FIGURE 19.1 Genetic pathways to colorectal carcinoma. All colorectal cancers (CRCs) arise within benign adenomatous precursors, fueled by mutations that serially enhance malignant behavior. Mutations that activate the Wnt signaling pathway seem to be necessary initiating events, following which two possible courses contribute to accumulation of additional mutations. A: Chromosomal instability is a feature of up to 80% of CRCs and is commonly associated with activating KRAS point mutations and loss of regions that encompass P53 and other tumor suppressors on 18q and 17p, often but not necessarily in that order. B: About 20% of CRCs are euploid but defective in DNA mismatch repair (MMR), resulting in high microsatellite instability (MSI-Hi). MMR defects may develop sporadically, associated with CpG island methylation (CIMP), or as a result of familial predisposition in hereditary nonpolyposis colorectal cancer (HNPCC). Mutations accumulate in the KRAS or BRAF oncogenes, p53 tumor suppressor, and in microsatellite-containing genes vulnerable to MMR defects, such as TGFβIIR. Epigenetic inactivation of the MMR gene MLH1 and activating BRAF point mutations are especially common in serrated adenomas, which progress in part through silencing of tumor suppressor genes by promoter hypermethylation. Progression from adenoma to CRC takes years to decades, a process that accelerates in the presence of MMR defects.
Global Events in Colorectal Cancer
In light of the central importance of somatic mutations, cellular conditions that elevate mutation rates might enable or accelerate tumor progression. Over 80% of CRCs display widespread chromosomal gains and losses, phenomena that favor amplification of oncogenes and loss of tumor suppressors.10 Chromosomal segregation defects may account for this background of chromosomal instability (CIN), as illustrated by the segregation factor Bub1 in mice,11 but few specific gene defects are implicated with confidence. The remaining fraction of CRCs appears euploid at the level of whole chromosomes but may carry thousands of point mutations, small deletions, and insertions near nucleotide repeat tracts, a defect known as microsatellite instability (MSI).12 Molecular determinants of progression in MSI+ adenomas differ from those associated with CIN; for example, BRAF V600E mutations occur more commonly in MSI+ serrated adenomas than in other subtypes.13 Hypermutability with CIN or MSI results in many inconsequential or detrimental “passenger” mutations, an important consideration that focuses attention on “driver” changes. Such changes are distinguished by their appearance in a significant proportion of tumor specimens and, ideally, by laboratory demonstration of their contribution toward malignancy.
Epigenetic mechanisms are probably as significant as mutations in cancer but also less well understood. Various covalent histone modifications and methylation of cytosine residues in DNA represent the principal means for gene regulation, the latter far better characterized in CRC than the former. The 5’-CpG-3’ dinucleotide pairs are particular targets for methylation in localized areas of high CpG content in promoters, where abundant methylation silences adjacent genes. Compared to normal tissue, CRCs show 8% to 15% lower total DNA methylation,14 even in colorectal adenomas.15 Reduced pericentromeric methylation might decrease the fidelity of chromosomal segregation, and altered methylation and loss of imprinting at the IGF2 locus are associated with increased CRC risk,16 suggesting broad effects of global hypomethylation on cell growth. However, because some animal models show increased tumor susceptibility with global hypomethylation,17 whereas ApcMin mice that lack or overexpress the de novo DNA methyltransferase DNMT3B show reduced or increased progression of small adenomas, respectively,18,19 its precise significance is unclear. Against the background of genomewide hypomethylation, a subset of CRCs show coordinate hypermethylation of characteristic CpG-rich promoter islands, conferring the CpG island methylator phenotype (CIMP), with transcriptional attenuation of associated genes, including tumor suppressors such as HIC1 and the secreted Wnt-inhibiting secreted Frizzled-related proteins (sFRPs).20,21 Adenomatous precursors of CIMP cancers show the distinctive histology of sessile serrated adenomas, with dysplasia within an architectural pattern typical of hyperplastic polyps.
There are few variations to the adenoma–carcinoma sequence. A tenfold elevated risk of CRC in patients with long-standing inflammatory bowel disease, especially ulcerative colitis (UC),22 probably reflects heightened mutation and tumorigenesis with repeated cycles of mucosal injury and repair. Such cancers arise not only from typical polyps but also within flat adenomatous plaques and nonadenomatous areas of dysplasia. A p53 gene mutation tends to occur earlier in the cancer sequence,23 and APC gene inactivation is less common than in sporadic CRC. Conversely, even in the absence of CIMP, methylation of the p16INK4a tumor suppressor gene, which is rare in sporadic CRC, is common in UC-associated cancers.24
EARLY EVENTS AND CRITICAL PATHWAYS IN COLORECTAL TUMORIGENESIS HIGHLIGHTED BY INHERITED SYNDROMES OF INCREASED CANCER RISK
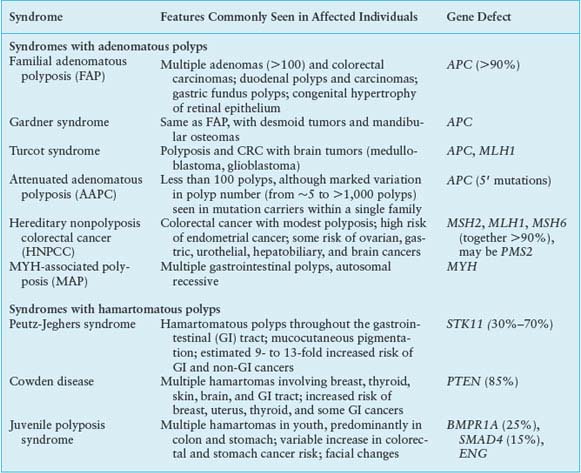
Two uncommon but highly penetrant inherited syndromes, familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC), together account for about 5% of all CRC cases. Other rare syndromes, familial juvenile polyposis (FJP), Peutz-Jeghers syndrome (PJS), and Cowden disease, each occurring in fewer than 1 in 200,000 births, also elevate the risk of CRC, and some genes responsible for these autosomal dominant disorders have been identified (Table 19.1). Elucidation of the corresponding molecular defects serves not only in accurate molecular diagnosis, risk assessment, and disease prevention in affected families but also informs understanding of the considerably larger proportion of sporadic cases.
