to phase 3 and to provide information about the best target population. The second key difference noted by Simon et al.12 is that the type I error “alpha level” for planning and analyzing the phase 2 trial can be increased from the two-sided 5% level used for phase 3 trials. By letting this alpha level increase to a one-sided 10%, meaningful savings in number of patients required can be achieved.
How large should a randomized phase 2 design comparing a new treatment to a control regimen be? Consider, for example, a randomized phase 3 trial comparing a new regimen to a control in a patient population in which the median time to progression on the control is 6 months and the median survival is 2 years. A 25% reduction in the hazard of death amounts to a 4-month prolongation of median survival with exponential distributions. A phase 3 trial with 90% statistical power for detecting this effect at a two-sided 5% significance level would require about 510 deaths (see Table 36.7). With an average follow-up time of 2 years, 50% of the patients would have events and so the number of patients required for randomization would be just in excess of 1,000. A randomized phase 2 trial with 90% power for detecting a 33% reduction in hazard of progression corresponding to a 2-month increase in median progression-free survival at a one-sided 10% significance level would require observing 164 progression events (Table 36.5). With an average follow-up time of 2 years, >90% would have progression events and so a sample size of 180 total randomized patients would suffice. Accrual to the randomized phase 2 study could potentially be stopped early based on futility monitoring if results are not promising for the new regimen. The results in Table 36.5 show that if an imbalanced randomization is used in which two-thirds of the patients are randomized to the new treatment, the number of progression events needed increases to 185 instead of 164. So although a larger total sample size would be required, somewhat fewer patients would receive the control regimen.
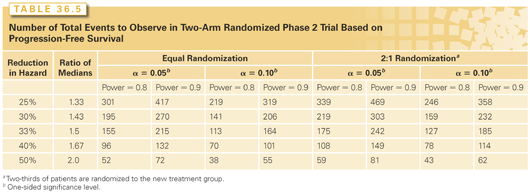
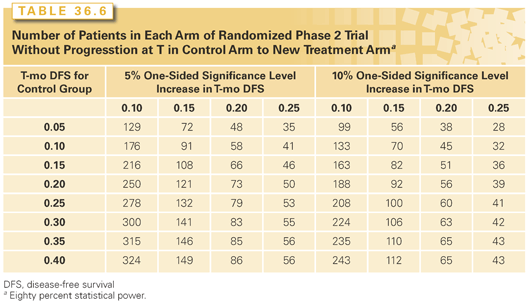
The randomized phase 2 design with control regimen has also been discussed by Korn et al.38 and by Rubinstein et al.39 Randomized phase 2 trials can require fewer patients than phase 3 trials, but they generally require more patients than single-arm phase 2 trials. Nevertheless, they are generally necessary for evaluating time to event end points or for evaluating combination regimens. Table 36.6 shows number of patients required for randomized phase 2 trials where the primary end point is either response rate or the proportion of patients without progression by a specified landmark time.
