Chronic Myeloid Leukemia
Michael W.N. Deininger
HISTORICAL PERSPECTIVE
Chronic myeloid leukemia (CML), although a rare disease, has disproportionately influenced hematology/oncology and modern medicine. Reports of what was probably CML date back to the middle of the 19th century. In 1845, John Hughes Bennett, an Edinburgh pathologist, described a “Case of Hypertrophy of the Spleen and Liver in which Death Took Place from Suppuration of the Blood.”1 It was only a few weeks later when Rudolf Virchow of Berlin reported on a very similar case in an article titled, “Weisses Blut” (white blood).2 It is likely that both patients involved indeed suffered from CML and presented with its most typical clinical features: leukocytosis and splenomegaly. Whereas Bennett believed that the new disease represented an infection, Virchow recognized the neoplastic nature of the process and coined the term “leukemia,” from the Greek words “λευκον αιµα,” which mean “white blood.” The ensuing academic dispute was eventually settled cordially: Virchow acknowledged Bennett’s priority of discovery and Bennett that leukemia is a neoplastic rather than infectious process. Ironically, both were actually preceded by Alfred Donné of Paris, Bennett’s previous mentor, whose description of leukemia in his 1844Cours de Microscopie had lacked sufficient detail and visibility to attract attention.3 The next leap forward came in 1872, when Ernst Neumann established the bone marrow as the origin of leukemia and of blood cells in general.4 The following 100 years saw progress in understanding leukemia, with perhaps the most notable advance being the recognition of myeloid versus lymphoid disease. In 1951, William Dameshek posited that CML belongs to a larger group of related disorders characterized by increased numbers of differentiated blood cells as the result of enhanced proliferation of the bone marrow, which he accordingly named myeloproliferative disorders.5
A fundamental experimental breakthrough by Philadelphia cytogeneticists Peter Nowell and David Hungerford followed in 1960. They observed an abnormally small G-group chromosome in metaphase spreads from several patients with CML.6 This first consistent karyotypic abnormality associated with cancer settled the debate as to whether DNA or proteins transmitted the neoplastic phenotype to the next generation of cells. The discovery team anticipated that this minute chromosome in CML might represent the first in a long list of cancer chromosomes, and thus referred to it as the Ph1 chromosome (now more commonly known as Ph). The presence of Ph in all CML cells was indicative of their clonal origin from a single parental cell. Formal proof for this notion was lacking until Fred Fialkow demonstrated X-chromosome inactivation in CML neutrophils by uniform glucose-6-phosphate dehydrogenase (G6PD) isoenzyme expression.7
In 1973, Janet Rowley recognized that Ph was not just a shortened chromosome 22, but was in fact the product of a reciprocal translocation between chromosomes 9 and 22.8 Over the following 25 years, the molecular anatomy and consequences of t(9;22) were revealed with increasing resolution. Work from several laboratories identified the genes juxtaposed by t(9;22) to form a fusion gene on Ph.9,10 The chromosome 9 partner was found to be ABL1 (formerly ABL), the human homolog of v-abl oncogene of the Abelson murine leukemia virus (A-MuLV).11 On the derivative chromosome 22, ABL1 sequences consistently translocated to the same genetic region, which became known as the “breakpoint cluster region” (BCR), a name that was subsequently used to describe the new gene fused upstream of ABL1. The next critical set of discoveries was that BCR-ABL1, similar to murine v-Abl, is a constitutively active tyrosine kinase and that kinase activity is required for cellular transformation.12 Lastly, retroviral expression of BCR-ABL1 in murine bone marrow cells was found to induce a CML-like disease in mice, another milestone in the history of cancer research.13
