Complement
Introduction
The complement system plays a major role in defense against many infectious organisms as part of both the innate and antibody-mediated adaptive immune responses. Named for some of the earliest observations of its activity—a heat-sensitive material in serum that “complemented” the ability of antibody to kill bacteria—we now know that complement comprises approximately 30 circulating and membrane-expressed proteins. Complement components are synthesized in the liver and by cells involved in the inflammatory response.
The biologic activities triggered by complement activation enhance pathways that remove microbial pathogens, and they also directly attack the pathogen itself. Because these activities are so powerful, however, they may also damage the host. Thus, under normal conditions, complement activation is tightly regulated. In this chapter we describe the different pathways of complement activation, complement’s key functions, and how complement activation is regulated. We also describe the clinical conditions that result from either inappropriate complement activation or deficiency of complement components.
Overview of Complement Activation
There are three pathways of complement activation: the classical, lectin, and alternative pathways. The key features of each are shown in Figure 14.1. Each pathway is initiated when a serum protein binds to the surface of a pathogen. The classical pathway is activated when complement component C1 binds to an antigen–antibody complex (most often, antibody bound to the surface of a pathogen such as a bacterium). The lectin pathway is activated when mannan-binding lectin (MBL) binds to terminal polysaccharide residues on the surface of many types of microbes (Gram-positive and Gram-negative bacteria, fungi, or yeast); the lectin pathway can also be activated by another serum protein, ficolin, which binds to acetylated molecules on microbial surfaces. The alternative pathway is activated when complement component C3b deposits on the surface of a pathogen.
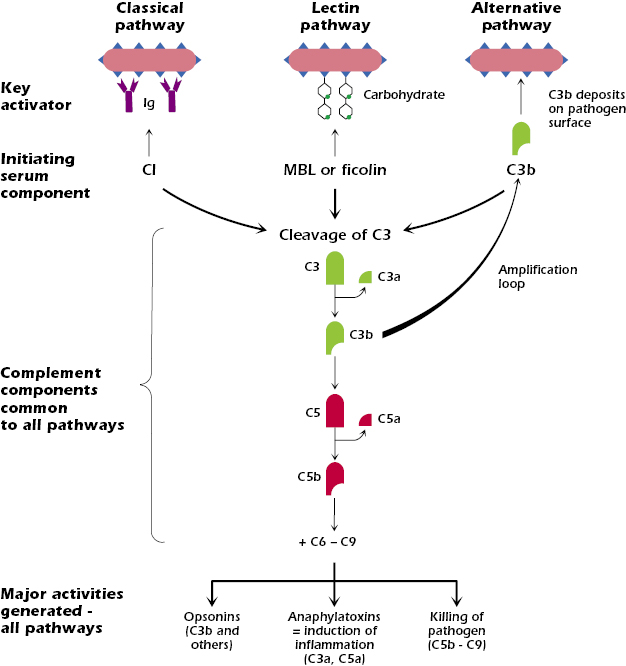
Although the three pathways are initiated by different activators, the early steps in each have a common general mechanism: complement components are sequentially activated on the surface of the pathogen. That is, activation of the component induces enzymatic function that acts on the next component in the cascade, splitting it into biologically active fragments, and so on. In addition, several activated complement components build up on the surface of the pathogen.
After the early steps, the pathways converge at the cleavage of complement component C3. Cleavage of C3 forms C3b and a small fragment, C3a. C3b covalently binds to the surface of the pathogen. C3b is an opsonin, which means that its deposition on the pathogen surface enhances pathogen uptake by phagocytic cells (see also Chapter 5). Thus opsonization of pathogens is one of the key functions of the complement activation pathways. C3a, released into the fluid phase, is an anaphylatoxin, a molecule that induces potent inflammatory responses by activating multiple cells. Thus, induction of inflammatory responses is a second key function resulting from complement activation.
As we describe in more detail below, in the alternative pathway the generation of C3b from C3 sets up an amplification loop that results in further triggering of the pathway.
After C3b has bound to the pathogen surface, the next component in the sequence, C5, is cleaved to produce C5b and C5a. C5b deposits on the surface of the pathogen, allowing the binding of components C6 through C9. These terminal components, C5b to C9, form a complex known as the membrane attack complex (MAC) on the surface of the pathogen that leads to the killing (lysis) of the pathogen. Thus, killing of pathogens is the third major function of complement activation. C5a, like C3a, is a small fluid-phase anaphylatoxin.
Thus, all three pathways of complement activation result in three major biologic activities: the production of opsonins on the pathogen surface, the synthesis of fluid-phase anaphylatoxins that enhance inflammatory responses, and the direct killing of the pathogen. All these activities lead to either rapid removal or direct destruction of the pathogen. We now describe each of the pathways and biological activities in more detail.
