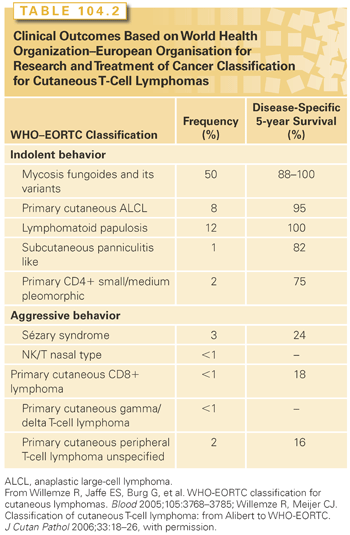
Further subgrouping based on clinical outcomes has been proposed for the cutaneous T-cell entities.2,3 The entities with indolent clinical behavior include mycosis fungoides (MF) and its variants, the cutaneous CD30+ entities, subcutaneous panniculitis-like T-cell lymphoma, and the primary cutaneous CD4+ small-/medium-sized pleomorphic T-cell lymphoma. Included in the aggressive group are Sézary syndrome (SS), the NK/T-cell disorders, gamma-delta positive disorders, CD8+ cutaneous diseases, and primary cutaneous peripheral T-cell lymphoma (PTCL). A similar classification for the cutaneous B-cell lymphomas has been proposed based on the histology (follicular or large cell type) and site of disease, with favorable outcomes seen in disease of the head or upper trunk and an unfavorable prognosis seen with either disseminated lesions or disease in the lower extremities (Table 104.3).
MYCOSIS FUNGOIDES AND THE SéZARY SYNDROME
MF was first reported by Alibert in 1806 as a common epidermotropic lymphoma with an indolent evolution characterized by cutaneous lesions in the forms of patches, plaques, or skin tumors. In 1980, Bunn et al.4 reported the presence of Sézary cells in the blood of patients with MF, and the Sézary syndrome (diffuse erythroderma, circulating Sézary cells, and involvement of lymph nodes and bone marrow) was thus identified.5–7 The International Society of Cutaneous Lymphoma (ISCL) established criteria for the diagnosis of SS, which include an absolute Sézary count of at least 1,000 cells/mm3 in the blood, immunophenotypic abnormalities (expanded CD4+ populations and/or loss of antigens such as CD2, CD3, CD5, or CD4), or the presence of a T-cell clone in the blood.8
Epidemiology and Etiology
According to SEER data, the incidence of MF–CTCL had increased 3.2-fold between 1973 and 1984. The overall incidence rate is approximately 4 per 1 million, with an incidence of 1,500 cases per year. The actual incidence rate may be an order of magnitude higher, given possible underreporting and the difficulty and confusion in making the diagnosis. The incidence of MF rises with age such that the majority of patients are between 40 and 60 years. The disease is 2.2 times more common in men than in women, and incidence rates are somewhat higher in African Americans than in Caucasians.
One hypothesis regarding the etiology of MF/SS is that it may possibly represent a clonal evolution from a chronic antigenic stimulus. Associations with exposure to occupational chemicals or pesticides have been proposed but not definitely demonstrated in epidemiologic studies.5,6 In other studies, an association with a Chlamydia infection of keratinocytes has been proposed, but data demonstrating Chlamydia proteins in affected skin lesions are equivocal.9,10 The association between human T-cell leukemia virus (HTLV) type 1 infection and adult T-cell leukemia-lymphoma (ATLL) or Epstein-Barr virus in conjunction with nasal NK/T-cell lymphoma is not reflected in the epidemiology of MF–CTCL, but there are reports of the detection of HTLV-like viral particles in affected skin lesions and antibodies to HTLV-1 tax protein in patients with MF/SS.11–14 These results suggest the association of perhaps a yet unknown retrovirus in some cases of MF/SS. Although there is no known geographical clustering and no evidence of maternal transmission of the disease, there are reports of multiple cases of MF/SS in a small number of families.