Therapy developed slowly. Arsenicals, in use for cancer treatment since ancient times, were the only CML therapy available in the 19th century, usually in the form of Fowler’s solution, which contained potassium arsenite. The German physician Heinrich Lissauer is credited with the first publication on the remarkable efficacy of Fowler’s solution in a patient with leukemia.14 In an 1882 Lancet paper, Conan Doyle, the author of the Sherlock Holmes detective stories, published on a patient with the clinical presentation of CML who achieved a partial response to arsenic.15 In the 1920s, splenic irradiation was used for symptomatic relief and remained the mainstay of therapy for the first half of the 20th century. In 1959, busulfan was introduced as the first drug that reliably controlled white blood cell counts in CML and, 10 years later, hydroxyurea premiered as the first intervention that significantly prolonged survival.16,17 Despite these advances, a cure remained elusive until the late 1970s, when the disappearance of the Ph+ clone in CML patients treated with allogeneic stem cell transplantation was reported.18 In the early 1980s, interferon-α (IFN) became the standard drug therapy. This agent induced complete cytogenetic responses (CCyRs) and long-term survival in 10% to 20% of patients.19
In 1992, Alexander Levitzki proposed inhibiting ABL1 with small molecules called tyrphostins as a potentially useful approach to treat leukemias driven by ABL1 oncogenes.20 At about the same time, Alois Matter, Jürg Zimmermann, and Nick Lydon at Ciba-Geigy had synthesized a compound called GCP57148B,21 now known as imatinib, that inhibited ABL1 and a restricted set of other tyrosine kinases at submicromolar concentrations. Clinical trials led by Brian Druker from Oregon Health & Science University, very much in the face of skepticism by the manufacturer, rapidly established imatinib’s unprecedented activity in patients with CML and revolutionized CML therapy.22,23,24 Dasatinib and nilotinib, tyrosine kinase inhibitors (TKIs) with increased activity against BCR-ABL1, were initially developed to overcome imatinib resistance, but have since gained approval for frontline therapy.25,26 However, once CML has progressed beyond the chronic phase, allogeneic stem cell transplant is still the recommendation for all eligible patients. There is compelling evidence that CML stem cells are not dependent on BCR-ABL1 kinase activity, implying that treatment must continue indefinitely in order to prevent recurrence of active leukemia. Current research efforts focus on improving the poor prognosis of advanced CML, controlling TKI resistance, and developing strategies to eradicate CML stem cells.
PATHOPHYSIOLOGY
It is thought that CML originates from a single hematopoietic cell that has acquired Ph. In contrast to an acute myeloid leukemia (AML)-associated fusion gene such as MOZ-TIF2, BCR-ABL1 does not confer self-renewal capacity, implying that the initial translocation event must occur in a multipotent hematopoietic stem cell already endowed with this critical property.27 BCR-ABL1 is detected in cells of all hematopoietic lineages.28 For unknown reasons, the BCR-ABL1-induced cellular expansion predominantly
targets the myeloid progenitor cell compartment, giving rise to the clinical phenotype, whereas many newly diagnosed patients have mostly Ph– stem cells.29,30 As a consequence, the hierarchical organization of hematopoiesis is maintained during the chronic phase, and a substantial number of Ph– stem cells are available to reconstitute the system when CML hematopoiesis is therapeutically suppressed. Both factors are important for the response to TKIs. The salient biologic features of CML cells are as follows: increased proliferation, resistance to apoptosis, perturbed interaction with bone marrow stromal cells, and genetic instability.31
targets the myeloid progenitor cell compartment, giving rise to the clinical phenotype, whereas many newly diagnosed patients have mostly Ph– stem cells.29,30 As a consequence, the hierarchical organization of hematopoiesis is maintained during the chronic phase, and a substantial number of Ph– stem cells are available to reconstitute the system when CML hematopoiesis is therapeutically suppressed. Both factors are important for the response to TKIs. The salient biologic features of CML cells are as follows: increased proliferation, resistance to apoptosis, perturbed interaction with bone marrow stromal cells, and genetic instability.31