Classical Pathway
The classical pathway was so named because it was the first complement pathway to be worked out. The component proteins are C1, C2, and so on, up to C9; the numbers designate the order in which the components were discovered, rather than their position in the activation sequence. Cleavage products are given lower case letters, such as C3a or C4b. Large fragments such as C3b and C4b can be cleaved further to yield products such as C3c, C3d, and so on.
Activators.
Antigen–antibody complexes are the major activators of the classical pathway, with antibody bound to the surface of a pathogen the predominant example. Antibody synthesis in response to pathogens is the key characteristic of the adaptive, humoral immune response. Thus, the classical complement pathway is a major effector mechanism of the adaptive immune response and leads to the elimination of pathogens.
Soluble antigen–antibody complexes also activate the classical pathway: Although they are normally removed by macrophages, they are found in autoimmune conditions such as systemic lupus erythematosus (SLE), which we discuss later in the chapter (see also Chapter 13). Other activators of the classical pathway include some viruses (including HIV-1, discussed later in this chapter), necrotic cells and subcellular membranes (e.g., from mitochondria), aggregated immunoglobulins, and beta amyloid, found in Alzheimer’s disease plaques. C-reactive protein (CRP)—a component of the inflammatory response (an “acute-phase reactant”)—also activates the classical pathway; CRP binds to the polysaccharide phosphocholine that is part of the cell wall of many bacteria, such as Streptococcus pneumonia.
Early Steps in the Classical Complement Pathway That Lead to C3 Cleavage.
Figure 14.2A shows the predominant way in which the classical pathway is initiated: C1 binds to the Fc region of two closely spaced IgG molecules or one IgM molecule (IgM not shown in the figure) bound to an antigen expressed on the surface of a bacterium. Thus, IgM and IgG—the IgG3 subtype in particular—are effective activators of the classical complement pathway. Remember that in Chapter 8 we described how IgM is synthesized early in the immune response, to both thymus-dependent and thymus-independent antigens. In addition, we noted in Chapter 11 that IgG3 is preferentially synthesized in antibody responses in which T cells synthesize interferon-γ, generally responses triggered by bacteria and viruses. Thus the synthesis of IgM or IgG3 in the adaptive humoral immune response results in the binding of these antibodies to the pathogen that elicited them, and via complement activation ultimately leads to the elimination of the pathogen.
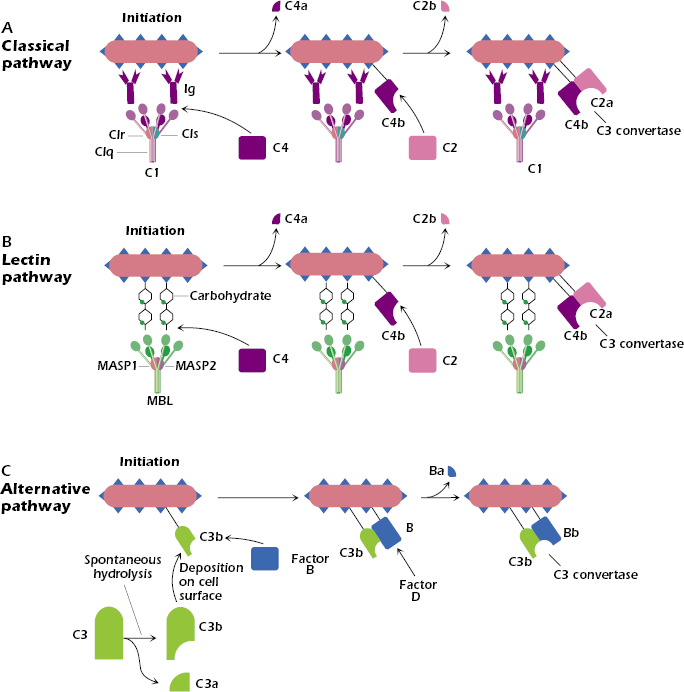
Not all classes of immunoglobulins (Igs) are equally effective at activating the classical complement pathway. Among human Igs, the ability to bind and activate C1 is, in decreasing order, IgM > IgG3 > IgG1 >> IgG2. Other antibody subtypes—IgG4, IgA, IgE, and IgD—do not bind or activate C1 and thus do not activate the classical complement pathway.