Pathobiology
The immunophenotypic profile of MF is one of clonal mature CD4+ CD45RO+ T cells with a marked homing capacity for the papillary dermis and epidermis. Some CTCL variants are CD8+ and different subtypes have distinct prognoses. Antigen loss is characteristic of the disease, with a loss of CD7, CD5, or CD2 and dim staining for CD3. Sézary cells express a TH2 phenotype, with secretion of interleukin (IL)-4, IL-5, IL-6, IL-10, and IL-13. The pruritus characteristic of the disease is related to secretion of IL-5 as well as other chemokines. One characteristic of the disease, even at its earliest stages, is profound immunosuppression with aberrant T-cell repertoires, cutaneous anergy, and increased susceptibility to bacterial and opportunistic infections.15,16
The homing to skin by CTCL cells appears to be mediated in part by expression of the surface glycoprotein cutaneous lymphoid antigen (CLA), an antigen whose expression is low or absent on normal infiltrating T cells.17,18 CLA mediates binding to E-selectin on endothelial cells of cutaneous venules, thereby facilitating their exit from the circulation and into the skin. CLA is the physiologic ligand of endothelial cell E-selectin, a cell adhesion molecule expressed on the surface of endothelial cells of cutaneous venules during chronic inflammation.19 Chemokine receptor CCR4 expressed by cells binds chemokine CCL17 that has adhered to the luminal side of the endothelium, facilitating T-cell leukocyte function antigen-1 binding to endothelial cell intracellular adhesion molecule-1 and fostering extravasation into the dermis.4
One of the most striking features of MF/SS is epidermotropism, or infiltration of the epidermis by malignant T cells. The pathognomonic feature of MF is the Pautrier’s microabscess, a collection of clonal malignant cells within the epidermis. The Pautrier’s microabscesses may be a consequence of the expression of intracellular adhesion molecule-1 (ICAM-1) on keratinocytes. ICAM expression is induced by interferon (IFN), which is produced by infiltrating T cells and is a ligand for leukocyte function antigen-1.20,21 In advanced disease or SS, the keratinocytes lose the ability to express ICAM-1 due to low levels of IFN-γ production, resulting in loss of epidermotropism.21 Although specimens from early lesions of MF have lymphocytes in both the epidermis and dermis, clonality studies on dissected cells demonstrated that virtually all of the lymphocytes found in the epidermis belong to the malignant clone, whereas the dermis contains a predominance of inflammatory cells and nonmalignant lymphocytes.
Although there is no characteristic chromosomal translocation in patients with MF and SS, significant chromosomal instability is noted and losses on 1p, 10q, 13q, and 17p and gains of 4, 17q, and 18 are commonly observed.22,23 Genetic instability is also evidenced by significant copy number alterations even in early disease.24 Recent studies have shown a high prevalence of deletions or translocations involving a gene, NAV3, at 12q2, which has helicase-like activity and might therefore contribute to genomic instability.25 Chromosomal amplification of JunB at 19p12 has also been detected in MF/SS and is thought to be contributory to the TH2 cytokine profile characteristic of Sézary cells.26 In Lin et al.,24 21 regions of amplification and 42 regions of deletion were identified, with significant amplifications of 8q (MYC) and 17q (signal transducer and activator of transcription 3 [STAT3]) and deletions of 17p (TP53) and 10 (phosphatase and tension homologue [PTEN], FAS).24
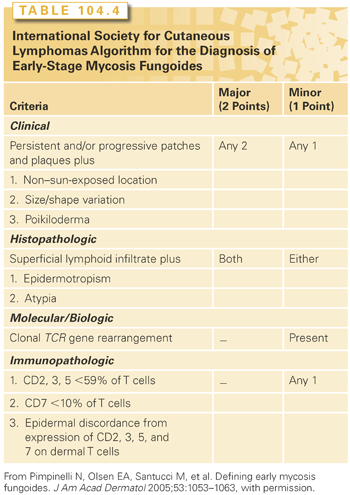
The diagnosis of MF depends on both clinical and histopathologic criteria. The skin manifestations can be in the form of patches, plaques, erythroderma, cutaneous tumors, or ulcers. Early patch and plaque lesions may be indistinguishable from those of benign dermatoses, including psoriasis, eczema, large plaque parapsoriasis, or drug eruptions. The distribution of the lesions favors non–sun-exposed areas such as the bathing trunk distribution. An early diagnosis can be difficult and may rely on multiple biopsies obtained from different lesions over time. The ISCL has developed criteria for the diagnosis of early-stage MF that relies on clinical, histopathologic, immunopathologic, and molecular criteria27 (Table 104.4). Of note, T-cell receptor clonality can be found in benign dermatoses and in lymphomatoid papulosis and pityriasis lichenoides (clonal dermatitis).28,29 Long-term follow-up of patients with clonal dermatitis reveals a significant risk of progression to overt MF, suggesting careful follow-up.
