Chronic myeloid leukemia
Jorge Cortes, MD  Richard T. Silver, MD
Richard T. Silver, MD  Hagop M. Kantarjian, MD
Hagop M. Kantarjian, MD
Overview
Chronic myeloid leukemia (CML) was the first malignancy where a unique chromosomal abnormality, the Philadelphia chromosome, was directly linked to it. The further unraveling of the molecular and physiologic consequences of this abnormality, with a fusion gene (BCR-ABL) translating into a chimeric protein with increased tyrosine kinase activity, led to the development of specific tyrosine kinase inhibitors. The use of such agents as initial therapy has resulted in a dramatic change in the natural history of the disease where patients properly managed are expected to have a life expectancy similar to that of the general population. Several treatment options are also available for patients who may not have optimal response to their initial therapy. Current research is focusing on approaches that may increase the probability of successful treatment discontinuation for more patients.
Chronic myeloid leukemia (CML) is a myoproliferative neoplasm affecting a pluripotent progenitor and involving myeloid, erythroid, megakaryocytic, B, and sometimes T, lymphoid cells, but not marrow fibroblasts. It is characterized by the presence of a unique chromosomal abnormality, the Philadelphia chromosome (Ph).
Historical perspective
In 1960, a minute chromosome was identified in patients with CML, an abnormality later identified as a balanced translocation between chromosomes 9 and 22.1, 2 Later studies demonstrated that this translocation resulted in the creation of a chimeric gene, BCR-ABL, that when transfected into mice can induce CML.3 Translation of this chimeric gene results in a fusion protein with constitutive tyrosine kinase activity.4 Interferon-alpha was the first therapy to induce the disappearance of the Philadelphia chromosome, establishing complete cytogenetic responses (CCyRs) as the gold standard of response to therapy.5 The knowledge of the activation of a tyrosine kinase led to the development of tyrosine kinase inhibitors (TKIs), initially imatinib6 and later second- and third-generation drugs that radically changed the natural history of the disease (Figure 1).7 Today, patients diagnosed with CML, if properly managed, have a life expectancy similar to that of the general population.
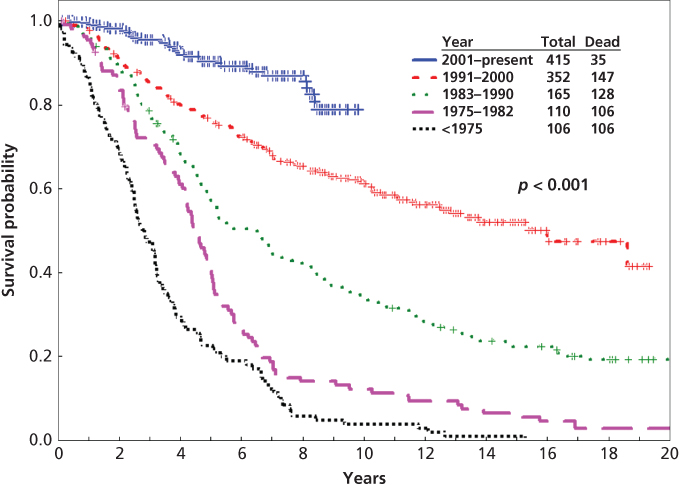
Figure 1 Survival of patients with newly diagnosed CML in chronic phase referred to MD Anderson Cancer Center (N = 1148; 1965–2010).
Source: Kantarjian et al. 2012.7 This research was originally published in Blood. © The American Society of Hematology.
Incidence and epidemiology
CML accounts for 15% of all leukemias.1, 2 The median age of onset of CML is 55–65 years, and the incidence increases with age with a slight male preponderance (ratio 1.8:1).8 It is estimated that 5980 new cases of CML will be diagnosed in the United States in 2014 with 810 patients dying from their disease.9 The incidence in the United States has remained constant at about 1.81:100,000. With modern therapy, the annual mortality has decreased from 15–20% to 2%, and the estimated median survival may exceed 20 years. Thus, the prevalence of CML in the United States in the next three decades may exceed 200,000 cases.10
Risk factors
In most patients with CML, a causative factor cannot be identified. Although ionizing radiation is leukemogenic, the most common leukemia following radiation is acute myeloid leukemia. CML has been reported following the atomic bomb catastrophe in Japan in 1945, and in earlier studies in radiologists and in patients with ankylosing spondylitis treated with radiation therapy.3, 4, 11, 12 No other known risk factors have been recognized.
Pathology
CML typically follows a biphasic or triphasic course with a chronic phase, followed by an intermediate accelerated phase, and eventually a frequently terminal blastic phase.13 Leukocytosis is common with white cell counts frequently greater than 100 ×109/L. Historically, the median survival for patients in chronic phase was 3–6 years, with an estimated annual risk of transformation to the blastic phase of 5–10% in the first 2 years and 15–20% subsequently. Accelerated phase is characterized by increasing maturation arrest. Different criteria have been used to define accelerated phase. One common classification defines accelerated phase as the presence of any of the following: ≥15% blasts, ≥30% blasts plus promyelocytes, ≥20% basophils, platelets <100×109/L unrelated to therapy, or cytogenetic clonal evolution.14 Other criteria have been proposed, such as the World Health Organization (WHO) proposal, but some of these classifications7, 15 have not been clinically validated.16 The median survival for patients in accelerated phase was 1–2 years,8, 17 but has greatly improved with the use of TKI.18 The blastic phase is defined by the presence of ≥30% blasts in the peripheral blood or bone marrow or by the presence of extramedullary disease with immature cells.19 The WHO proposed this to be changed to ≥20% blasts, but this change is not justified based on clinical data.10, 16 The blastic phase can be classified according to the immunophenotype as myeloid, lymphoid, biphenotypic, or mixed lineage (lymphoblastic–myeloblastic). Lymphoid blastic phase occurs in 20–30% of patients, myeloid in 50%, and undifferentiated in 25%.11, 20 The median survival in blastic phase is 3–6 months. Patients with lymphoid blastic phase have a better prognosis, with a response rate of ≥90% and median survival of >18 months with TKI combined with chemotherapy.21
Laboratory features in the chronic phase include leukocytosis with left maturation shift and frequently basophilia and eosinophilia (Figure 2). Thrombocytosis is common but thrombotic phenomena are unusual. Some degree of anemia is common. There is a reduction in leukocyte alkaline phosphatase (LAP) activity and a marked elevation of serum B12 levels.