BCR-ABL1 Translocation and Fusion Gene
Ph is the result of a reciprocal translocation between the long arms of chromosomes 9 and 22 [t(9;22)(q34;q11)].8 As a result of this translocation, genetic sequences from the ABL1 gene on 9q34 are fused downstream of the BCR gene on 22q11 (Fig. 81.1A,B). With rare exceptions, breaks in chromosome 22 localize to one of three BCRs and determine the portions of BCR retained in the BCR-ABL1 fusion mRNA and protein. In contrast, the chromosome 9 breaks can occur over a large genetic region, 5′ of ABL1 exon Ib, 3′ of ABL1 exon Ia, or most commonly between the two alternative first ABL1 exons. Rare exceptions aside, splicing consistently leads to fusion mRNAs that encompass ABL1 exons 2 to 11 (Fig. 81.2C).32 Breakpoints in the minor BCR (m-BCR) give rise to an e1a2 fusion mRNA and p190BCR-ABL, which is found in 2/3 of Ph+ acute lymphoblastic leukemia (ALL) cases. The very rare p190BCR-ABL positive CML is associated with monocytosis and exhibits a more aggressive clinical course.33,34 In an overwhelming majority of CML patients, the break occurs in the major BCR (M-BCR), generating e13a2 or e14a2 fusion mRNAs (formerly referred to as b2a2 and b3a2) and a p210BCR-ABL fusion protein. p230BCR-ABL, the largest of the fusion proteins, corresponds to a break in the micro-BCR (µ–BCR), an e19a2 fusion mRNA, and is associated with neutrophilic predominance and possibly less aggressive disease.35 In addition to the major types of BCR-ABL1 fusion mRNAs, there are a number of rare variants whose main clinical significance is that they can give rise to misleading reverse transcription polymerase chain reaction (RT-PCR) results.36, 37 and 38 Approximately two-thirds of CML cases also express the reciprocal ABL1-BCR mRNA, but there is no evidence that this influences disease biology or prognosis.39,40
BCR and ABL1 Proteins and Their Contribution to Cellular Transformation
BCR is an ubiquitously expressed 160 kD cytoplasmic protein with several functional domains (Fig. 81.2A). The N-terminus contains a coiled-coil motif that allows for dimerization, which is critical for activation of ABL1 kinase in the BCR-ABL1 fusion protein.41 Further 3′ there are: a serine/threonine kinase motif whose only recognized substrate is Bap-1, a member of the 14-3-3 family of proteins,42 and dbl-like and pleckstrin-homology (PH) domains that stimulate the exchange of GTP for GDP on Rho guanidine exchange factors.43 The C-terminus contains a putative site for calcium-dependent lipid binding (CaLB) and a GTPase activating function (RAC-GAP). The latter regulates activity of RAC, a small GTPase of the RAS superfamily that regulates actin polymerization and an NADPH oxidase in phagocytic cells.44,45 Despite these diverse functions, Bcr null mice are viable and fertile and the only recognizable defect is an increased oxidative burst in neutrophils, suggesting redundancy of signaling pathways.46 BCR can be phosphorylated on several tyrosine residues, most importantly tyrosine 177, which binds GRB2, an adapter molecule involved in activation of the RAS pathway in CML cells.47,48,49 The Rho-GEF and RAC-GAP functions of BCR that are retained in p210BCR-ABL but missing from p190BCR-ABL are thought to attenuate the disease and cause a phenotypic shift from lymphoid to myeloid
leukemia.43,50,51 It is possible that this reflects the fact that hematopoietic stem cells are susceptible to transformation by p210BCR-ABL, and p190BCR-ABL targets pre-B-cells.52 It has been suggested that BCR may negatively regulate BCR-ABL1-induced leukemogenesis.53 However, the fact that incidence and biology of p190BCR-ABL-induced leukemia are identical in Bcr-/- mice compared to wildtype mice indicates that BCR is not essential for Ph+ leukemia.54
leukemia.43,50,51 It is possible that this reflects the fact that hematopoietic stem cells are susceptible to transformation by p210BCR-ABL, and p190BCR-ABL targets pre-B-cells.52 It has been suggested that BCR may negatively regulate BCR-ABL1-induced leukemogenesis.53 However, the fact that incidence and biology of p190BCR-ABL-induced leukemia are identical in Bcr-/- mice compared to wildtype mice indicates that BCR is not essential for Ph+ leukemia.54