Early lesions of MF may be asymptomatic, such as scaling erythematous macular eruptions often in sun-shielded areas (Fig. 104.1). A patch is defined as a lesion that is not elevated or indurated and that may be hyper- or hypopigmented. A plaque is raised or indurated and may be associated with scaling, crusting, or ulceration. A tumor is a lesion that is more than 1 cm with evidence of depth or vertical growth. Erythroderma is defined as diffuse erythema involving more than 80% of the skin surface with or without scaling.8
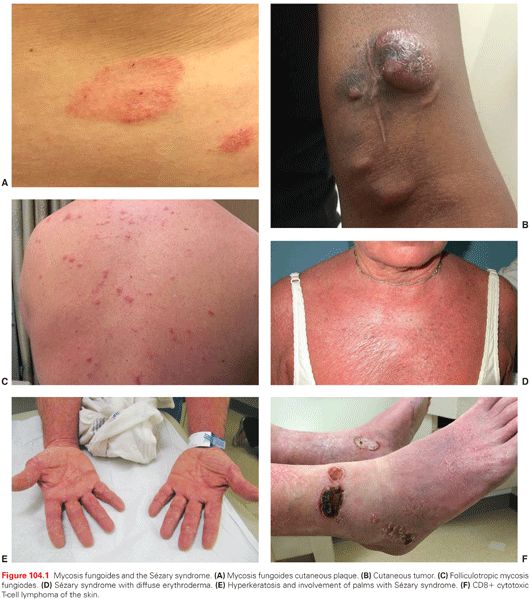
Painful and/or pruritic erythroderma may arise de novo or during any of the earlier described phases and is not always associated with frank T-cell leukemia (as in SS). Infrequently, MF presents with cutaneous tumor nodules in the absence of patches or plaques (as in tumor d’emblée). Patients may also present with or progress to involvement of visceral organs.
The Sézary Syndrome
The diagnostic criteria for SS are dependent on the presence of a circulating Sézary cell count of at least 1,000 cells/mm3. The phenotype is typically that of a mature, memory CD4+ T cell with a frequent loss of normal T-cell antigens (CD3, CD5, CD2, CD7, and CD26).8 The CD4/CD8 ratio is elevated, usually more than 10, and a T-cell clone is detected in the blood by polymerase chain reaction (PCR). The presence of more than 1,000 Sézary cells/mm3 is not absolutely diagnostic of SS in the absence of other clinical features of the disease, because these cells may be seen in about 5% of patients with benign dermatoses manifested by erythroderma.30,31 Histopathologic features in skin biopsies of patients with SS can be nonspecific, and there is a loss of epidermotropism in up to 70% of cases. Cytogenetic studies demonstrate unbalanced translocations and deletions, often involving 1p, 10q, 14q, and 15q, with evidence of clonal evolution and chromosomal instability over time.32 A differential diagnosis includes viral or drug-induced exanthems, atopic dermatitis, or psoriasis.