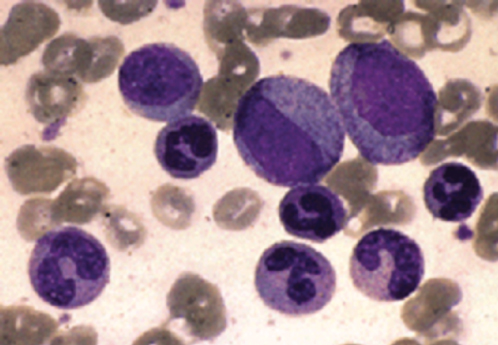
Figure 2 Chronic myeloid leukemia. Leukocytosis with myelocytes, metamyelocytes, band cells, and polymorphonuclear leukocytes are characteristics of the peripheral blood in the chronic phase of this disease.
The bone marrow is hypercellular. In chronic phase, all stages of differentiation are present, but the myelocytes predominate, whereas myeloblasts and promyelocytes account for <10% of cells (Figures 3 and 4). Megakaryocytes may be increased, and there might be increased reticulin fibrosis, which may worsen with disease progression, and is reversed with TKI.22
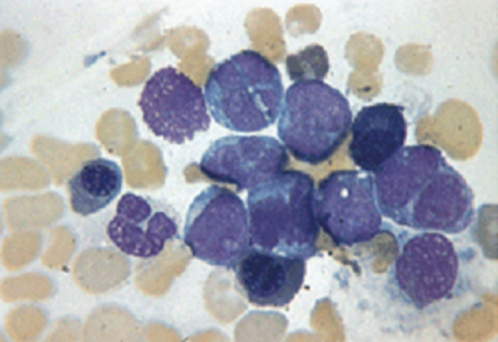
Figure 3 Chronic myeloid leukemia, myeloid blast crisis. The marrow aspiration shows predominance of blast forms, which have a myeloid appearance.
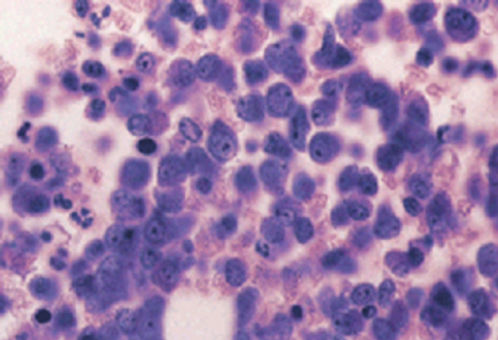
Figure 4 Chronic myeloid leukemia, blast phase. This bone marrow biopsy shows “blasts” with prominent nucleoli comprising about 75% of the marrow cells.
In the blastic phase, lymphoid blasts contain terminal deoxynucleotidyl transferase (TdT). Lymphoblasts usually express CD10, CD19, and CD22 or other B-cell markers20; T-cell blastic phase is less common. The myeloid blastic phase may mimic acute myeloid leukemia. The myeloblasts stain with myeloperoxidase and express myeloid markers including CD13, CD33, and CD117.
Rarely, patients may be present in lymphoid or myeloid blastic phase without a recognized antecedent chronic phase. The differentiation between this presentation and Ph-positive acute lymphoblastic (ALL) or myeloid (AML) leukemias may be impossible, but the distinction is semantic as the treatment and prognosis are the same. Megakaryoblastic, erythroblastic, and basophilic transformations are uncommon.
Prognostic classification
Prognosis in CML is variable. Risk classifications have been proposed to stratify patients and assist in treatment decisions. The Sokal model is most frequently used23 and defines three risk groups: low (about 40–50% of all patients), intermediate (about 30%), and high risk (10–20%), with median survivals of 4.5, 3.5, and 2.5 years, respectively, with busulfan or hydroxyurea. The model still predicts response to therapy and progression-free survival with imatinib therapy, although outcomes for all risk groups are significantly better than in the past. Other classifications have been proposed such as the Harford score (more applicable to interferon-treated patients),24 the simpler but not universally validated EUTOS score (based only on percentage of basophils and size of spleen),25–27 or the Gratwohl score for SCT (stem cell transplant).20, 28 With TKI therapy, the significance of several prognostic factors (older age, marrow fibrosis, deletion of derivative 9q, complex Ph chromosome) has been reduced or eliminated.21–23, 29–32 Interestingly, adolescents and young adults may have a worse outcome, arguably because of poor adherence to therapy.33
Pathogenesis
The primary biologic defect in CML is unregulated proliferation with discordant maturation, reduced apoptosis, and defective adherence to the bone marrow stroma.34–36
Cytogenetics
The hallmark of CML is the Ph chromosome. This results from a reciprocal translocation between chromosome 9 and chromosome 22, t(9;22)(q34.1;q11.21) that transposes the 3′ segment of the ABL gene to the 5′ segment of the BCR gene.2 This abnormality creates the chimeric BCR-ABL oncogene (Figure 5).