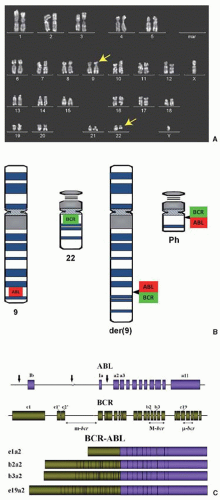 FIGURE 81.1. A: Metaphase karyogram of a newly diagnosed man with CML. The yellow arrows indicate the derivative chromosome 9 and the Philadelphia chromosome (Ph). B: Schematic of chromosomes 9, 22 and the derivatives resulting from t(9;22)(q34;q11). C: Molecular anatomy of the BCR-ABL1 fusion gene. The upper panel shows the ABL1 gene, which contains 11 exons, including two alternative first exons. Arrows indicate the breakpoints at the genomic level. The middle panel depicts the BCR gene with the three breakpoint cluster regions (BCRs). Breaks in the minor BCR (m-BCR) give rise to p190BCR-ABL, which is typical of Ph+ ALL, whereas breaks in the major BCR (M-BCR) generate p210BCR-ABL, typical of CML and also found in 1/3 of Ph+ ALL patients. Lastly, breaks in the micro-BCR (µ–BCR) lead to p230BCR-ABL, which is associated with neutrophilia. The lower panel shows the structure of the corresponding mRNAs. Note that BCR exons b2 and b3 are now commonly referred to as exons e13 and e14, respectively. |
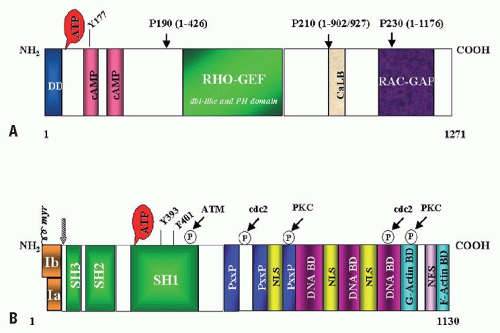 FIGURE 81.2. A: Structure of the BCR protein. Note the dimerization domain (DD) and the two cAMP kinase homologous domains at the N-terminus. Y177 is the autophosphorylation site crucial for binding to GRB2. The center of the molecule contains a region homologous to Rho guanidine nucleotide exchange factors (Rho-GEF) as well as dbl-like and pleckstrin-homology (PH) domains. Toward the C-terminus, a putative site for calcium-dependent lipid binding (CaLB) and a domain with activating function for RAC-GTPase (RAC-GAP) are found. Arrowheads indicate the position of the breakpoints in the BCR-ABL1 fusion proteins. B: Structure of the ABL1 protein. The type Ia isoform is slightly shorter than type Ib, which contains a myristoylation (myr) site for attachment to the plasma membrane. Note the three SRC-homology (SH) domains situated toward the NH2 terminus. Y393 is the major site of autophosphorylation within the kinase domain, and phenylalanine 401 (F401) is highly conserved in PTKs containing SH3 domains. The middle of the protein is dominated by proline-rich regions (PxxP) capable of binding to SH3 domains and also harbors one of three nuclear localization signals (NLS). The C-terminus contains DNA binding as well as G- and F-actin binding domains. Phosphorylation sites by ATM, cdc2, and PKC are shown. The arrowhead indicates the position of the breakpoint in the BCR-ABL1 fusion protein. |
ABL1, the human homolog of the v-abl oncogene of the A-MuLV,55 encodes a nonreceptor tyrosine kinase.56 The 145-kDa ABL1 protein is ubiquitously expressed and exhibits two isoforms arising from alternative splicing of the first exon.57 The N-terminal region contains three SRC-homology domains (SH1 to SH3). SH1 carries the tyrosine kinase function, whereas the SH2 and SH3 domains engage in protein-protein interactions (Fig. 81.2B).58 In addition, proline-rich sequences in the center of ABL1 interact with SH3 domains of other proteins such as Crk.59,60 The C-terminus contains nuclear localization, DNA binding,61 and actin-binding motifs.62 Diverse functions have been attributed to ABL1, which include negative regulation of cell cycle and proliferation,63,64 response to genotoxic stress,65,66,67,68,69 and integrin signaling.70 The emerging picture is complex and perhaps ABL1 could best be characterized as a cellular module that integrates signals from various extra- and intracellular sources to influence decisions in regard to cell cycle and apoptosis. It must be stressed, however, that many results are based primarily on in vitro studies in fibroblasts, not hematopoietic cells. Unfortunately, the generation of Abl1 null mice failed to shed light on the physiologic function of ABL1. These mice have a severe phenotype, including high perinatal mortality, runting, and skeletal and immune system defects.71,72 Mice null for Abl1 and the Abl-related gene (formerly Arg, now Abl2) are embryonically lethal due to absence of neurulation.73 The latter observation raised considerable concerns about the potential side effects of ABL inhibitors that were fortunately not confirmed clinically.