Clinical features of the SS include extensive skin involvement with erythroderma, which may progress to lichenification, palmoplantar hyperkeratosis, and diffuse exfoliation. Skin edema, hypoalbuminemia due to insensible fluid loss related to impaired skin integument, and intense pruritus are frequently observed in patients with advanced disease. Lymphadenopathy, histopathologically effaced nodes, and bone marrow involvement are common. Significant immunosuppression occurs related to impaired T-helper function as well as T-cell repertoire skewing, leading to a high incidence of infections, particularly related to indwelling intravenous catheters. The overall prognosis is poor, with a median survival of 2 to 4 years.33
Staging and Prognosis of Mycosis Fungoides and the Sézary Syndrome
Staging systems for MF have been developed based on clinical features of skin involvement as well as infiltration of lymph nodes and viscera. Skin involvement is defined on the basis of the type of lesions and extent. T1 and T2 diseases are patches or plaques involving less than or more than 10% of the skin surface, respectively. T3 disease is the presence of at least one cutaneous tumor. T4 disease is erythroderma. Lymph node involvement has been classified based on the degree of infiltration with malignant cells. The dermatopathic node demonstrates typically many atypical lymphocytes in three to six cell clusters (LN2) or larger aggregates of atypical lymphocytes with nodal architecture preserved (LN3) clusters of T cells often with expansion of the parafollicular zones. LN4 nodes are effaced by tumor cells, and typically such effacement is by atypical lymphocytes or neoplastic cells.5 T-cell receptor rearrangement (TCRR) is found in half of patients with LN3 nodes and rarely in those with LN2 histology.34 Bone marrow involvement has been shown to have prognostic significance based on the degree of involvement, with cytologically atypical lymphoid aggregates and infiltrative disease associated with inferior survival.35 In retrospective studies, bone marrow involvement was associated with blood involvement and advanced lymph node disease.7,36–38
The initial staging system for MF/SS was proposed by the MF Cooperative Group in 1979 and was based on skin involvement, palpable nodes, and visceral involvement.39 More recently, the ISCL further stratifies patients based on the extent of blood involvement (Table 104.5).40 In this new system, patients with significant blood involvement are identified in the erythroderma, or stage III group, and patients with stage IVA disease are further categorized based on the degree of lymph node and blood infiltration. Early stage (TI/T2) disease has been proposed to be divided into patch alone versus both patch and plaque disease. These changes have been validated by Agar et al.,41 who analyzed the outcome of 1,502 MF/SS patients at their institution.
Overall outcome in MF/SS is correlated with clinical stage, and retrospective studies have identified the extent of skin involvement as well as visceral disease as the most important prognostic factors.7,14,38 Patients with limited patch/plaque disease covering less than 10% of their skin surface have a prognosis indistinguishable from that of age-, sex-, and race-matched controls.42 The 10-year disease-specific survival for patients with more extensive skin involvement with patches or plaques is 83%, whereas those with tumors or histologically documented lymph node involvement had survivals of 42% or 20%, respectively.38 Patients with effaced lymph nodes or the presence of large cell transformation had a uniformly poor prognosis.43,44 Other poor prognostic factors include blood involvement and loss of T-cell markers CD5 and CD7.45 Even in patients with B0 disease, the presence of T-cell clonality has been shown to portend a worse prognosis.41
CLINICAL EVALUATION OF PATIENTS WITH CUTANEOUS LYMPHOMA
An initial evaluation should include a careful assessment of the number and distribution of each type of skin lesion. The Skin Weighted Assessment Tool divides the body surface into areas that are assigned a value based on the percent of total body surface area represented.46 The observer then estimates the percent of each body area involved with disease based on the estimation that the palm of the hand is 1%. The involvement is weighted based on whether the lesions are patch, plaque, or tumor. The sum is the skin score, which can be recorded and monitored during therapy.
Skin biopsies at multiple sites may be necessary to establish the diagnosis because lesion morphology varies even for different lesions from the same patient and the quality and quantity of infiltrating cells may be affected by topical therapies, including topical steroids. In addition, most of the cells in the underlying, often much more impressive dermal infiltrate are nonneoplastic reactive CD4+ and CD8+ T lymphocytes. Features of pleomorphism and the presence of large cells should be noted. Transformation to a large cell phenotype in patients with MF/SS is associated with a poor prognosis. Immunophenotyping and molecular studies for TCRR should be performed on skin biopsies.
Laboratory studies should include flow cytometry to detect circulating neoplastic cells. In investigational settings, it is possible to use monoclonal antibodies directed against TCR-Vb families to detect and precisely quantitate the levels of circulating leukemia cells. In most instances, the level of circulating CTCL cells is actually much higher than estimated by less-sensitive techniques such as by evaluation of the peripheral smear for atypical cells.21 In many patients, the expansion of the neoplastic T-cell clone is accompanied by depression of normal T cells to levels comparable with those observed in advanced AIDS. Such a de facto T-cell deficiency may both explain the susceptibility of erythrodermic CTCL patients to infection by bacterial, viral, and fungal pathogens and contribute to the progression of the disease, which is often held in check by host immune mechanisms.47
Flow cytometry should be performed with antibodies to the CD4, CD8, CD3, CD45R0, and CD26 antigens. The ratio of CD4+ to CD8+ cells is normally 0.5:3.5; elevations in this ratio correlate with total leukocyte count and with extent of skin disease in CTCL patients. An elevated ratio of CD4+ to CD8+ cells above 4.5:1.0 strongly suggests significant levels of circulating CTCL cells. Dual color staining with CD4 and other antigens can detect low or absent expression of CD3, CD7, or CD26 as a feature of Sézary cells.