Familial Adenomatous Polyposis and the Central Importance of Wnt Signaling
In FAP, an autosomal dominant monogenic disorder that underlies about 0.5% of all CRCs, individuals develop hundreds to thousands of colonic polyps by their teens or early 20s, and the lifetime risk of progression to invasive cancer approaches 100%, with cancer diagnosed at a median age of 39. Extraintestinal manifestations include duodenal and gastric adenomas; congenital hypertrophy of the retinal pigmented epithelium; osteomas and mesenteric desmoid tumors in the Gardner syndrome variant25; and less commonly, brain tumors in the Turcot syndrome variant,26 cutaneous cysts, thyroid tumors, and adrenal adenomas. Although most features are benign, rare patients develop hepatoblastoma or thyroid cancer. Reflecting the similar regulation of small bowel and colonic epithelia, patients have a 5% to 10% risk of developing duodenal or ampullary adenocarcinoma, mandating close endoscopic monitoring of the upper intestine after prophylactic colectomy.27
The gene affected by mutations in this disorder, adenomatous polyposis coli (APC) on chromosome 5q21, encodes a 2842-residue protein. Germline mutations occur throughout the locus but cluster in the 5’ half and exon 15,28 mostly introducing stop codons or frame shifts that truncate the protein. Although a few mutations correlate with phenotypic severity or specific extraintestinal manifestations, identical mutations can produce different clinical features. In the attenuated APC (AAPC) variant, disease onset is delayed, individuals develop fewer colonic polyps or cancers, and mutations cluster in the extreme 5’ or 3’ ends of APC exons.29 The I1307K allele, present in the Ashkenazi Jewish population, barely doubles the lifetime risk of CRC and does not affect APC protein function but replaces an (A)3T(A)4 coding sequence with an extended (A)8 tract that is occasionally targeted for nearby truncating mutations.30 Identification of an APC mutation in a proband allows reliable testing of family members. Carriers should have screening colonoscopy annually after age 10, gastroduodenoscopy after age 25, and treatment with nonsteroidal anti-inflammatory drugs to reduce the risk of progression to cancer.31 Prophylactic colectomy is highly recommended, with subsequent monitoring of the rectal stump and other at-risk tissues.
TABLE 19.1
GENETICS OF INHERITED COLORECTAL TUMOR SYNDROMES
The larger significance of the APC gene derives from its somatic inactivation in about 80% of sporadic CRCs and early colorectal adenomas.32 Indeed, somatic APC mutations are found in tiny adenomas, containing few dysplastic glands. Attesting to the tumor suppressor function, tumors arising sporadically or in FAP patients show biallelic APC gene inactivation and loss of heterozygosity, with one copy usually lost by deletion. Except for the small bowel, APC mutations are rare in other cancers, including those in other digestive organs. APC gene inactivation is a rate-limiting step for development of adenomas, and its designation as a gatekeeper gene in CRC is now well supported by knowledge of its cellular functions. APC encodes several functional domains and proteins truncated by mutation that could in principle interfere with a wide range of cellular activities. Disruption of its known role in chromosome segregation might, for example, contribute to CIN.33 However, attention on APC centers rightfully on its control of the Wnt signaling pathway. About half the sporadic CRCs with intact APC function carry activating point mutations in the CTNNB1 gene,34,35 which encodes β-catenin, a transcriptional effector of Wnt signaling. Moreover, acute loss of APC function in mice produces intestinal defects identical to those observed upon Wnt pathway activation.36
The Wnt glycoproteins are secreted morphogens with diverse functions in development and homeostasis. In the absence of Wnt signaling, cells use a complex containing APC, Axin2, and other cytoplasmic proteins to promote phosphorylation, by casein kinase I and glycogen synthase kinase (GSK)-3β, of several conserved serine and threonine residues in the β-catenin N-terminus, thereby targeting β-catenin for ubiquitin-mediated proteasomal degradation.37 When Wnt ligands bind a surface protein complex that contains a member of the Frizzled protein family and the obligate coreceptor LRP5/6, they antagonize APC/Axin2 activity and thereby stabilize β-catenin. CTNNB1 mutations in CRC alter consensus residues for N-terminal phosphorylation and render the mutant protein resistant to degradation. Thus, inactivating APC or activating CTNNB1 mutations, two alternative lesions in CRC, have the same effect: constitutive, Wnt-independent stabilization of β-catenin (Fig. 19.2). Accumulated β-catenin translocates to the cell nucleus, where it acts as a transcriptional coactivator for the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors. Nuclear β-catenin provides TCF/LEF proteins with an activating partner, resulting in transcription of target genes.38 Of the four known TCF/LEF proteins, TCF4 is the most important in normal bowel epithelium and CRC.34,39 Among the many components of the Wnt signaling cascade, rare AXIN2 mutations of uncertain significance are reported in MSI+ cases,40 but mutations in CRC are otherwise found only in APC and CTNNB1.