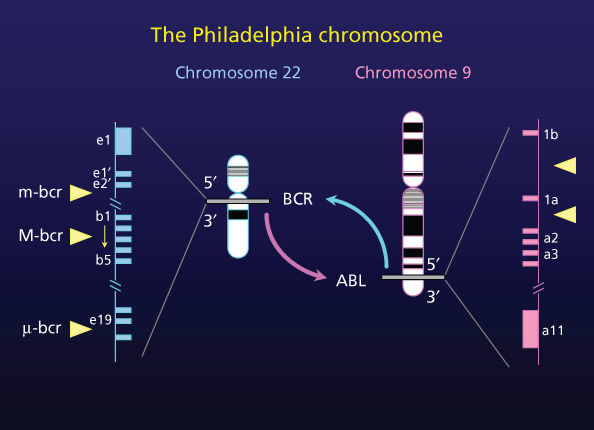
Figure 5 Schematic of 9;22 chromosome translocation.
The Ph chromosome is found in ∼95% of patients with CML. It is also observed in 5% of children and 15–30% of adults with acute lymphoblastic leukemia and in 2% of patients with newly diagnosed acute myeloid leukemia.37 Some patients have variant translocations, which may be simple (involving chromosome 22 and one additional chromosome other than chromosome 9) or complex (involving chromosomes 22, 9, and at least one other chromosome).38 These patients historically had an inferior outcome, but with imatinib therapy they have a similar prognosis to patients with the classic Ph chromosome.31, 32
Progression of CML is frequently accompanied by additional cytogenetic abnormalities. The most common abnormalities include a second Ph chromosome, isochromosome 17, trisomy 8, trisomy 19, and deletion 20q.39 The molecular consequences of these abnormalities are not known. Mutations or deletions of tumor suppressor genes, such as p16 and TP53, and methylation of ABL, BCR, p15, and cadherin-13 may contribute to transformation.40–44 Activation of JAK–STAT pathway might be responsible for the survival of the leukemic stem cell.45, 46 In blast phase, granulocyte-macrophage progenitors are the candidate stem cells, and the activation of β-catenin may enhance the self-renewal activity of these cells.47
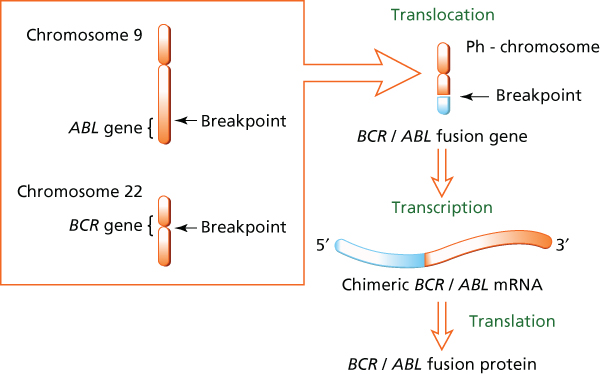
Figure 6 Summary of the cytogenetic and molecular effects of the Ph chromosome.
After successful treatment with TKI, chromosomal abnormalities are found in the Ph-negative cells in 10–15% of patients, most frequently trisomy 8, monosomy 7 or 5, and deletion 20q.48 These may regress spontaneously in some cases, but in rare instances (<1%) may lead to the development of a myelodysplastic syndrome or AML.40–42, 49–51
Molecular biology
The reciprocal 9;22 translocation results in a chimeric oncogene, BCR-ABL, that codes for a Bcr-Abl oncoprotein with constitutional tyrosine kinase activity.46, 52
There is some heterogeneity in the breakpoints on chromosomes 9 and 22. On chromosome 9, breaks may occur in a region 200 kb or more in length, resulting in most of the c-abl gene being translocated.46 The breakpoints within the ABL gene occur either upstream of exon Ib, downstream of exon Ia, or, more frequently, between exons Ib and Ia. Breakpoints in BCR occur most frequently in the major breakpointcluster region that includes exons e12–e16 (formerly b1–b5), resulting in either b2a2 (e13a2) or b3a2 (e14b2) fusion transcripts, both of them generating a 210-kDa protein (p210BCR-ABL1). In few patients with CML (but more frequently in Ph-positive ALL), the breakpoint may occur in the minor breakpoint cluster region, resulting in an e1a2 fusion (translated into a 190-kDa protein).53 Less frequently, the breakpoint may occur in a different, more distal, breakpoint region μ-bcr.
Transcription of the fusion BCR-ABL oncogene results in a chimeric BCR-ABL mRNA that is translated into three fusion proteins of varying sizes p190Bcr-Abl, p210Bcr-Abl, and p230Bcr-Abl, according to the breakpoint on BCR (Figure 6). In all instances, the fusion protein has unregulated constitutive tyrosine kinase activity that triggers intracellular signaling pathways such as STAT, RAS, RAF, JUN kinase, MYC, AKT, and BCL-2, which confer the malignant phenotype.52
Most patients with typical chronic phase CML express either the b2a2 (e13a2) or b3a2 (e14a2) rearrangements. The clinical features, response to treatment, and prognosis are similar in both groups. Patients with Ph-positive acute lymphoblastic leukemia may express either p210Bcr-Abl (30–50%) or p190Bcr-Abl (50–70%). Rare patients with CML in chronic phase express e1a2 (p190Bcr-Abl) and have a worse prognosis.53, 54 The p230Bcr-Abl is associated with a more indolent disease and a phenotype similar to chronic neutrophilic leukemia.