Imaging studies (computed tomography scan or magnetic resonance imaging) are recommended at the initial evaluation, especially for those with advanced disease, as well as during follow-up, to detect enlargement of thoracic, abdominal, or pelvic nodes. Positron emission tomography has been performed for patients with CTCL, but there is not enough experience to reliably determine the sensitivity and specificity in cutaneous lymphoma.48,49 Pathologically enlarged lymph nodes should be biopsied at the initial staging and subsequently if enlargement is detected on the physical examination or imaging studies because a proportion of patients with CTCL may have other lymphomas (B or T cell; e.g., Hodgkin’s) concurrently. A bone marrow biopsy should be obtained in patients with advanced disease, including those with SS, as well as in patients with compromised hematologic function. Biopsies of visceral organs such as the liver should be dictated based on clinical indication or to confirm findings on imaging studies.
PRINCIPLES OF THERAPY OF MYCOSIS FUNGOIDES AND THE SÉZARY SYNDROME
Treatment approaches for MF/SS depend on the clinical stage of disease. Early-stage disease that is localized to the skin (patch or plaque disease) has an excellent chance of cure or long-term control with therapies directed to the skin alone. In contrast, tumor stage disease, extensive plaque stage disease that is refractory to topical therapies, and nodal or visceral disease can be palliated but rarely cured. Over the past 15 years, a number of novel agents have shown activity in MF/SS (Table 104.6). Because MF/SS is immunosuppressive and an immunologically responsive disease, initial therapies for many patients involve cutaneous and biologic approaches, which act directly on CTCL cells (e.g., they are directly cytotoxic) but also have indirect effects (e.g., alter the cutaneous environment) that may play a role in disease control.50
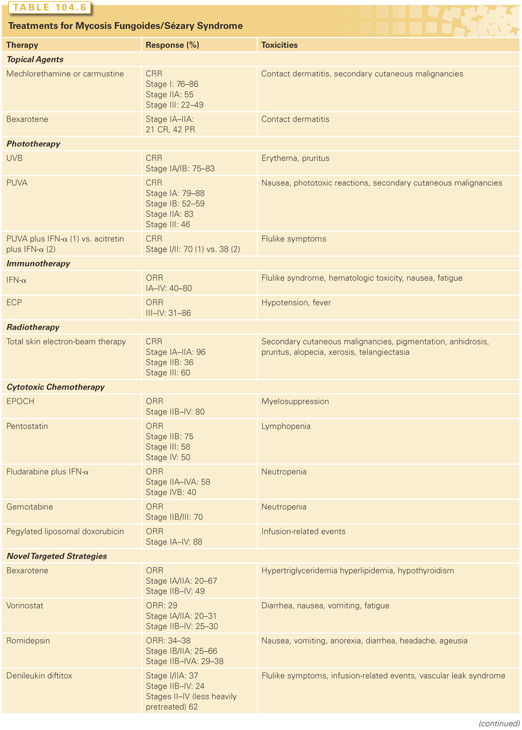
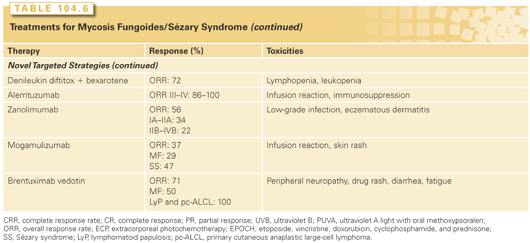
Skin-Directed Therapy
Skin-directed modalities include those for localized disease (radiotherapy, bexarotene, and carmustine) and those applicable to total skin therapy (topical chemotherapy with nitrogen mustard [NM], phototherapy, and total skin electron-beam therapy [TSEBT]; Table 104.6). All skin-directed therapies exert their primary effects on disease confined to the skin by inducing apoptosis of tumor cells and interfering with the local production of cytokines by epithelial and stromal cells necessary for neoplastic T-cell survival and proliferation.51
Approximately 7% of patients with stage I disease present with a solitary cutaneous lesion or several in proximity. Wilson et al.52 found that the rate of clinical remission after local external-beam radiotherapy is very high (approximately 95%) in these patients and may be the treatment of choice. In one study, a total of 21 patients were treated with electron-beam radiation to a median dose of 20 Gy. With a median follow-up of 36 months, the actuarial disease-free survival rates at 5 and 10 years were 75% and 64%, respectively, with a local control rate of 83% at 10 years.