Constitutive Kinase Activation in the BCR-ABL1 Fusion Protein
In contrast to ABL1, a tightly regulated nuclear kinase, BCR-ABL1 is constitutively active and localized in the cytoplasm. The mechanism by which the replacement of the ABL1 N-terminus with BCR sequences leads to kinase activation has been probed by mutagenesis and X-ray crystallography.74 A critical feature is the coiled-coil domain of BCR that promotes dimerization, which allows for an initial transphosphorylation event, followed by autophosphorylation of additional tyrosine residues to activate the kinase fully.75 Moreover, the N-terminal “cap” region of ABL1, when myristoylated, binds a hydrophobic pocket at the base of the kinase domain, thereby forming a latch that maintains ABL1 in an inactive state.76, 77 and 78 Replacement of the cap by BCR sequences abolishes this negative regulation. Unexpectedly, recent studies have shown that certain residues within the SH2 domain participate in the regulation of kinase activity, an indication of the complexity of kinase regulation.79 Lastly, trans-acting binding partners such as the ABL1 interacting proteins have been implicated as physiologic inhibitors whose down-regulation upon disease progression may contribute to BCR-ABL1 activation.80,81
Signaling Pathways in BCR-ABL1 Transformed Cells
Two decades of intense research have identified multiple BCR-ABL1 substrates, binding partners, and downstream signaling molecules involved in the process of cellular transformation (Fig. 81.3). Much effort has been directed at linking these pathways to the specific pathologic features that characterize CML cells, such as increased proliferation or genetic instability leading to disease progression. A comprehensive review of the multiple pathways implicated in BCR-ABL1 transformation is beyond the scope of this chapter. Studies of mice with homozygous deletions of signaling proteins have identified very few pathway components with a truly essential function, speaking to the extensive redundancy of the transformation network.
Phosphatidylinositol-3 Kinase
In BCR-ABL1-expressing cells, phosphatidylinositol-3 kinase (PI3K) is activated by at least two different mechanisms. The first requires autophosphorylation of BCR-ABL1 tyrosine 177 (Y177), which generates a docking site for the GRB2 adapter protein, which in turn recruits GAB2, another adapter, into a complex that activates PI3K.82,83 Consistent with a critical role for the Y177/GRB2/GAB2 axis, mutation of tyrosine 177 to phenylalanine or lack of GAB2 abrogates myeloid leukemia in murine models.83 Alternatively, PI3K can be activated by complex formation between the p85 regulatory subunit of PI3K, CBL, and CRKL, which bind to the SH2 and proline-rich domains of BCR-ABL1.84 The main downstream outlet of PI3K is the serine-threonine kinase AKT, a major conduit for oncogenic signals in different cancer types. In CML cells, AKT enhances survival by suppressing activity of the forkhead O transcription factors (FOXO), as well as phosphorylation and inactivation of proapoptotic proteins such as BAD.85,86,87 Additionally, PI3K promotes proteasomal degradation of p27, a cyclin-dependent kinase inhibitor, through up-regulation of SKP2, the F-Box recognition protein of SCFSKP2 E3 ubiquitin ligase. Accordingly, absence of SKP2 from leukemia cells prolongs survival in a murine CML model.88 Furthermore, AKT activates mTOR, which phosphorylates ribosomal proteins p70S6 kinase (S6K) and 4E-BP1. S6K activation promotes cell growth and proliferation by yet undefined mechanisms. However, phosphorylation by mTOR has been shown to inactivate 4EBP-1, thereby activating the translation initiation factor eIF4E to enhance protein synthesis.89,90
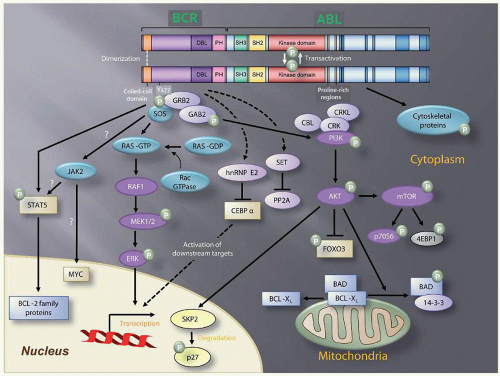 FIGURE 81.3. Simplified representation of molecular signaling pathways activated in chronic myeloid leukemia (CML) cells. Autophosphorylation generates docking sites on BCR-ABL1 that facilitate interaction with intermediary proteins such as GRB2. CRKL and CBL are also direct substrates of BCR-ABL1 that are part of a multimeric complex. Additionally, BCR-ABL-dependent signaling complexes activate multiple pathways, which modulate the function of critical proteins (blue) that inhibit apoptosis, enhance proliferation, perturb adhesion and migration, and impart genomic instability. For details see text. |
RAS/Mitogen-activated Protein Kinase Pathways
Phosphorylation of tyrosine 177 with recruitment of GRB2 and SOS promotes exchange of GTP for GDP on RAS.82,91 In its GTP-bound form, RAS activates the serine-threonine kinase RAF-1,92,93 which subsequently activates mitogen-activated protein kinase (MAPK). Other small GTPases such as RAC1/2 have also been implicated in BCR-ABL1 signaling.94 Lack of RAC1/2 has been shown to delay BCR-ABL1 leukemia in a murine model.95
Janus Kinase/Signal Transducer and Activator of Transcription Pathway