In approximately 5–10% of morphologically typical cases of CML, the Ph chromosome cannot be identified. In one-third of these, the BCR-ABL rearrangement is present. These patients have similar characteristics, response to treatment, and prognosis as Ph-positive CML.16, 55 The other two-thirds lack the BCR-ABL rearrangement (called “atypical CML” in the WHO classification) with different clinical and hematologic features, including lower initial white cell and platelet counts, with progression characterized by progressive cytopenias and organomegaly rather than blastic transformation and a median survival of 18–24 months.17, 56, 57 This type of leukemia is not discussed further in this chapter. Reverse transcriptase polymerase chain reaction (RT-PCR) can detect the BCR-ABL transcript with a sensitivity of 10−5.48, 58 BCR-ABL rearrangement has been identified in up to 25–30% of normal adults using RT-PCR approximately 3-log more sensitive (sensitivity ∼10−8) than the one used clinically.59 This suggests that clonal disease requires escape from immune surveillance and/or a second oncogenic event to become clinically relevant.
A subset of patients treated with TKI develop resistance. Several mechanisms of resistance have been identified; the most common are mutations in the BCR-ABL kinase domain (KD). More than 50 different mutations have been reported and involve many of the domains in the BCR-ABL structure, including the P-loop (the domain where ATP binds), the activation loop, and the catalytic domain, as well as the amino acids where imatinib makes contact with BCR-ABL.60 Different mutations vary in their sensitivity to different TKI. Some mutations are inhibited by slightly higher concentrations of TKI than that is required to inhibit the wild-type form; others are completely insensitive.61, 62 Mutational analysis is useful in patients with resistance to TKI to identify the TKI predicted to inhibit a given mutation as determined by the IC50 for particular agents. For example, F317L and V299L are insensitive to dasatinib, V299L is insensitive to bosutinib, and F359V and E255K/V are insensitive to nilotinib, while T315I is sensitive only to ponatinib (a third-generation TKI effective against all mutations).63–65
Diagnosis
Approximately 90% of patients are diagnosed in the chronic phase, which is usually asymptomatic.13 Symptoms usually develop insidiously and are usually due to splenomegaly (pain, abdominal fullness, early satiety) or anemia (fatigue). Less commonly, patients may be present with gout, anorexia, weight loss, unexplained fever, or signs of platelet dysfunction (e.g., ecchymoses or hemorrhage). Patients with very high WBC may have signs of hyperviscosity (priapism, cerebrovascular accidents, tinnitus, confusion, retinal hemorrhage). Symptoms associated with the accelerated phase may include fever, night sweats, weight loss, or bleeding associated with thrombocytopenia. Occasionally, blood and bone marrow features of accelerated phase occur in patients who are asymptomatic and identified only during routine follow-up. The blastic phase of CML is commonly associated with constitutional symptoms (night sweats, weight loss, fever, bone pain), anemia, an increased risk of infections, and/or bleeding.
When the diagnosis of CML is suspected, a bone marrow aspiration is mandatory. Although the diagnosis of CML can be made in peripheral blood, proper staging and recognition of all features of the disease can only be made with the evaluation of the bone marrow and peripheral blood. The bone marrow aspiration should include the following: (1) cell differential for proper staging; (2) assessment of fibrosis and other characteristics; and (3) cytogenetic analysis by G-banding to confirm the presence of the Ph chromosome and possibly additional chromosomal abnormalities; at least 20 metaphases are required for a proper interpretation of the karyotype and assessment of response. In addition, RT-PCR is recommended at the time of diagnosis. Although the number of BCR-ABL transcripts has no bearing on the prognosis (and is not reliable at baseline when using ABL as the control gene), it permits investigation of unusual transcripts (e.g., e19a2, b2a3, b3a3) not detected by standard PCR.
During the course of treatment, a bone marrow aspiration with cytogenetic analysis should be performed every 6 months until a CCyR is confirmed. Thus, a bone marrow is needed less frequently or perhaps not at all in most patients. It may be appropriate to continue doing karyotype analysis in patients with recognized chromosomal abnormalities in Ph-negative metaphases. FISH can be used to assess achievement of a cytogenetic response, and it can be done in peripheral blood, but it does not provide information on the presence of additional chromosomal abnormalities whether in Ph-positive cells (i.e., clonal evolution) or in Ph-chromosome-negative cells. A bone marrow aspiration should be performed in all patients with unexplained changes in peripheral blood counts or in those with loss of major molecular response (MMR) (and definitely if levels are close to 1% using the international scale). During treatment, patients should also be monitored with RT-PCR at 3, 6, and 12 months after the start of therapy and every 6 months after that.
Assessment for mutations is unnecessary at the time of diagnosis in patients in the chronic phase as mutations have not been found in this setting with the standard methodology. A search for mutations should be done when there is loss of CCyR. Patients who do not meet the recommended response criteria at the given times as per the European Leukemia Net (ELN) recommendations may have a mutation analysis performed, but mutations are more commonly found among patients with secondary resistance (i.e., loss of cytogenetic response) compared to those with primary resistance (i.e., not achieving optimal response).