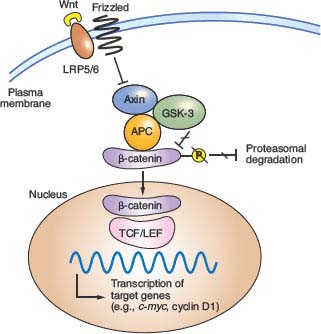
FIGURE 19.2 Outline of Wnt signaling, the key driver pathway in colorectal cancer. Members of the Wnt family of glycoprotein morphogens bind the cell surface co-receptors Frizzled and LRP5/6. In the absence of Wnt binding, normal cells use a complex containing adenomatous polyposis coli (APC), Axin, and other cytoplasmic proteins to promote glycogen synthase kinase (GSK)-3β-mediated phosphorylation of the β-catenin N-terminus, which targets β-catenin for proteasomal degradation (from ref. 37.). Binding of a Wnt ligand to Frizzled and its obligate coreceptor LRP5/6 antagonizes the APC/Axin destruction complex, stabilizing β-catenin (CTNNB1), which moves into the nucleus and coactivates genes through T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors. Either of the two principal gatekeeper events in colorectal cancer, inactivating APC or activating CTNNB1 mutations, results in constitutive, Wnt-independent stabilization of β-catenin and unregulated activation of the cognate transcriptional program. Wnt signaling in the intestine is normally confined to crypt progenitors, and its aberrant activation by APC or CTNNB1 mutations confers a permanent cryptlike state that favors cell replication.
Although Wnt ligands signal in many tissues, intestinal homeostasis is particularly dependent on this pathway. Wnt signaling in the intestine is confined to proliferative crypt stem and progenitor cells. In mice, cycling crypt cells that express the surface marker LGR5 are far more susceptible to Wnt-induced transformation than their differentiated progeny, implying that CRC arises in a primitive stem or progenitor cell and not in mature descendants.41 Moreover, the Wnt-dependent transcriptional program in CRC cell lines overlaps materially with that in intestinal crypts.42 Wnt signaling is hence necessary for intestinal epithelial self-renewal, and constitutive, ligand-independent activation imposed by APC or CTNNB1 mutations induces and sustains adenomas. Among the diverse transcriptional targets of the TCF-β-catenin complex identified to date, CMYC seems especially important because its absence in mice abrogates the effects of acute APC loss in the intestine.43,44 Mice that lack CD44, another prominent Wnt-pathway target, develop fewer adenomas in an APC-deficient background.45 Gene expression profiling offers an expanded list of over 100 candidate transcriptional targets,42,46 but the individual significance of each will take many years to investigate.
Hereditary Nonpolyposis Colorectal Cancer and the Role of DNA Mismatch Repair
HNPCC, an autosomal dominant disorder that confers a nearly 80% lifetime risk of developing CRC, usually before age 50, is estimated to account for 2% to 4% of all CRC cases in the United States.47 Affected individuals do not lack intestinal polyps (nearly all CRCs, syndromic or sporadic, arise within adenomatous precursors) but develop many fewer colonic polyps than patients with FAP, a condition that must be excluded to satisfy criteria for diagnosis of HNPCC (Table 19.2).48 Cancers tend to develop in the ascending colon, and patients are further predisposed to develop tumors of the endometrium, small intestine, stomach, upper urothelium, ovary, biliary tract, and brain, a spectrum reflected in the revised Amsterdam II criteria (Table 19.2). The lifetime risk of endometrial cancer, in particular, is 35% to 50% and that of urologic and ovarian cancers is 7% to 8%.49 Cancers in HNPCC show pronounced variation in the lengths of microsatellite DNA sequences in tumors compared with unaffected tissues. Cancers showing such MSI at two or more among a panel of five mono- and dinucleotide tracts (BAT26, BAT25, D5S346, D2S123, and D17S250) carry the MSI-Hi designation. Most other CRCs harbor CIN and show microsatellite stability (MSS) or, in a small fraction, instability at only one of the five test tracts (MSI-Lo), a finding of uncertain significance.
TABLE 19.2
CRITERIA FOR CLINICAL DIAGNOSIS OF HEREDITARY NONPOLYPOSIS COLORECTAL CANCER

HNPCC results from germline mutations in any of several genes that enable DNA mismatch repair (MMR), a proofreading process that corrects base-pair mismatches and short insertions and deletions in the normal course of DNA replication. MMR in mammalian cells is mediated by homologs of bacterial and yeast repair proteins: MutS homologs (MSH) 1-6, MutL homologs (MLH) 1-3, and PMS1 and PMS2. MLH1 and PMS2 are recruited to sites of DNA mismatch as a MutLα complex and in turn recruit MSH2-MSH6 (MutSα) or MSH2-MSH3 (MutSβ) heterodimers to sites of 1-bp or 2- to 4-bp errors, respectively. These proteins excise the strand that carries the mismatch, and they resynthesize and ligate the repaired DNA. Germline mutations in MSH2, MLH1, and MSH6 together explain more than 90% of kindreds50,51; the significance of mutations in other canonical MMR pathway genes, PMS1 and PMS2, is less certain.52
MSI-Hi colorectal cancers commonly show lymphocytic infiltrates, mucinous signet-ring differentiation, and a medullary growth pattern; Bethesda guidelines (Table 19.2) combine clinical and phenotypic features to facilitate diagnosis of HNPCC.53 When these criteria are met, tumor DNA should either be tested for MSI in a simple, PCR-based assay or by immunohistochemistry for absence of the commonly implicated MLH1, MSH2, and MSH6 proteins.54 A positive result should prompt genetic testing for MLH1, MSH2, or MSH6 mutations; the personal and family history can predict the probability of identifying mutations.55 Genetic testing identifies the mutant allele and carriers, allowing targeting of a recommendation for biannual colonoscopic screening between the ages of 20 and 40, with annual screening thereafter. Women should undergo annual endometrial evaluation soon after age 25, and carriers should consider prophylactic subtotal colectomy, hysterectomy, and oophorectomy.