C1 is a complex of three different proteins: C1q (comprising six identical subunits) combined with two molecules each of C1r and C1s (Figure 14.2A). As a consequence of C1q binding to the Fc region of the IgM or IgG bound to the antigen, C1s becomes enzymatically active. This enzymatically active form, known as C1s esterase, cleaves the next component in the classical pathway, C4, into two pieces, C4a and C4b. C4a, the smaller piece, remains in the fluid phase, while C4b binds covalently to the surface of the pathogen. C4b bound to the cell surface then binds C2, which is cleaved by C1s. Cleavage of C2 generates the fragments C2b, which remains in the fluid phase, and C2a. C2a binds to C4b on the surface of the cell to form a complex, C4b2a. The C4b2a complex is known as the classical pathway C3 convertase; as we describe below, this enzyme cleaves the next component in the pathway, C3.
Lectin Pathway
Activators.
Terminal mannose residues expressed by Gram-positive (Staphylococcus aureus) and Gram-negative bacteria (Klebsiella, Escherichia coli, and Haemophilus influenzae type b), fungi (Candida and Aspergillus fumigatus), and yeast particles activate the lectin pathway by binding MBL. Many microbes, including the pathogen Streptococcus pneumoniae, express acetylated or neutral carbohydrate structures as part of extended polysaccharides, such as 1,3-beta-D-glucan; these are all bound by ficolin. Because the lectin pathway is activated by the molecular patterns expressed by pathogens (PAMPs, see Chapter 2) in the absence of antibody, it is part of the innate immune defenses and is involved in the rapid response to pathogens.
The terminal carbohydrate structures that activate the lectin pathway are generally not expressed on the surface of mammalian cells, so the lectin pathway of complement activation may be thought of as yet another way that the body discriminates between self and nonself. We referred to this critical concept earlier in the book, applicable in both innate immunity (pattern recognition receptors for pathogens expressed on cells of the innate immune system) and adaptive immunity (T and B cells respond to nonself antigens but do not respond to self-antigens).
Early Steps in the Lectin Pathway That Lead to C3 Cleavage.
The lectin pathway is initiated when MBL binds to the terminal polysaccharide residues of a pathogen such as a bacterium (Figure 14.2B). MBL is structurally homologous to C1q in the classical pathway. Ficolin (not shown in the figure) has a structure similar to MBL and also binds carbohydrates, such as N-acetylglucosamine or N-acetylgalactosamine, on microbial surfaces.
MBL and ficolin are found in the circulation complexed with proteases, known as the mannose-associated serine proteases (MASPs). Once bound to the bacterium, one of the proteases, MASP-2, sequentially cleaves C4 and C2 to form C4b2a on the surface of the bacterium. As we discussed previously, C4b2a is also formed in the classical pathway; it is the C3 convertase that cleaves the next component in the pathway, C3. Thus, the lectin and classical pathways converge at this point.
Alternative Pathway
Activators.
The alternative pathway of complement activation can be triggered by almost any foreign substance, and in the absence of specific antibody. Thus, the alternative pathway is a key part of the innate immune defenses, involved early in the response to pathogens. The most widely studied activators include lipopolysaccharides from the cell walls of Gram-negative bacteria (which are endotoxins); the cell walls of some yeasts; and cobra venom factor, a protein present in some snakes. Some agents that activate the classical pathway—viruses, aggregated immunoglobulins, and necrotic cells—also trigger the alternative pathway.
Early Steps in Alternative Pathway That Lead to C3 Cleavage.
The deposition of C3b on the cell surface initiates the alternative pathway (Figure 14.2C). C3b is generated in the circulation in small amounts by the spontaneous cleavage of a reactive thiol group in C3; this “preformed” C3b can bind to proteins and carbohydrates expressed on cell surfaces, either of a pathogen or of a host (mammalian) cell. (If C3b does not bind to one of these surfaces, it is rapidly inactivated.)
Thus, in a sense, the alternative pathway is always “on,” and continual activation could damage cells of the host. However, as we describe in more detail subsequently, mammalian cells regulate the progression of the alternative pathway. Microbial cells lack such regulators and cannot prevent the development of subsequent steps in the alternative pathway.
Following the deposition of C3b, the serum protein factor B combines with C3b on the cell surface to form a complex, C3bB. Factor D then cleaves factor B in the cell surface-associated C3bB complex, generating fragments Ba, which is released into the fluid phase, and Bb, which remains attached to C3b. C3bBb is the alternative pathway C3 convertase, which cleaves C3 into C3a and C3b.
Steps Shared by All Pathways: Activation of C3 and C5
C3 cleavage is the first step that is common to all three complement pathways (Figure 14.3). In the classical and lectin pathways Figure 14.3A), the C3 convertase C4b2a cleaves C3 into two fragments, C3a and C3b. In the alternative pathway (Figure 14.3B), the C3 convertase C3bBb cleaves C3 into the same two fragments. The smaller fragment, C3a, is released into the fluid phase, and the larger one, C3b, continues the complement activation cascade by binding covalently to the cell surface around the site of complement activation.