Topical Chemotherapies
Topical NM is one of the first treatments for cutaneous manifestations of MF. The NM liquid can be applied to the skin as an aqueous solution of 10 mg/dL or applied in an ointment base. Long-term effects include induction of second cutaneous malignancies (e.g., squamous cell carcinomas) and hyperpigmentation and hypopigmentation. Between 64% and 90% of NM-treated patients with T1 and T2 CTCL can achieve a complete response to therapy.
Topical Bexarotene Gel
Bexarotene (Targretin) is a novel RXR retinoid (retinoid X receptor) that has been shown to be effective both systemically and topically for patients with MF/SS. The overall response rate to topical bexarotene in clinical trials was 44%. The drug is not absorbed to any significant levels. The irritant dermatitis induced by the retinoid limits the use of the gel to patients with body surface area (BSA) of less than 15% because of discomfort. In many cases, topical bexarotene gel is used, alternating with topical steroids to minimize the irritant effect.
Phototherapy
Phototherapy has been effective for patients with MF/SS because keratinocytes are resistant to ultraviolet (UV) light–induced injuries, whereas lymphocytes are extremely sensitive to light in the form of either UVA (320 to 400 nm), UVB (290 to 320 nm), or narrow-band UVB (311 nm). Currently, narrow-band UVB is used most commonly because of the low risk of secondary skin neoplasms. Patients typically are treated three to four times per week for approximately 30 to 40 treatments to achieve a remission, and then treatment frequency is decreased to a maintenance schedule at weekly intervals. Broad-band UVB has the same treatment schedule.
Photochemotherapy with orally administered PUVA (ultraviolet A light with oral methoxypsoralen) has been an effective therapy for patients with patch and plaque stage MF. The intensity of the light and frequency of administration are titrated based on patient response and tolerability. Maintenance treatment is often necessary to prevent disease recurrence.
Combination Regimens Involving PUVA Photochemotherapy
Several well-conducted trials have assessed the role of PUVA in combination with various systemic agents, notably IFN-α and retinoids. Phase I and II studies of PUVA (three times weekly) combined with variable doses of IFN-α (maximum tolerated dose of 12 MU/m2 three times weekly) in 39 patients with MF (all stages) and SS have reported an overall response rate of 100%.53 The median duration of response (DOR) was 28 months, with a median survival of 62 months.
A randomized controlled trial compared PUVA (two to five times weekly) plus IFN-α (9 MU three times weekly) with IFN-α plus a retinoic acid receptor (RAR) retinoid, acitretin (25 to 50 mg per day), in 98 patients with a maximum duration of treatment in both groups of 48 weeks.54 In 82 patients with stage I/II diseases, complete response rates were 70% in the PUVA/IFN group compared with 38% in the IFN/acitretin group. Time to response was 18.6 weeks in the PUVA/IFN group, compared with 21.8 weeks in the IFN/acitretin group.
Total Skin Electron-Beam Therapy
Total skin electron-bean therapy (TSEB) involves the use of electrons ranging in energy between 4 and 7 MeV applied homogeneously to the skin surface.55 Structures below the deep dermis are relatively spared because most of the dose (80%) is typically administered within the first 10 mm of depth, and less than 5% beyond 20 mm depth. Generally, doses to skin target are in the range of 30 to 36 Gy. Blood and superficial lymph nodes may receive 20% to 40% of the skin surface dose, and this may be clinically important.
TSEBT may be administered as just one in a sequence of treatments for CTCL in a particular patient. For example, TSEBT is excellent treatment for patients with diffuse involvement with thick plaques or cutaneous tumors and is also suitable for patients with symptomatic erythroderma–T4 disease.56 TSEBT is also an excellent alternative for patients with extensive patches or thin plaques refractory to PUVA or other skin-directed therapies.57 Subsequently, TSEBT may be administered to a patient several times using a variety of dose schedules, as clinically required to help control progressive disease.