Staging and prognostic factors
CML has three stages: chronic, accelerated, and blast phases. The features that define each phase are described earlier in the section titled “Pathology”. For patients in the chronic phase, risk classifications have been proposed. These include the Sokal,23 Hasford (Euro),24 and EUTOS25 classifications. These classifications are used mostly for prognostic purposes; treatment recommendations generally apply equally to all risk groups. The Sokal score is the most commonly used and is obtained through the formula: exp (0.0116 × (age [years] − 43.4)) + (0.0345 × (spleen size [cm] − 7.51) + (0.188 × ((platelets [109/L]/700)2 − 0.563)) + (0.0887 × (blasts [%] − 2.10)). Based on the score, three risk groups are identified: low (score < 0.8), intermediate (score 0.8–1.2), and high risk (>1.2). In the United States, only 10–15% of patients have a high-risk score at the time of diagnosis, whereas in other areas of the world, these can represent up to one-third of all patients.
Treatment
Initial treatment for all patients with CML is with TKI. Imatinib mesylate, a selective Bcr-Abl TKI introduced in the 1990s, has changed the natural history of CML.6, 66–70 Patients in the chronic phase at the time of diagnosis treated with TKI have a life expectancy that matches that of the general population.71
Imatinib mesylate
Imatinib is a potent inhibitor of Bcr-Abl and few other tyrosine kinases such as c-kit and PDGFR.6, 72 It was first used in CML after failure or intolerance to interferon-alpha.73, 74 Imatinib 400 mg taken orally daily, given to 454 patients, resulted in a CCyR of 57%. The estimated 5-year survival rate was 76%.73
The efficacy of imatinib in previously untreated patients was demonstrated in a multicenter randomized trial comparing imatinib to the combination of interferon-alpha and low-dose cytarabine [IRIS (Insulin Resistance Intervention after Stroke) trial].75, 76 With 8 years of follow-up, a CCyR was achieved by 83% of all patients, resulting in an event-free survival of 81%, transformation-free survival of 92%, and overall survival of 85%.77 The standard starting dose of imatinib in the chronic phase is 400 mg daily. Higher doses of imatinib (600–800 mg daily) may result in improved response rates that are achieved earlier.78, 79 It is controversial whether higher doses result in improved long-term outcome of patients.65, 80–82 To date, 400 mg daily remains the standard initial dose. Among patients in chronic phase who progress on imatinib 400 mg daily therapy, dose escalation to 600 or 800 mg daily recaptured some CCyRs.83, 84
Second-generation tyrosine kinase inhibitors
Dasatinib and nilotinib are second-generation TKI that are approximately 300-fold85 and 30-fold86 more potent than imatinib, respectively. They were first investigated and approved for treatment of patients who have experienced resistance or intolerance to imatinib. Subsequently, trials demonstrated that both of these agents resulted in more, deeper and faster responses than those seen with imatinib.87–89 This translated in a lower rate of transformation to accelerated and blast phase compared to imatinib. Thus, both agents are approved and considered standard for the initial treatment of patients with chronic phase CML.
In the randomized trial of dasatinib versus imatinib, the rate of confirmed CCyR by 12 months was 77% with dasatinib versus 66% with imatinib, and the cumulative rate of MMR by 36 months was 69% versus 55%, respectively. At 3 months, 84% of patients treated with dasatinib achieved BCR-ABL/ABL transcript <10% compared to 64% with imatinib. Transformation to accelerated and blast phase occurred in 3% and 5%, respectively. No difference in event-free or overall survival has been reported up to 4 years of minimum follow-up.90, 91 The standard starting dose of dasatinib for patients with newly diagnosed CML in chronic phase is 100 mg once daily.
A similar randomized trial investigated two different dose schedules of nilotinib (300 mg twice daily and 400 mg twice daily) compared to imatinib. By 12 months, the rate of CCyR was 80% with nilotinib 300 mg twice daily and 78% with 400 mg twice daily compared to 65% with imatinib. By 3 years, the rates of MMR were 73%, 70%, and 53%, respectively. BCR-ABL/ABL levels of <10% were achieved by 91% treated with nilotinib 300 mg twice daily, 89% with nilotinib 400 mg twice daily, and 67% with imatinib; the rates of freedom from progression to accelerated or blast phase at 4 years were 96.7%, 97.8%, and 93.1%, respectively. The 4-year estimated progression-free survivals were 92.7%, 96.3%, and 92%, and overall survivals were 94.3%, 96.7%, and 93.3%.92, 93 Nilotinib 300 mg twice daily is the standard therapy for newly diagnosed CML in chronic phase.
Bosutinib has also been investigated as an initial therapy for CML. There is an increase rate of responses, with deeper and faster responses resulting in fewer transformations to the accelerated and blast phases. However, bosutinib is not currently approved as an initial therapy for CML.94
Treatment algorithm
Achievement of a CCyR is associated with near eradication of the risk of transformation to accelerated and blast phases and a significant survival benefit, with a 10-year survival probability of 70–80% equivalent to that of the general population. Achieving molecular responses increases the probability of long-term durable responses although such responses have not resulted in improved survival among patients with CCyR. The 7-year probability of event-free survival for patients that achieve MMR is 95% compared to 86% among those with CCyR but no MMR. There is no difference in survival free from transformation to accelerated or blast phase or in overall survival. Deeper responses may offer the possibility of considering treatment discontinuation, something that today should be considered only through clinical trials.