Randomized Screening Designs
Phase 2 trials are generally viewed as a means of determining whether a particular regimen is worthy of phase 3 evaluation. They can, however, be viewed as way to screen a wide range of new regimens in order to select the most promising for phase 3 evaluation. Traditional single-arm phase 2 designs are problematic for screening when there is substantial interstudy variation in patient selection and outcome evaluation. Simon, Wittes, and Ellenberg40 proposed the randomized phase 2 design in which multiple new regimens are randomized against each other as one way of avoiding such interstudy variablility in prioritizing the candidate regimens.40 This randomized design can provide more interpretable results if it also incorporates a control arm. This design is more efficient than separate randomized phase 2 trials because the control arm does not have to be replicated in all of the randomized phase 2 trials. Using the example described previously, if it takes 90 patients per arm to conduct a randomized phase 2 trial, instead of 180 × 5 = 900 patients to conduct randomized phase 2 trials of five new regimens, one would require only 90 × 6 = 540 patients, a savings of 40%. The savings in number of patients can be even more dramatic if one takes the position that the objective is not to evaluate all five new regimens, but rather to select the best one and determine whether it is worthy of phase 3 evaluation. For this selection objective, one does not require 90 patients per arm.40 These designs have been discussed and extended by others.41–44
Simon et al.12 showed that one can take advantage of the nontoxic nature of some molecularly targeted drugs to efficiently evaluate multiple regimens in the same study. They propose using a factorial design in which concurrent randomizations are made for each drugs. For example, if there are three drugs (A, B, C) being evaluated, then some patients will receive all three, some will receive pairs (AB, AC, or BC), some will receive single drugs (A, B, C), and one group will receive none of the drugs. In evaluating each drug, the time to progression for all patients receiving that drug are compared to the times for all patients not receiving that drug. The trial can be sized as if it were a single two-arm trial. The design is effective as long as there are not negative interactions among drugs. Negative interactions would result from the toxicity of one drug interfering with the full-dose administration of other drugs, which may not be a problem for many molecularly targeted drugs. The design is also useful for attempting to identify combinations that are therapeutically synergistic, a circumstance of particular importance with molecularly targeted drugs.
Rosner, Stadler, and Ratain45 describe a “randomized discontinuation design” for phase 2 studies of therapeutically targeted drugs. All eligible patients are started on the drug and given two to four courses of treatment. Patients are then evaluated: Those with progression are removed from study, those with objective tumor response are continued on treatment, and the remaining patients are randomized to either continue or discontinue the drug. The continued and discontinued groups of randomized patients are compared with regard to time to progression. Freidlin and Simon46 evaluated and further developed this design. It may require as large a number of patients started on treatment as a straightforward randomized phase 2 design. The advantage of the design is that because all patients start on the new regimen, accrual rate may be better with the randomized discontinuation design.
Seamless Phase 2/3 Designs
Hunsberger, Zhao, and Simon47 developed a design for a seamless phase 2/3 design. Patients are randomized between a new regimen and control. An interim analysis is performed using a phase 2 end point such as response rate or time to progression to decide whether the results with the new treatment as sufficiently promising to continue to a phase 3 sample size. If accrual continues, then the final analysis is performed using an acceptable phase 3 end point. A similar approach was described by Goldmanm LeBlanc, and Crowley.48 Phase 2/3 designs using Bayesian methods have been reviewed by Thall.49 Sher and Heller50 proposed conducting phase 3 trials with multiple experimental regimens, a control arm, and early termination of all experimental arms that are not promising. They used the statistical design of Schaid, Wieand, and Therneau51 for time to event data. Thall, Simon, and Ellenberg52 had studied such designs when the end point was binary. A similar approach was recommended by Parmar et al.53 Freidlin et al.54 have discussed statistical and practical aspects of conducting clinical trials with a control arm and multiple new treatment arms. Freidlin, McShane, and Polley55 have also introduced a design for a randomized phase 2 design of a new drug with a candidate predictive biomarker for determining whether the drug is entirely inactive, active only in the marker positive group, or active regardless of the biomarker status. This design enables investigators to appropriately plan whether to continue biomarker development into phase 3 development.
DESIGN OF PHASE 3 CLINICAL TRIALS
Good therapeutic research requires asking important questions and getting reliable answers. The most important clinical trials are often the most difficult to conduct.56 They may involve withholding a treatment established by tradition, transferring patient management responsibility across specialties, standardizing procedures among physicians, and sharing recognition with a large group of collaborators.
End Points
Phase 3 trials attempt to provide guidance to practicing physicians to help them make treatment decisions with their patients. Consequently, the trials should provide reliable information concerning end points of relevance to the patients. The major end points for evaluating the effectiveness of a treatment should be direct measures of patient welfare. Survival and symptom control are two such end points. The latter is not routinely used because of the difficulty of measuring it reliably and because it may be influenced by concomitant treatments.