The transcription factor signal transducer and activator of transcription 5 (STAT5) is constitutively tyrosine-phosphorylated in CML cells. Initial experiments in mice failed to demonstrate a critical role for STAT5.96 However, it was subsequently established that the mice used in this study were not null for Stat5, but expressed an N-terminally deleted protein with partially retained function. Although STAT5 is not required for the initiation of CML, there is convincing evidence that it plays a rate-limiting role in leukemogenesis.97,98 In fact, absence of only STAT5A or overexpression of a dominant-negative STAT5 mutant in the bone marrow attenuates CML.99 The principal consequence of STAT5 activation is inhibition of apoptosis by enhancing transcription of antiapoptotic proteins such as MCL-1 and BCL-XL.100 BCR-ABL1 may activate STAT5 through direct phosphorylation or indirectly by promoting phosphorylation by HCK or Janus kinase 2 (JAK2).101,102 There is controversy about the role of JAK2 in BCR-ABL1-induced leukemia. Previous studies had suggested that JAK2 plays an important if not essential role,103 but a recent report using conditional knockout technology showed that murine CML does not require JAK2.104
Cytoskeletal Proteins
BCR-ABL1 phosphorylates several proteins involved in adhesion and migration, including focal adhesion kinase (FAK), CRKL, paxillin, p130CAS, and HEF1.105, 106, 107, 108, 109 and 110 Although details remain to be elucidated, it is thought that this may explain why integrin-mediated adhesion of CML progenitors to bone marrow stroma and extracellular matrix is defective. Alternatively, integrin functions may be compromised through activation of RAS or BCR-ABL1 binding to F-actin.82,111 Given that adhesion to integrins inhibits proliferation of hematopoietic progenitors, the defect in integrin function may contribute to the premature circulation as well as the abnormal proliferation of Ph+ progenitor cells.112
DNA Damage Surveillance and Repair Pathways
The fact that untreated CML-CP invariably progresses to blast phase CML (CML-BP) is testimony to the profound dysregulation of DNA repair in CML. A central finding is the two- to sixfold increase in reactive oxygen species (ROS) in CML CD34+ cells compared with normal controls, which is particularly pronounced in CML-BP cells.113,114,115 ROS generate oxidized bases and double-strand breaks (DSB). It was estimated that CML cells may contain up to eight times more oxidized bases and DSBs than normal cells.116 Although PI3K activation is primarily responsible for ROS production,117 an additional mechanism operational in lymphoid blast phase cells is activation of activation-induced cytidine deaminase (AID), which promotes point mutations.118 The consequences of increased DNA damage are greatly aggravated by the impairment of DNA damage surveillance and repair. Multiple mechanisms have been implicated, and many but not all of these mechanisms are BCR-ABL1-dependent. For example, BCR-ABL1 has been shown to impair the intra-S-phase cell cycle checkpoint through suppression of checkpoint kinase 1 (CHK1), either by inhibition of the nuclear protein kinase ATR119 or down-regulation of BRCA1, a substrate of ataxia telangiectasia mutated (ATM).120 Nonhomologous end-joining (NHEJ) and homologous recombination (HR), critical DSB repair pathways, are also deregulated in CML. For example, the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) is down-regulated in CD34+ CML progenitors, leading to error-prone NHEJ.121 Furthermore, BCR-ABL1 up-regulates RAD51.122 As a result, DSB repair upon challenge with cytotoxic agents and induced ROS is shifted toward a rapid but low fidelity pathway, thus promoting chronic oxidative DNA damage. Lastly, telomere length is correlated with time to disease progression.123 Whether TKIs themselves may cause DNA damage is controversial. Imatinib was reported to induce centrosome abnormalities in vitro.124 Given that the overall risk of cancer is not increased in patients on long-term imatinib,125 the significance of these findings is not clear. Furthermore, the fact that clonal cytogenetic abnormalities in Ph-negative cells (CCA/Ph–) appear in some CML patients with a cytogenetic response to TKI therapy is consistent in principle with genomic instability prior to acquisition of Ph.126 Indeed, some patients with CCA/Ph– have progressed to MDS or AML, especially those with monosomy 7.127 However, the overall prognosis of patients with CCA/Ph– is identical to that of other CML patients with a comparable cytogenetic response, which argues against genomic instability predating Ph.128 The mechanistic underpinning of genomic instability in CML is of considerable complexity.115,116
A wealth of data on BCR-ABL1 signaling has been amassed, yet a complete picture is still elusive. As conventional approaches have typically focused on single pathways, efforts are now underway to establish a more comprehensive picture of BCR-ABL1 signaling complexes by quantitative genomics.129 These data suggest that cellular processes in CML rely on integrated networks rather than single pathways, which co-operate to realize the leukemogenic potential of BCR-ABL1 fully.