In addition to the depth of response, the time to response is important to improve long-term outcomes. Patients with BCR-ABL/ABL transcripts <10% at 3 months from the start of therapy have significantly better probability of EFS (approximately 95%) compared to those with >10% transcripts (approximately 80%).95–98 There is also a significant but smaller difference in the overall survival. This has resulted in recommendations of what is considered optimal response to therapy that include BCR-ABL/ABL <10% by RT-PCR and/or Ph <35% by standard karyotype at 3 months, BCR-ABL/ABL <1% and/or Ph 0% at 6 months, and BCR-ABL/ABL <0.1% at 12 months. Levels above these are defined as warning or failure (Table 1).99
Table 1 Suggested management of most common adverse events associated with imatinib
| Adverse events | Management |
| Nausea/vomiting | Take with food, fluids |
| Antiemetics | |
| Diarrhea | Loperamide |
| Diphenoxylate atropine | |
| Peripheral edema | Diuretics |
| Periorbital edema | Steroid-containing cream |
| Skin rash | Avoid sun exposure |
| Topical steroids | |
| Systemic steroids (early intervention important) | |
| Muscle cramps | Tonic water or quinine |
| Electrolyte replacement as needed | |
| Calcium gluconate | |
| Arthralgia, bone pain | Nonsteroidal anti-inflammatory agents |
| Elevated transaminases (uncommon) | Hold therapy and monitor closely |
| Dose reduction upon resolution | |
| Myelosuppression | |
| Anemia | Treatment interruption/dose reduction usually not indicated |
| Consider erythropoietin or darbepoietina | |
| Neutropenia | Hold therapy if grade ≥3 (i.e., ANC < 1 × 109/L) |
| Consider filgrastima if recurrent/persistent, or sepsis | |
| Thrombocytopenia | Hold therapy if grade ≥3 (i.e., platelets <50 × 109/L) |
| Consider IL 11a 10 mcg/kg 3–7 days/week |
a The use of erythropoietin, darbepoietin, filgrastim, and interleukin-11 (IL-11) in this setting is not standard and should be considered investigational.
Patients who achieve an optimal therapy can continue therapy unchanged. The current algorithm involves therapy to continue indefinitely although studies are ongoing to determine whether treatment may be discontinued in some patients. Those who meet the definition of warning can continue therapy but adherence should be assessed, therapy optimized, and patients monitored rigorously every 3 months with treatment change considered if failure is identified. Once the definition of failure is met, treatment changes should be implemented.99
Intolerance is another reason why treatment change may be needed. Despite the excellent overall tolerability of TKI, they all have adverse events that need monitoring and adequate management. With proper management100 that may include transient treatment interruptions, dose adjustments, medical management of adverse events, and supportive care, most patients can continue therapy as an experience and adequate response. In general, patients should not be changed to a different TKI based on the first occurrence of an adverse event unless this is life threatening (e.g., Stevens–Johnson’s, myocardial infarction, stroke). True intolerance to TKI occurs in only approximately 5% of all patients. Suggestions for the management of the most common adverse events are presented in Table 2.
Table 2 Response criteria according to the European Leukemia Net.
| Time (mo) | Response | ||
| Failure | Warning | Optimal | |
| 3 | No CHR, and/or Ph+ >95% | BCR-ABL >10%, and/or Ph+ 36–95% | BCR-ABL ≤10%, and/or Ph+ ≤35% |
| 6 | BCR-ABL >10% and/or Ph+ >35% | BCR-ABL 1–10%, and/or Ph+ 1–35% | BCR-ABL <1%, and/or Ph+ 0% |
| 12 | BCR-ABL >1% and/or Ph+ >0% | BCR-ABL >0.1–1% | BCR-ABL <0.1% |
| Any | Loss of CHR Loss of CCyR Confirmed MMR loss Mutations CCA/Ph+ | CCA/Ph– (−7, or 7q–) | BCR-ABL <0.1% |
Abbreviations: CHR, complete hematologic response; Ph+, percentage of metaphases with presence of the Philadelphia chromosome (a minimum of 20 required for full assessment); CCyR, complete cytogenetic response; MMR, major molecular response; CCA/Ph+, clonal chromosomal abnormalities in cell with the Philadelphia chromosome.
Source:Adapted from Quintas-Cardama et al. 2009. This research was originally published in Blood. © The American Society of Hematology.
Treatment options after failure of prior TKI
Several TKI are available for the management of patients with resistance (as defined by the ELN99) or intolerance to prior TKI. These include dasatinib, nilotinib, bosutinib, and ponatinib. In addition, omacetaxine, a protein-synthesis inhibitor (not a TKI), is also approved for patients who have received at least two prior TKI.
Among patients who have received imatinib as their only prior TKI and experienced resistance or intolerance, dasatinib at a dose of 100 mg once daily induced a CCyR in 44% of patients with resistance and 67% of those with intolerance. A MMR occurred in 37% of patients with resistance or intolerance, with a 6-year probability of progression-free survival of 49% and overall survival of 71%.101, 102 Similarly, nilotinib, at a dose of 400 mg twice daily (the standard dose for second-line therapy), induced CCyR in 41% of patients with resistance to imatinib and 51% of those with intolerance for a 4-year progression-free survival of 57% and overall survival of 78%.103 Bosutinib is also a second-generation TKI with an activity against Src and Abl, 30–50 times more potent than imatinib against Abl but, in contrast to other TKI used for CML, with minimal inhibitory activity against c-kit or PDGF-R. Bosutinib has also been effective in this patient population inducing a CCyR in 48% of patients with resistance and 52% of those with intolerance to imatinib with MMR in 64% and 65% of those achieving CCyR, respectively. This resulted in a progression-free survival of 73% and 95%, respectively.104 None of these agents is active in patients with the T315I mutation.