Although durable complete regression of metastatic disease is usually a good surrogate for prolonged survival, partial tumor shrinkage usually is not an appropriate end point for phase 3 trials. Torri et al.57 performed a meta-analysis of the relationship between difference in response rates and difference in median survivals for randomized clinical trials of advanced ovarian carcinoma. They found that large improvements in response rates corresponded to very small improvements in median survival. Hence, use of response rate as an end point may result in giving patients increasingly intensive and toxic therapy with little or no net benefit to them. Proper validation of an end point as a surrogate for clinical benefit requires a series of randomized clinical trials in which treatment differences with regard to the candidate surrogate are related to treatment differences with regard to clinical benefit.58–60 It is not sufficient to show that clinical outcome is related to the candidate surrogate measured on the same treatment arm as this may just reflect the known responder versus nonresponder bias.
Disease-free survival is often accepted as an important measure of clinical benefit to be used as an end point for adjuvant treatment trials. There is more controversy, however, about the use of time to progression in metastatic disease trials. The controversy relates to whether prolonged time to progression provides clinical benefit and whether it can be measured without bias. With unblinded evaluation of time to progression, there could be a reduced threshold for declaring progression for control patients so that they can cross over to the new treatment.61 Central party blinded review of progression is often used to avoid such potential bias. Because the review is not performed in real-time, however, it can introduce additional biases of “informative censoring.” Freidlin et al.62 proposed an approach to adjusting for increased surveillance of the control group. Dodd et al.63 proposed that central review be performed only for a subset of patients to evaluate whether local assessments were biased, not to replace local assessments.
Patient Eligibility
To ensure that the results of phase 3 trials are applicable to patients seen in the community outside of clinical research settings, the trials often involve numerous centers and extensive community participation. In order to ensure broad generalization of conclusions, most multicenter phase 3 trials have employed broad eligibility criteria. In the United Kingdom, many trials have been designed using the uncertainty principle, an approach that leaves much of the decision making about eligibility to the treating physician. There may be guidelines for eligibility, but the ultimate decision is made by the treating physician; if he or she is uncertain about which treatment is more appropriate for the patient, the patient is eligible.
There is a growing recognition, however, that the one of the key hallmarks of cancer is intertumor heterogeneity. Tumors that arise in the same primary site are often quite different with regard to their oncogenesis, pathophysiology, and drug sensitivity. Consequently, conducting broad eligibility clinical trials with drugs only expected to be effective for an identifiable subset of patients is often no longer an appropriate research strategy.64–66 Particularly with molecularly targeted drugs, effectiveness is likely to be limited to a sensitive subset of tumors that may be characterized based on whether the molecular target of the drug is deregulated in the tumor. Even with cytotoxics, many patients are generally treated for each patient who benefits. The high costs of many molecularly targeted drugs make the traditional broad eligibility trial approach increasingly unsustainable.
Clinical trials can be conducted with fewer patients if patients are selected based on assays that identify the tumors likely to be sensitive to the drug in question. Simon and Maitournam67,68 and others69,70 have evaluated the efficiency of such targeted designs. When fewer than half of the patients “test positive” and when the new treatment has little benefit for patients who test negative, the required sample size can be dramatically reduced by restricting eligibility to patients who test positive. Simon and Zhao have made available a web-based computer program to enable investigators to compare such designs to standard broad eligibility designs (http://linus.nci.nih.gov/brb).
This targeted approach was effectively used for the development of trastuzumab in patients with metastatic breast cancer. In that case, about 450 patients whose tumors overexpressed HER-2 participated in a randomized clinical trial that provided convincing evidence that trastuzumab prolonged survival. Had the study been conducted without evaluating HER-2 expression, >8,000 patients would have been needed for similar statistical power.67 Even had a huge study of unselected patients been conducted and given a statistically significant result, the size of the benefit would have been very small as the benefit in the 25% of patients with HER-2 overexpression would have been diluted by lack of benefit from the remaining 75%. It is questionable whether such a small benefit overall would have justified approval or use of a drug with clear and serious toxicities.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