Transformation to Accelerated and Blastic Phase
It could be argued that the key objective in CML therapy is the prevention of transformation to blastic phase (BP), an acute leukemia with either myeloid (M-BP) or pre-B-cell phenotype (L-BP). A host of genetic alterations has been associated with transformation to accelerated phase (AP)/BP. CCA/Ph+ is present in 70% to 80% of cases and includes a number of nonrandom abnormalities, the most common of which are +8 (34% of cases with CCA/Ph+), +Ph (30%), i(17q) (20%), +19 (13%), -Y (8% of males), +21 (7%), +17 (5%), and monosomy 7 (5%). These frequent chromosomal changes are sometimes referred to as major route abnormalities,130 although this is not a universally accepted definition. Many other cytogenetic abnormalities have been described in smaller subsets of patients and multiple different mutations have been detected at the molecular level. For example, in M-BP, inactivating mutations of p53 and RUNX1 occur in 20% to 30% and 38% of cases, respectively, whereas L-BP is characterized by inactivating mutations in CDKN2A/B (50%) or IKZF1 (55%) (reviewed in Ref. 116).
BCR-ABL1 expression and activity increase with progression to BP. In some patients, rising BCR-ABL1 mRNA levels are associated with duplication of Ph, but in the majority of cases this seems to result from enhanced transcription due to as yet unknown mechanisms.131 Increased BCR-ABL1 promotes expression of SET, which in turn inhibits PP2A, a tumor suppressor phosphatase that dephosphorylates BCR-ABL1 and other important substrates by activating SHP-1.132 Thus, reduced PP2A activity will increase BCR-ABL1 activity and further enhance the signal in a positive feedback loop. Consistent with the importance of this pathway, reduced SHP-1activity has been associated with resistance to TKIs.133 In addition to SET-mediated inhibition of PP2A, cancerous inhibitor of PP2A (CIP2A) was also shown to inhibit this phosphatase, and high CIP2A activity predicts a high risk of transformation to BP.134
The central feature of blastic transformation is the loss of terminal differentiation capacity. Based on the paradigm that AML requires at least two types of mutations, one driving proliferation and one blocking differentiation, it follows that the BP in CML should be associated with inactivating mutations in myeloid differentiation genes.135 Consistent with this, some patients acquire AML-type translocations such as CBFβ-MYH11 at the time of transformation, but these are rare.136 In the majority of patients, myeloid differentiation is impaired due to suppression of CAAT enhancer binding protein α (C/EBPα), which occurs as a result of increased BCR-ABL1 expression and activity.137 Mechanistically, this is due to enhanced expression of the RNA-binding protein heterogeneous nuclear ribonucleoprotein E2 (hnRNP-E2), which inhibits C/EBPα translation.137 Down-regulation of miR-328 is also required for this to occur, as miR-328 would otherwise bind hnRNP-E2, thereby blocking its activity.138 Repression of other myeloid transcription factors such as C/EBPβ and overexpression of EVI-1 have also been implicated in blastic transformation.139,140,141 In addition to the loss of differentiation capacity, the organization of leukemic hematopoiesis seems to change upon transformation, as granulocyte-macrophage progenitor cells acquire self-renewal capacity, possibly by activation of nuclear β-catenin, undermining the hierarchical structure of CML-CP hematopoiesis.142 Various mechanisms have been implicated in β-catenin activation, including stabilization by BCR-ABL1 tyrosine phosphorylation and inactivation of glycogen synthase kinase 3β by missplicing.143,144 Unsurprisingly, CML-BP responses to therapy are usually short-lived.