In incipient cancers, random events first disrupt function of the wild type allele of a mutant MMR gene, and the resulting “mutator phenotype” induces brisk accumulation of DNA replication errors.50 Consequently, adenomas progress into carcinomas over 3 to 5 years instead of two or more decades.56 Paradoxically, the prognosis for patients with MSI-Hi colorectal cancers is better than for those with MSS disease, perhaps because many resulting somatic mutations are disadvantageous. Cancer develops, of course, because some mutations activate oncogenes or inactivate tumor suppressor genes. One frequently inactivated tumor suppressor gene in MSI-Hi colorectal cancers, TGFβRII, encodes the type II transforming growth factor-beta (TGF-β) receptor and contains a vulnerable mononucleotide tract in the coding sequence.57 TGF-β inhibits proliferation of many normal epithelial cells, including intestinal cells, and biallelic TGFβRII inactivation is detected in over 90% of MSI-Hi and 15% of MSS, sporadic CRCs.58 Other genes mutated in MSI-Hi colorectal cancers encode proapoptotic molecules, such as CASP5 and BAX,59 transcription factors, including TCF4,60 and the epidermal growth factor receptor.61 Notably, all CRCs, regardless of MMR status, seem to require mutational deregulation of the gatekeeper APC-β-catenin pathway.62
Another recessively inherited syndrome of multiple adenomas and CRC, MUTYH-associated polyposis (MAP), is caused by germline mutations in MYH, the human homolog of Escherichia coli MutY, another base excision-repair gene.63 Disease begins later in life than in FAP, polyp numbers vary widely, and extracolonic tumors are less frequent than in HNPCC. Because MYH encodes a DNA glycosylase that mediates oxidative DNA damage, tumors are not associated with MSI but with somatic G:C to T:A mutations, including in the APC gene. Two alleles, Y165C and G382D, account for most cases, and cancers develop in homozygotes or compound heterozygotes; monoallelic carriers show no increased risk.64 Surveillance recommendations are similar to those in HNPCC.
The cancer spectrum in HNPCC and the particular predilection for CRC remain unexplained. Colonic, endometrial, and selected other epithelial cells may be especially sensitive to the class of mutations that occur in the MMR setting; loss of the wild type MMR allele may occur more readily in these tissues; or they may lack repair safeguards that protect other cell types. MSI-Hi is observed in 12% to 15% of sporadic cases of CRC.65 Most such tumors are believed to arise from serrated adenomas in the ascending colon and do not reflect unrecognized germline mutation or somatic disruption of a known MMR gene.66 Rather, most sporadic MSI-Hi cases reflect epigenetic inactivation of the MLH1 gene by promoter hypermethylation, often in association with the CIMP phenotype and with activating BRAF mutations.67–69
Familial Juvenile Polyposis (FJP)
Patients with FJP develop three to ten or more premalignant hamartomatous polyps in the stomach or small or large intestine in childhood or adolescence.70,71 Although affected individuals often have the appropriate family history, a significant minority represents the first case in their families. Germline mutations in genes that encode the bone morphogenetic protein (BMP) receptor BMPR1A, the accessory TGF-β receptor endoglin, ENG, or the SMAD4 signal transducer point to the TGF-β signaling pathway in disease pathogenesis.72,73 Indeed, sporadic CRCs are frequently insensitive to the growth inhibitory effects of TGF-β, and loss of BMP function in mice expands stem and progenitor cells, leading to polyposis or ectopic crypts.74–76 Not all patients carry these mutations, indicating that additional genes remain undiscovered. Notably, conditional loss of Smad4 in mouse intestinal cells does not affect growth, whereas selective loss in T lymphocytes causes intestinal mucosal thickening and polyposis.77 These results complicate interpretation of TGF-β functions and implicate stromal inflammation in intestinal tumorigenesis.
Peutz-Jeghers Syndrome
Patients with PJS develop hamartomatous polyps, benign tumors containing differentiated but disorganized cells, mainly in the small intestine but also in the colon or stomach; these grow to variable size, often leading to hemorrhage or intussusception. PJS shows autosomal dominant inheritance and is associated with macular lesions on the skin and buccal mucosa; bladder and bronchial polyps; and a propensity to develop a range of cancers, including those of the lung, breast, and female reproductive organs. The lifetime risk for all cancers exceeds 90% and the incidence of digestive tract cancers, especially in the small intestine, stomach, and pancreas, is elevated 50- to 500-fold over the general population; CRC risk is nearly 100 times greater.78
Serine–threonine kinase 11 (STK11) or LKB1 is a recessive oncogene that shows loss of heterozygosity in PJS tumors and somatic mutations in sporadic pulmonary, pancreatic and biliary carcinomas, and melanoma.79 Its product is a complex protein that functions at the intersection of several cellular pathways and points to the central importance of cell polarity and metabolism in cancer. Although LKB1 is implicated in diverse functions, its principal activity, exerted through the adenosine monophosphate (AMP)-activated protein kinase AMPK, seems to be in linking nutrient and energy utilization to controls over cellular structure and, in particular, polarity.80 LKB1 also modulates the Rheb-GDP:Rheb-GTP cycle and downstream activity of the tuberous sclerosis gene TSC2 and the mammalian target of rapamycin (mTOR),81 key regulators of protein synthesis and cell growth. Determining how LKB1’s roles in cell polarity and metabolism contribute to tumor suppression is an intense and exciting area of cancer biology, likely to hold many surprises and useful clues for rational therapy.