Bosutinib and ponatinib have been investigated among patients who have received two or more TKI. With bosutinib 500 mg once daily, a major cytogenetic response was achieved in 30–35% of 118 patients who had received imatinib and were then resistant or intolerant to dasatinib, or resistant to nilotinib. The corresponding rates of CCyR were 14%, 28%, and 27%, respectively, and 2-year estimates of progression-free survival 65%, 81%, and 77%.105 Ponatinib is a potent inhibitor of Abl tyrosine kinase activity as well as other kinases including c-kit, FLT3 and VEGFR. Importantly, it has potent inhibitory activity against unmutated BCR-ABL or in the presence of any of the KD mutations tested, including the multiresistant T315I.106 With ponatinib at a dose of 45 mg daily, among 267 patients of which 93% had received at least two prior TKI (60% had received at least three), a major cytogenetic response was achieved in 56% (complete in 46%) and a MMR in 34%. Major cytogenetic response was sustained for at least 12 months in 91%, and the overall survival was 94% at 12 months. For patients with T315I, the response rates were 70%, 66%, and 56%, respectively, with similar durability of response and overall survival.107, 108 With omacetaxine, 20% of patients treated in chronic phase after resistance to at least two prior TKI achieved a major cytogenetic response, with a median overall survival of 33.9 months.109 Responses can be seen regardless of mutation status including patients with T315I.110 Allogeneic SCT should also be considered in patients who have experienced resistance to TKI.
Stem cell transplantation
Allogeneic SCT may be curative in 40–80% of patients who receive a transplant from an HLA-identical sibling or an unrelated donor.111 Among patients transplanted in first chronic phase, 3514 from a match-related sibling and 1052 from an unrelated donor, the 5-year survival probability was 63% and 55%, respectively, and the leukemia-free survival was 55% and 50%, respectively. The 5-year cumulative incidence of relapse is 12–14%.111 However, for patients who have been alive and in continuous complete remission for ≥5 years, the survival rate at 15 years was 88% for those receiving sibling donor cells and 87% from those with unrelated donors, with cumulative incidence of relapse of 8% and 2%, respectively, with relapses documented as late as 18 years after transplant.112 Patients who were treated with stem cell transplant had a higher probability of death for the first 14 years after transplant compared to individuals in the general population of similar age, sex, and race.112 The availability of donors has improved in recent years because of the increasingly successful results with transplant using cord blood or haploidentical donors, and the reach has extended to higher age groups with the use of non-myeloablative conditioning regimens. In addition, early mortality rates have decreased with improved supportive care and GVHD (graft-versus-host disease) prophylaxis and management. However, with the availability of TKI, the role of SCT has changed significantly. Several reports have confirmed that previous exposure to TKI does not adversely affect the outcome after SCT although time from diagnosis to SCT remains an important prognostic factor for the outcome after transplant.113, 114 Similarly, patients transplanted in chronic phase have a significantly better outcome than those transplanted in advanced phase.111 Until the advent of ponatinib and omacetaxine, SCT was the only available therapy for patients with T315I mutation, with 2-year survival probability of 59% for patients transplanted in chronic phase.115, 116
Patients transplanted in accelerated or blastic phase have significantly worse outcomes, with 5-year survival probabilities of 40% accelerated phase and 10–15% blastic phase. Patients in blastic phase transplanted after achievement of a second chronic phase may have long-term outcomes similar to that of the accelerated phase.28, 117, 118
Treatment recommendations in CML in 2015
The results with imatinib to date have been excellent and durable, and with shorter follow-up, the results with dasatinib and nilotinib appear superior with earlier and deeper responses and with lower rate of transformation, although these outcomes have not translated into a significant difference in event-free or overall survival. The life expectancy of patients treated with TKI is similar to that of the general population, particularly for those patients achieving a CCyR. Based on this, TKIs are standard initial therapy for all patients with CML in chronic phase. Because of the improved outcome with dasatinib or nilotinib in randomized trials compared to imatinib,90–93 these agents are preferred in many instances. However, imatinib is likely to remain the treatment of choice for a large percentage (perhaps the majority) of patients throughout the world. It should be emphasized that imatinib is also adequate treatment. The most important aspect to offer a patient the best possible long-term outcome is the proper management of the patient. This includes a close follow-up to recognize and manage adverse events. All TKI have adverse events associated with them, but most of these are manageable through transient treatment interruptions, dose adjustments, and/or medical interventions. Very few patients (around 5%) are truly intolerant to a given drug. Until ongoing studies demonstrate a long-term benefit of early change in therapy, patients not having a major cytogenetic response (or not having BCR-ABL/ABL transcripts <10%) at 3 months may continue therapy unchanged but they need to be checked again at 6 months.99 For patients still not achieving a major cytogenetic response (or still with BCR-ABL/ABL transcripts >10%) after 6 months of therapy treatment change may be considered, although there are also no studies that have shown that change of therapy in this setting alters long-term outcome. Patients need to continue with monitoring at least every 6 months as detailed earlier. A change of therapy is clearly indicated when the criteria for failure is met (as defined by the ELN). In these instances (i.e., failure), randomized trials have demonstrated that a change of therapy from imatinib to a second-generation TKI improves the outcome compared to an increase in the dose of imatinib.119 A mutation analysis is always indicated when the criteria of failure are met, and a bone marrow aspiration is also needed to determine the cytogenetics and stage of the patient before change. If a mutation is identified, this can guide which TKI is expected to induce a response (e.g., if F317L or V299L identified, consider nilotinib or ponatinib; if F359V, consider bosutinib, dasatinib, or ponatinib; for T315I, ponatinib or omacetaxine may be considered).61, 62 If there is no mutation, or there is a mutation for which there is no available information or no meaningful difference between the various TKI, then other considerations may help in the decision of what drug to use, such as comorbidities that might expose the patient more to adverse events associated with one drug or another, the adverse event profile that might be more acceptable for a given patient, and the dose schedule that a patient might find more convenient or acceptable to optimize adherence. Patients with resistance to at least two TKI can be considered for SCT. SCT may also be considered in other settings where long-term therapy might not be optimal or feasible (e.g., for cost considerations).