Clinical complete response rates for patients with T1 or T2 (patch or plaque) disease range from 71% to 98% and are higher in patients with less-extensive disease. Patients with T1 and T2 disease treated with TSEBT have disease-free and overall survivals of 50% to 65% and 80% to 90%, respectively, at 5 years, although patients with antecedent or coexisting lymphomatoid papulosis or alopecia mucinosa–follicular mucinosis appear to have a shorter disease-free survival after TSEBT than those who do not. Patients with more advanced T3 and T4 disease fare significantly worse, with 5-year disease-free and overall survivals of approximately 20% and 50%, respectively. However, those T3 patients with less than 10% of the total skin surface involved by CTCL have significantly better disease-free and overall survival after TSEBT than those with more extensive disease. For patients with erythrodermic MF (T4) who are managed with TSEBT alone (32 to 40 Gy), without concomitant or neoadjuvant therapy, the complete response rate is approximately 70%. The 5-year progression-free, cause-specific, and overall survivals are 26%, 52%, and 38%, respectively.56 Based on data from Stanford lower dose TSEBT can also be considered. Overall response rates were in excess of 90% in thoise T2-T4 disease receiving 5–19 Gy. For those who received 1–<20 gy and 20 to <30 Gy, overall response was in excess of 95%.58
Palliation of adenopathy or visceral involvement in patients with N3 disease can be accomplished by the use of appropriate high-energy orthovoltage or megavoltage photons to doses of 20 to 30 Gy. Even 6 to 8 Gy in three fractions are sufficient (e.g., when combined with TSEBT). Combinations of TSEBT with total nodal radiation have been investigated. Although feasible, such combinations do not appear to prolong survival and may be associated with hematologic toxicities not observed with TSEBT alone.
TSEBT is well tolerated by most patients; acute sequelae either during or within the initial 6 months after treatment may include pruritus, desquamation, alopecia, epilation, hypohidrosis, xerosis, erythema, lower extremity edema, bullae of the feet, and onychoptosis. Chronic changes can include atrophy of the skin, telangiectasia, alopecia, hypohidrosis, and xerosis. Second malignancies such as squamous and basal cell carcinomas, as well as malignant melanomas, have been observed in patients treated with TSEBT, particularly in patients exposed to multiple therapies that are themselves known to be mutagenic, such as PUVA and mechlorethamine.57,59
For patients who suffer diffuse cutaneous recurrences after TSEBT not amenable to other skin-directed therapies, a second course of TSEBT is both feasible and worthwhile. At Yale University, a total of 14 patients have received two courses and 5 patients received three courses of TSEBT. The median total dose after these additional courses was 57 Gy, and 86% of the patients achieved a complete response after the second course, with a median disease-free interval of 11.5 months. The median dose was 36 Gy for the first course, 18 Gy for the second, and 12 Gy for the third.60 A similar experience was reported from Stanford University, where 15 patients were identified who had been treated with a second course of TSEBT (median dose of 20 Gy), with a complete response rate of 40%.61 Nine of these patients had a partial response to therapy, and the median total dose for the entire group was 56 Gy. In both series, repeat courses were relatively well tolerated, and sequelae were similar to those observed during and after the first course of therapy.
Combined and Sequential Therapy
The adjuvant use of PUVA after TSEBT in patients with T1 and T2 disease significantly decreased cutaneous relapse. Patients treated with adjuvant PUVA after TSEBT had a 5-year disease-free survival of 85%, compared with 50% for those not receiving PUVA (p <0.02). The median disease-free survival for the T1 patients receiving adjuvant PUVA was not reached at 103 months, versus 66 months for the non-PUVA group (p <0.01). For those with T2 disease, the disease-free survival figures were 60 and 20 months, respectively (p <0.03).62 Adjuvant topical NM also appears able to delay cutaneous recurrence after TSEBT. In 1999, Chinn et al.63 from Stanford University showed that TSEBT with or without NM provided improved response rates compared with mustard alone for those patients with T2 and T3 level disease (76% versus 39%, p <0.03 for T2; 44% versus 8%, p <0.05 for T3).63
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