Cowden Syndrome
In Cowden syndrome, the lifetime risk of developing colon or thyroid cancer approaches 10%, whereas the risk of breast cancer is nearly 50%. A diverse array of oral and gastrointestinal mucosal lesions, including lipomas, fibromas, ganglioneuromas, and hamartomas, occur together with specific cutaneous lesions (facial trichilemmomas and acral verrucous papules) and benign breast fibroadenomas, neurofibromas, lipomas, uterine leiomyomas, and meningiomas.82 Cowden syndrome results from germline mutations in PTEN, the tumor suppressor gene encoding the phosphatase and tensin homolog deleted on chromosome 10.83 PTEN is a lipid phosphatase that dephosphorylates key phosphoinositide signaling molecules84 and is accordingly a negative regulator of intracellular growth signaling through PI-3 kinase and its downstream effectors AKT and mTOR. PTEN is the second most frequently mutated gene in cancers, after TP53. Although mutations are rarely seen in sporadic CRC, PTEN immunostaining is lost in about 40% of cases, often as a result of promoter hypermethylation in the MSI-Hi setting,85 emphasizing its tumor suppressor function in this disease.
Significance of Inherited Syndromes of Elevated Colorectal Cancer Risk
Following a clinical diagnosis of the above syndromes, known germline mutations should be tested and patients provided with genetic counseling and advice on cancer prevention and screening. Elucidation of the corresponding molecular defects profoundly informs understanding of sporadic CRC, revealing in particular that APC gene inactivation or CTNNB1 activation are early, rate-limiting events and the seminal role of the Wnt signaling pathway. Likewise, LKB1 and PTEN loss in inherited and sporadic CRCs shed light on vital molecular pathways, and HNPCC helps in classifying the disease and appreciating the significance of MSI, a feature seen in 12% to 15% of sporadic cases. Even in the absence of a well-recognized predisposition syndrome, individuals with a history of CRC in a first-degree relative are up to four times more likely to develop CRC than those without a family history.
Specific environmental factors that compound the risk of developing CRC are complex and insufficiently characterized but might include obesity, excessive consumption of red meat, lack of exercise, and vitamin D deficiency.86 As many of these factors converge on insulin signaling, some experts propose that insulin and insulinlike growth factors play a seminal role in CRC.87 However, three of every four CRCs arise in individuals lacking a well-defined risk factor, and it is unknown to what extent particular genotypes confer sensitivity to environmental variables.
Insights from Genomewide Association Studies
The quarter or more of sporadic CRC cases with a familial component88,89 probably have diverse molecular etiologies, with low risk conferred by some common genetic variants and interaction of individual risk alleles with other genes and with environmental factors. One early study linked colon cancer to chromosomal segment 9q22-31,90 which reinforced the idea that single risk loci might contribute to susceptibility in nonsyndromic forms of familial disease; the authors recently narrowed the interval to a 7.7-cM distance covering five genes.91 Genomewide association studies (GWAS) have interrogated thousands of genomes to uncover statistical association of CRC risk with at least seven distinct loci, including those linked to single nucleotide polymorphisms (SNPs) rs6983267 at 8q24.21, rs4939827 on 18q21, and rs3802842 at 11q23 (Table 19.3). Risk alleles typically elevate the rate of CRC 10% to 25% above the background in persons with the nonrisk allele.92,93 Homozygosity for some risk alleles and combinations of risk alleles at different loci compound the risk, to 50% to 250% over the background.
TABLE 19.3
SINGLE NUCLEOTIDE POLYMORPHISMS (SNPS) CONFERRING INCREASED RISK OF COLORECTAL CANCER
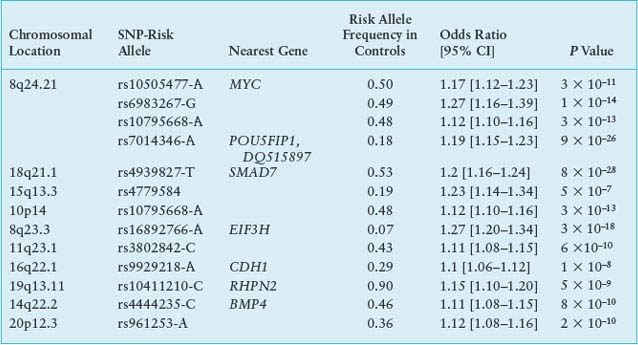
The causal significance of most of these DNA sequence variants is unclear, and many localize in gene deserts or poorly characterized regions of the genome. Addressing the role of TGF-β in CRC, one group of risk polymorphisms on chromosome 18q21 is linked to the SMAD7 locus.94 Particularly strong association occurs with the SNP rs6983267 at 8q24.21, which lies in a gene desert near low-risk susceptibility alleles for breast and prostate cancers. Molecular studies indicate that each of the culpable regions acts as a tissue-specific enhancer, controlling expression of the nearest neighboring structural gene, CMYC,95 a highly plausible factor in disease susceptibility. In summary, common predisposition alleles confer some risk of CRC with low penetrance. Their pathophysiologic functions should be studied in greater detail to inform prevention and screening strategies and to determine how risk alleles interact with particular habits or environmental factors.