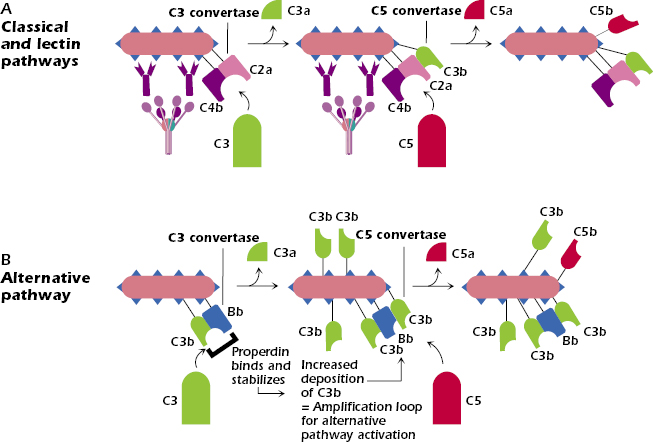
Note a unique feature of the alternative pathway shown in Figure 14.3B: binding of the serum protein properdin (also known as factor P) stabilizes C3bBb on the pathogen surface. As a result, C3bBb rapidly cleaves further C3 molecules, resulting in the huge buildup of C3b on the surface of the pathogen. As we described above, the deposition of C3b on a cell surface is the initiating step in the activation of the alternative pathway. Thus, the deposition on the cell surface of these rapidly produced and increased levels of C3b results in an almost explosive triggering of the alternative pathway. As we describe below, properdin’s ability to activate this amplification loop is balanced by negative or regulatory molecules. Consequently, under normal conditions, the alternative pathway is not continually activated.
C3b binding to either the classical/lectin or alternative pathway C3 convertases allows the next component in the sequence, C5, to bind and be cleaved (middle section of Figure 14.3A,B). For this reason, the C3 convertases with bound C3b are referred to as C5 convertases—C4b2a3b in the classical/lectin pathways, C3bBb3b in the alternative pathway. The cleavage of C5 produces two fragments: C5a is released into the fluid phase and has potent anaphylatoxic properties, and C5b binds to the cell surface and forms the nucleus for the binding of the terminal complement components.
Terminal Pathway
The terminal components of the complement cascades—C5b, C6, C7, C8, and C9—are common to the three complement activation pathways. These components bind to one another and form a membrane attack complex (MAC) that results in the death (lysis) of the cell on which they deposit (Figure 14.4).
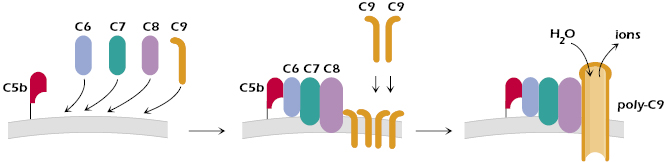
The first step in MAC formation is C6 binding to C5b on the cell surface. C7 then binds to C5b and C6, with C7 inserting into the outer membrane of the cell. The subsequent binding of C8 to C5b67 results in the complex penetrating deeper into the cell’s membrane. C5b-C8 on the cell membrane acts as a receptor for C9, a perforin-like molecule (see Chapter 11) that binds to C8. Additional C9 molecules interact with the C9 molecule in the complex to form polymerized C9 (poly-C9). Poly-C9 forms a transmembrane channel that disturbs the cell’s osmotic equilibrium: Ions pass through the channel and water enters the cell. The cell swells and the membrane becomes permeable to macromolecules, which then escape from the cell. The result is cell lysis.
Regulation of Complement Activity
Uncontrolled complement activation can rapidly deplete complement components, leaving the host unable to defend against subsequent invasion by infectious agents. In addition, the fragments generated by complement activation (especially the cleavage products of C3, C4, and C5) induce potent inflammatory responses, which may damage the host. Indeed, complement activation is believed to play an adverse role in autoinflammatory conditions such as rheumatoid arthritis and in myocardial infarctions (heart attacks) in which complement is activated by necrotic tissue (discussed later in the chapter). In addition, dysregulation of complement function in the eye has been suggested as playing a major role in age-related macular degeneration, the leading cause of visual impairment and blindness in the USA among individuals over 60 years of age.
Normally, inappropriate activation of complement does not occur, because many steps in the complement pathways are negatively regulated by specific inhibitors. Some of these negative regulators are specific for one complement activation pathway, but many inhibit all the pathways. The importance of these complement regulators is underscored by the clinical conditions that arise when regulatory molecules are lacking: The individual may be either damaged by inflammatory responses or become susceptible to infectious diseases. Some of these conditions are described later in the chapter.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