CLINICAL FEATURES
Epidemiology
With an annual incidence of 1.3 to 1.6/105 population, CML accounts for 15% to 20% of cases of leukemia in adults. Males are more commonly affected, but there is no geographic, ethnic, or familial predisposition. The only well-established risk factor is exposure to ionizing radiation, evident by the increased CML incidence in survivors of the atomic bomb explosions in Japan; patients who were treated with radiotherapy or exposed to thorotrast, an α-emitting contrast medium used in the 1930s,145, 146 and 147 have an increased CML risk as well. From 2004 to 2009, the median age at diagnosis in the United States was 64 years.148 The prevalence of CML is increasing due to reduced mortality.149 For the United States, estimates
are 70,000 in 2010, 144,000 in 2030, and 181,000 in 2050, when prevalence will approach a plateau.150
are 70,000 in 2010, 144,000 in 2030, and 181,000 in 2050, when prevalence will approach a plateau.150
Clinical Presentation
Signs and Symptoms
CML may present with fatigue, weight loss, fever, night sweats, bone pain, abdominal pain, and fullness.151 Rare patients with excessively high white blood cell counts have symptoms related to leukostasis, such as priapism and neurologic deficits. Physical exam may reveal splenomegaly and sometimes hepatomegaly, although involvement of other extramedullary sites is uncommon. None of these signs or symptoms is typical or even pathognomonic for CML, and the clinical presentation has changed considerably due to earlier diagnosis. In the United States and other developed countries, the majority of patients are diagnosed incidentally, when an abnormal complete blood count obtained for an unrelated reason leads to a diagnostic work-up.
Disease Phases
CML evolves in stages termed chronic phase (CML-CP), accelerated phase (CML-AP), and blastic phase (CML-BP). In the Western world, most patients present in CML-CP, whereas more advanced disease at diagnosis is common in developing countries. CML-CP is characterized by left-shifted granulocytosis with maintained terminal differentiation, frequently accompanied by basophilia and sometimes eosinophilia. Thrombocytosis and mild anemia are also common. Dysplasia is not a feature of CML and raises the question of alternative diagnoses, such as atypical CML.152 Bone marrow aspirate and histology are hypercellular for age, but demonstrate complete cellular maturation (Fig. 81.4A). A fairly typical finding is micromegakaryocytes, which are small monolobated megakaryocytes. Mild reticulin fibrosis may be present, but severe reticulin fibrosis is uncommon and may be associated with poorer outcomes.153 Without effective therapy, CML-CP inexorably progresses to CML-BP, which can exhibit a pre-B-lymphoid (25%), myeloid (70%), or indeterminate (5%) phenotype (Fig. 81.4B).154 CML-BP can develop rapidly or over a period of time through the intermediary stage of CML-AP. A variety of clinical and laboratory features has been used to define CML-AP, making comparison of results between different studies difficult. Today, the criteria used in the clinical trials leading to approval of imatinib are widely accepted (Table 81.1).155 The exception is clonal cytogenetic evolution, i.e., the presence of additional cytogenetic abnormalities in the Ph+ cell clone (CCA/Ph+). Although there is agreement that CCA/Ph+ on treatment is diagnostic of CML-AP and therapy failure,156 this is not universally accepted for newly diagnosed patients. Recent studies showed that these patients have inferior outcomes even in the absence of any other features of CML-AP, raising the question of whether the criteria should be changed.157 A blast count of ≥30% in the blood or bone marrow or extramedullary blastic leukemic infiltrates (chloroma) in tissues other than liver or spleen define CML-BP. Gene expression profiling has revealed that CML-AP and CML-BP are closely related to each other, suggesting that CML is a two-phase rather than a threephase disease.158 In the future, molecular markers and response to treatment may replace conventional diagnostic criteria used to define the phases of CML.159
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