TABLE 19.4
SOMATIC MUTATIONS IN ONCOGENES AND TUMOR SUPPRESSOR GENES
ONCOGENE AND TUMOR SUPPRESSOR GENE MUTATIONS IN COLORECTAL CANCER PROGRESSION
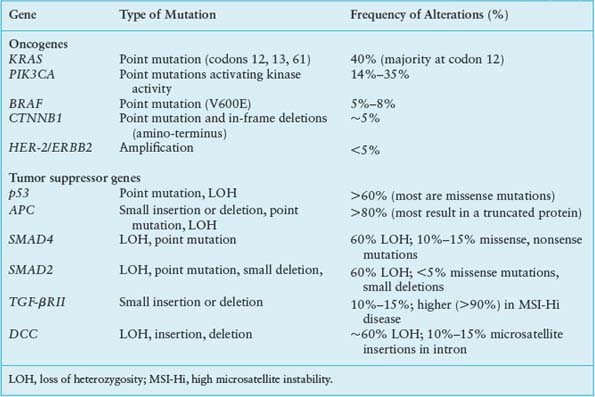
Building on the foundation established upon loss of the APC gatekeeper function, somatic mutations in cellular protooncogenes and tumor suppressor genes contribute cumulatively to the acquisition of malignant properties. The limited spectrum of recurring mutations in CRC provides a context for refined appreciation of the adenoma–carcinoma sequence and points to rational targets for therapy. The most frequent genetic events (Table 19.4) are discussed below, with reference to clinical associations and their impact on cell growth and differentiation.
KRAS, BRAF, and PIK3CA Oncogene Mutations
The Ras family of small G-proteins transduces growth factor signals and is aberrantly activated in a wide variety of cancers. KRAS gene mutations are detected in about 40% of colorectal carcinomas,96 mostly clustered in codons 12 and 13, with fewer than 10% occurring at codon 61. These mutations can be present in small polyps, and their frequency increases with lesion size.97 The significance of the same mutations in lesions of low malignant potential, such as hyperplastic polyps or aberrant crypt foci lacking dysplasia, is uncertain.98 KRAS mutation is not required to initiate adenomas but almost certainly contributes to their progression, and disruption of mutant KRAS alleles in colon cancer cells impedes growth.99,100
Oncogenic KRAS mutations have the potential to deregulate several effector pathways for cell proliferation, survival, and metastasis (Fig. 19.3). Constitutive phosphorylation of extracellular signal-regulated kinases, ERK-1 and ERK2, frequently accompanies KRAS mutation, reflecting activation of the mitogen-activated protein kinase (MAPK) pathway. This KRAS-mediated growth factor signaling cascade recruits RAF kinases to the plasma membrane and triggers ERK kinases MEK1 and MEK2 to activate ERK1 and ERK2, which phosphorylate factors that control the G1-S cell cycle transition, among other substrates.101 Although other, non-KRAS-mediated growth factor pathways also activate the MAPK cascade, signaling in cancer is most often deregulated through activating mutations in KRAS or BRAF. BRAF is mutated in 5% to 8% of CRCs, especially those associated with MSI-Hi or CIMP.102,103 The most common BRAF mutation in colorectal and other cancers such as melanoma, V599E, affects a residue within the activation loop of the kinase domain and constitutively activates the kinase function, perhaps as a phosphomimetic.104 Like mutant KRAS, activated BRAF also phosphorylates MEKs and ERKs, leading to dysregulated growth. Notably, KRAS and BRAF mutations are mutually exclusive in CRC,102 reinforcing the role of the two oncogenes in a common cellular pathway and possibly reflecting alternative routes to the same end. However, patients with BRAF-mutant CRCs have a worse prognosis than those with KRAS-mutant tumors,9 indicating the presence of distinctive features.
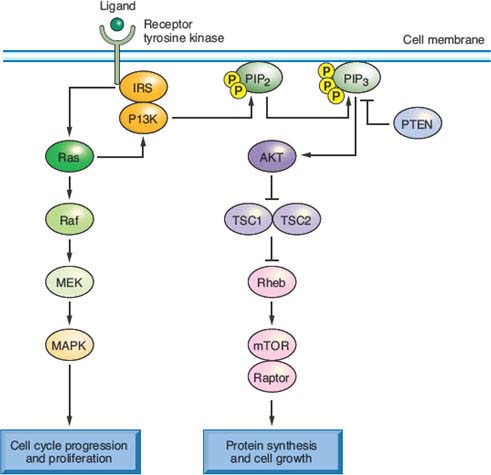
FIGURE 19.3 Signaling pathways, oncogenic mutations, and therapeutic opportunities in colorectal cancer (CRC). It is instructive to consider common genetic alterations in CRC in light of a common canonical outline of signaling through receptor tyrosine kinases, among which the epidermal growth factor receptor is a prime example. KRAS, the oncogene mutated in up to 40% of CRCs, signals receptor activation through RAF proteins (including BRAF, which is mutated in 5% to 8% of CRCs) and phosphatidylinositol 3-kinase (PI3K), whose catalytic PIK3CA subunit is mutated in 15% to 20% of CRCs. These transducers in turn activate the intracellular mitogen-activated protein kinase and AKT or mammalian target of rapamycin (mTOR) pathways, respectively. Common mutations hence confer growth factor independence on cells, resulting in dysregulated proliferation, protein synthesis, and metabolism. They also represent promising targets for therapeutic interference with aberrantly activated signaling cascades.