Patients with accelerated phase features at the time of diagnosis can be treated with a TKI, particularly dasatinib or nilotinib, as their prognosis in this setting is nearly identical to that of patients in chronic phase.120 SCT for these patients is not required unless not responding adequately. Patients who progress to accelerated phase while on treatment with TKI for chronic phase should receive a different TKI and considered for SCT.121 Patients with blast phase CML should receive TKI, usually in combination with chemotherapy,21, 122, 123 and SCT should be strongly considered. Although results for SCT in this setting are best with the least residual disease, once the patient achieves a hematologic complete remission transplant can proceed as further chemotherapy may cause comorbidities that may make SCT impossible or of greater risk, and response may not improve further.
Suvivorship and follow-up
Patients with CML should continue therapy and have close follow-up indefinitely. This includes not only monitoring of the status of the disease (with PCR in peripheral blood every 6 months) but also attention to co-morbidities and possible side effects. There is increased awareness of adverse events that will need continued assessment. These include arteriothrombotic events such as ischemic heart disease (including angina and myocardial infarction), ischemic heart disease (including transient ischemic attacks and strokes), and peripheral arterial occlusive disease, particularly with ponatinib and nilotinib. Attention should be paid to monitoring and managing risk factors such as hypertension (frequently caused or aggravated by ponatinib), diabetes (frequently caused or aggravated by nilotinib), hyperlipidemia (also associated with TKI therapy), and others. Recent analyses also suggest there might be an impairment of renal function with TKI, particularly with imatinib. Patients on dasatinib with respiratory symptoms should be evaluated for pleural effusion or pulmonary hypertension. Although most adverse events occur early during the course of the disease, for some (such as pleural effusion or arteriothrombotic events) the incidence is constant and a first event may occur after several years of treatment.
Research is ongoing to explore treatment discontinuation in suitable patients.124, 125 Among patients with sustained undetectable transcripts (with PCR sensitivity of at least 4.5 logs) for at least 2 years, treatment discontinuation yielded a sustained remission in approximately 40% of patients after 3–4 years of follow-up. This approach at the moment should be considered only in clinical trials and patients should continue with close monitoring, at least monthly for the first 6 months, then every 2–3 months for 2 years, and then every 6 months indefinitely.
Conclusion
The outcome of patients with CML has improved significantly since the introduction of TKI. It is expected that a patient with CML diagnosed today may have a life expectancy similar to that of the general population. For a patient to have such favorable outcome, adequate management is important including adequate dose optimization, proper management of adverse events, continued periodic monitoring of disease response, timely intervention when indicated, and continued support throughout the life of the patient. With this approach, transformation to advanced stages is uncommon and few patients will die of CML.
Summary
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia chromosome. This results in the creation of the BCR-ABL fusion gene, which is in turn translated into a tyrosine kinase with constitutive kinase activity. The disease evolves in three stages: chronic, accelerated, and blast phases. Most patients are diagnosed in the chronic phase and are asymptomatic at the time of diagnosis. Treatment with TKIs has changed the natural history of the disease with a life expectancy that is similar to that of the general population. Imatinib was the first TKI to be used and is still a standard and an effective therapy for frontline therapy. Most patients will achieve a complete cytogenetic response (CCyR) making transformation to accelerated and blast phases very rarely. Higher doses of imatinib have been suggested to provide higher rates of response including deeper molecular responses. Dasatinib and nilotinib are also the approved treatment options for frontline therapy and may improve the rate of complete cytogenetic and major molecular (as well as deeper molecular) responses, with fewer instances of transformation to the accelerated and blast phases, but have not resulted in improved event-free or overall survival compared to imatinib in randomized trials. For patients who experience resistance or intolerance to one TKIs, alternative inhibitors can be used. In this setting, bosutinib and ponatinib are additional options. Overall, approximately 40% of patients with resistance or intolerance to initial therapy might achieve a CCyR with a subsequent TKI. The most common mechanism of resistance is the emergence of mutations of the KD. Different mutations may have differing sensitivities to the various inhibitors, and the presence of such mutations, when present, may allow selection of the most appropriate therapy. Although generally well-tolerated TKIs may have caused adverse events, some of them are potentially serious, which require proper identification and management; this may include the change of therapy in some instances. SCT remains a useful and potentially curative treatment modality for patients who have not experienced adequate response to various lines of therapy. With adequate access to treatment and proper management, patients diagnosed with CML should be able to have a normal life expectancy.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