Besides the MAPK cascade, KRAS signals through the phosphatidylinositol 3-kinase (PI3K) pathway,105 which phosphorylates the intracellular signaling lipid phosphatidylinositol-4,5-bisphosphate at the 3 position, triggering a cascade that promotes cell growth and survival.106 Between 15% and 35% of CRCs carry activating mutations in PIK3CA, the gene encoding the catalytic p110 subunit of PI3K; most mutations cluster in exons 9 and 20 and seem to arise late in the adenoma–carcinoma sequence, perhaps coincident with invasion.107 Curiously, although both PI3K and BRAF act downstream of KRAS, only the BRAF–KRAS mutant pair is mutually exclusive; PIK3CA mutations appear in nearly 10% of KRAS-mutant CRCs, implying that activation of both oncogenes may not be redundant. One reason could be that mutant KRAS activates PI3K signaling inefficiently108; more likely, oncogenic signaling pathways are less strictly linear than is convenient to depict. Indeed, the overtly parallel streams of KRAS signaling through RAF-MEK and PI3K (Fig. 19.3) interact extensively with one another and, in particular, both streams modulate mTOR, which coordinates cell growth with nutrient responses.109 Such considerations notwithstanding, the recurrence of KRAS, BRAF, and PIK3CA mutations in CRC, at a collective frequency that rivals growth factor pathway aberrations in any carcinoma, appropriately places these oncogenes and their effectors at the center of research and drug development efforts.
The p53 Tumor Suppressor
Allelic loss of chromosome 17p is observed in three of four colorectal carcinomas but fewer than 10% of adenomatous polyps.97 The remaining p53 allele is inactivated in most tumors with 17p loss of heterozygosity (LOH), most often at codons 175, 245, 248, 273, or 282.110 LOH of 17p and mutations in p53 thus appear to arise during the transition from adenoma to carcinoma, perhaps facilitating progression. When faced with stress from DNA damage, hypoxia, reduced nutrient access, and aneuploidy, cells with intact p53 function undergo cell cycle arrest and apoptosis. The loss of p53 may allow cells to overcome such barriers to tumor survival and progression.
Chromosome 18q Loss of Heterozygosity
LOH of chromosome 18q is rare in adenomas, except for large villous adenomas, observed in over 60% of primary CRCs, and present in nearly all liver metastases from colorectal primary tumors lacking MSI.111 This sequence implicates loss of one or more genes on chromosome 18q in disease progression. Recent studies112 challenge early ideas that 18q LOH conferred a poor prognosis.113 The minimal common region of LOH on 18q21 contains two candidate tumor suppressor genes.114 One of these genes, SMAD4 (DPC4), is deleted in about one-third of cases and the remainder show loss of the other candidate gene, DCC (deleted in colorectal cancer), which encodes a cell surface receptor for the netrin family of axonal guidance proteins. SMAD4/DPC4 and SMAD2 are implicated as positive and negative regulators of TGF-β signaling, respectively, and lie close together on chromosome 18q. Somatic SMAD4 deletions or localized mutations are present in 10% to 15% of CRCs, with 18q LOH and germline mutations noted in some FJP kindreds115; but SMAD2 is somatically inactivated in fewer than 5% of CRCs.116 Although DCC messenger RNA and protein are lost in more than 50% of CRCs,117 few specific DCC coding mutations are known. Together with the low frequency of SMAD4 or SMAD2 mutations in 18q-deficient tumors, these findings suggest a complex, multifactorial basis for selection of 18q LOH in CRC.
INFREQUENT CHANGES AND CURRENT VIEWS OF THE MUTATIONAL LANDSCAPE OF COLORECTAL CANCER
Few cellular oncogenes besides KRAS, BRAF, and PIK3CA are common targets of point mutations in CRC (Table 19.4). Sequencing of the kinase domains in 133 tyrosine kinase (TK) and TK-like genes and 340 serine-threonine kinase (STK) genes revealed few recurrent nonsynonymous somatic mutations, including a handful in the NTRK2, NTRK3, and FES receptor TK genes118 and the STK genes MAPK24 and MYLK2.119 Analysis of tyrosine phosphatases similarly identified scattered mutations of uncertain pathogenic significance in up to a quarter of CRCs.120 In principle, increased expression by epigenetic mechanisms or gene amplification can have the same effect as activating point mutations, but oncogenes such as HER-2 (ErbB2), MYC, MYB, and CCND1 are amplified in fewer than 5% of cases in aggregate.
Most studies summarized in this chapter suffer from biased or opportunistic testing of genes, gene families, and pathways. To avoid such bias, groups have assessed copy number alterations (CNAs) by array hybridization and begun to sequence all exons and full genomes. Analysis of more than 13,000 genes in 11 CRCs revealed a substantially larger number of mutations than expected, with a conservative average estimate of 81 per tumor, but few of these mutations recur frequently and most may make small, if any, contribution to the neoplastic process.121 The results are nevertheless informative because they reveal, for example, substantive differences in the nature of mutations in different tumor types, with CRCs showing a genomewide bias toward C:G to T:A transitions at 5’-CpG-3’ sites and a lower frequency of mutations in 5’-TpC-3’ dinucleotides than observed in breast cancer.121 Such findings may find value in linking specific environmental contributors to the mutational spectrum in initiation and progression of disease. For nearly half of the 50 CNAs—28 amplifications and 22 deletions—identified in one study, 15 of them were recurrent and encompassed fewer than 12 genes122 and were also detected in other cancers, presumably reflecting selection for common genetic events. A subsequent study found that CRCs carry an average of 17 genes that are deleted or amplified to 12 or more copies per cell.123 Pathways controlling cell adhesion, signaling, DNA topology, and cell cycle were most commonly affected by these changes or by point mutations, confirming that CRCs reflect disturbances in selected pathways for replicative and tissue homeostasis. Although affected cellular processes may be common to most cancers, the specific molecular defects in CRC identify the best targets for rational, directed therapy.
Prognostic and Predictive Value of Molecular Properties and Tumor Genotypes
Phenotypes and specific genetic alterations in a cancer can hold useful clues about clinical behavior, prognosis, and response to therapy. Aneuploidy and tetraploidy confer a poor prognosis and MSI-Hi imparts a relatively favorable outcome in early stage CRC.124 Benefit from adjuvant 5-fluorouracil monotherapy may be confined to patients with MSS tumors.125,126 As nearly all CRCs have constitutive Wnt pathway activity, this factor alone has limited prognostic value, and outcomes seem unaffected by whether Wnt activity was stimulated by APC or CTNNB1 mutations. The presence of KRAS or BRAF mutations and possibly also PIK3CA mutation or loss of PTEN expression predicts a lack of response to EGFR monoclonal antibodies.8,127 These observations are important because they direct treatment decisions and because, unlike acute leukemia or breast cancer, CRC had previously resisted subgrouping on the basis of specific genetic alterations. For example, mutations in the two most frequently affected oncogenes, KRAS or PIK3CA, seem not to impact survival in stage III or IV disease treated with current drug regimens.9,128 Patients with metastatic BRAF-mutant disease have especially low survival and respond poorly to current chemotherapy regimens.9
Mouse Models of Colorectal Cancer
The laboratory mouse has immense value in genetic analysis of CRC. The most informative mouse line, multiple intestinal neoplasia (Min), carries a mutagen-induced nonsense mutation in the murine homolog of APC and phenocopies human FAP with respect to intestinal adenomatosis,129 although species’ differences place the burden mainly in the small intestine instead of the colon. APCMin mice serve as a cornerstone for genetic analysis of intestinal tumors and deregulated Wnt signaling. Modifications of the murine APC locus influence disease phenotypes, as detailed in other reviews130; the D716 allele increases the number of adenomas, all showing LOH of the wild type copy,131 whereas the APC1638N mutation results in fewer adenomas, more of which appear in the colon.132 ApcPirc rats carry a stop codon at position 1137 and develop more than half the tumors in the colon, similar to humans.133 Deletion of the N-terminal degradation domain of β-catenin also stabilizes the protein and produces widespread intestinal polyposis in mice.134 Expression of KrasG12D or KrasG12V in the intestinal epithelium of otherwise intact mice has subtle consequences on cell signaling and proliferation, whereas expression in mutant Apc backgrounds expands progenitor cell numbers and hastens adenoma progression,135 modeling the sequence of mutations in human colon cancer. Finally, the in vivo requirements of each MMR gene have been analyzed by introducing inactivating mutations in all murine mutS and mutL homologs.136 These mice generally develop lymphomas more often than intestinal tumors, and some mutants show neither MSI nor intestinal tumors, highlighting species differences. Nevertheless, the defects point to an essential role for some MMR genes in meiosis and underscore the importance of MMR in protecting cells from mutation and malignancy.
Selected References
The full list of references for this chapter appears in the online version.
1. Noffsinger AE. Serrated polyps and colorectal cancer: new pathway to malignancy. Annu Rev Pathol 2009;4:343.
3. Levin B, Lieberman DA, McFarland B, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008;134:1570.
5. Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 2008;105:4283.
6. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10:789.
8. Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408.
9. Souglakos J, Philips J, Wang R, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009;101:465.
12. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993;363:558.
14. Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985;228:187.
17. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003;300:455.
20. Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681.
22. Jess T, Loftus EV Jr, Velayos FS, et al. Risk of intestinal cancer in inflammatory bowel disease: a population-based study from Olmsted county, Minnesota. Gastroenterology 2006;130:1039.
34. Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275:1787.
36. Sansom OJ, Reed KR, Hayes AJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 2004;18:1385.
37. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 2006;127:469.
41. Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608.
42. van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002;111:241.
43. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509.
44. Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007;446:676.
48. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453.
50. Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev 1995;5:382.
53. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261.
57. Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995;268:1336.
62. Huang J, Papadopoulos N, McKinley AJ, et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci U S A 1996;93:9049.
63. Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003;348:791.
65. Thibodeau SN, French AJ, Cunningham JM, et al. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998;58:1713.
66. Liu B, Nicolaides NC, Markowitz S, et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 1995;9:48.
69. Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006;38:787.
73. Sweet K, Willis J, Zhou XP, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. J Am Med Assoc 2005;294:2465.
79. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391:184.
81. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004;6:91.
82. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 2007;21:2525.
83. Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997;16:64.
85. Goel A, Arnold CN, Niedzwiecki D, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability–high sporadic colorectal cancers. Cancer Res 2004;64:3014.
87. Slattery ML, Fitzpatrick FA. Convergence of hormones, inflammation, and energy-related factors: a novel pathway of cancer etiology. Cancer Prev Res 2009;2:922.
90. Wiesner GL, Daley D, Lewis S, et al. A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2-31.2. Proc Natl Acad Sci U S A 2003;100:12961.
92. Tomlinson I, Webb E, Carvajal-Carmona L, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 2007;39:984.
95. Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 2010;107:9742.
96. Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987;327:293.
97. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319:525.
101. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003;3:11.
102. Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002;418:934.
104. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949.
107. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554.
110. Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989;244:217.
115. Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;280:1086.
121. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268.
123. Leary RJ, Lin JC, Cummins J, et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc Natl Acad Sci U S A 2008;105:16224.
127. Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One 2009;4:e7287.
129. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322.
134. Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 1999;18:5931.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


