INTRODUCTION
SUMMARY
The chronic myelogenous leukemias (CMLs) include BCR rearrangement-positive CML, chronic myelomonocytic leukemia, juvenile myelomonocytic leukemia, chronic neutrophilic leukemia, chronic eosinophilic leukemia, and chronic basophilic leukemia. The term chronic, in contrast to acute, once had prognostic implications. However, although the terms remain useful for nosology, they no longer reflect an invariable difference in prognosis. For example, acute myelogenous leukemia in children and young adults has higher remission and cure rates than juvenile or chronic myelomonocytic leukemia in children or adults, respectively. BCR rearrangement-positive CML presents with anemia, exaggerated granulocytosis, a large proportion of myelocytes and mature neutrophils, absolute basophilia, normal or elevated platelet counts, and, frequently, splenomegaly. The marrow is intensely hypercellular, and marrow cells contain the Philadelphia (Ph) chromosome in approximately 90 percent of cases by cytogenetic analysis. A rearrangement of the BCR gene on chromosome 22 is present by molecular diagnostic analysis in approximately 96 percent of cases that have a classic morphologic appearance. The BCR-rearranged form of the disease usually responds to a tyrosine kinase inhibitor, and median survival has been extended significantly. Allogeneic hematopoietic stem cell transplantation can cure the disease, especially if the transplantation is applied early in the chronic phase, although this approach is now uncommon as a result of the effect of tyrosine kinase inhibitor therapy. The effect of stem cell transplantation is related in part to a robust graft-versus-leukemia effect, engendered by donor T lymphocytes. The natural history of the chronic phase is to evolve into an accelerated phase that often terminates in acute leukemia (blast crisis), but the frequency of this progression has been markedly decreased by the advent of tyrosine kinase inhibitors. Blast crisis results in a myelogenous leukemic phenotype in 75 percent of cases and a lymphoblastic leukemic phenotype in approximately 25 percent of cases. Ph chromosome–positive acute myeloblastic leukemia (AML) may appear de novo in approximately 1 percent of cases of AML, and Ph chromosome–positive acute lymphocytic leukemia (ALL) may occur de novo in approximately 20 percent of cases of adult ALL and approximately 5 percent of childhood ALL cases. In Ph chromosome–positive ALL, the translocation between chromosomes 9 and 22 results in the fusion gene encoding a mutant tyrosine kinase oncoprotein that may be identical in size to that in classic CML (210 kDa) in approximately one-third of cases. A smaller mutant tyrosine kinase (190 kDa) is encoded in approximately two-thirds of cases. In children, the cells in approximately 90 percent of cases contain a 190-kDa mutant tyrosine kinase. These acute leukemias may reflect (1) the presentation of CML in acute blastic transformation without a preceding chronic phase or (2) de novo cases resulting from a BCR-ABL1 mutation occurring in a different early hematopoietic cell from the event in CML or with as yet unidentified modifying gene alterations. Chronic myelomonocytic leukemia has variable presenting features. Anemia may be accompanied by mildly or moderately elevated leukocyte counts; an elevated total monocyte count; a low, normal, or elevated platelet count; and sometimes splenomegaly. Although cytogenetic abnormalities may be present, there is no specific genetic marker of the disease. In a very small proportion of cases, a translocation involving the platelet-derived growth factor receptor (PDGFR)-β gene is associated with eosinophilia and is responsive to a tyrosine kinase inhibitor. Juvenile myelomonocytic leukemia occurs in infancy or very early childhood. Anemia, thrombocytopenia, and leukocytosis with monocytosis are usual. The disease is refractory to treatment and, even with current maximal therapy and stem cell rescue, cures are uncommon. Chronic neutrophilic leukemia presents with mild anemia and exaggerated neutrophilia, with very few immature cells in the blood. Splenomegaly is common. The disease usually occurs after age 60 years. Chronic and juvenile myelomonocytic leukemia and chronic neutrophilic leukemia have a propensity to evolve into acute myelogenous leukemia. Prior to that evolution, morbidity and mortality are related to infection, hemorrhage, and complicating medical conditions. Chronic eosinophilic leukemia represents the major subset of the hypereosinophilic syndrome. It is a clonal disorder with a striking absolute eosinophilia, often neurologic and cardiac manifestations secondary to toxic effects of eosinophil granules, and sometimes a translocation involving the PDGFR-α gene that encodes a mutant tyrosine kinase, imparting sensitivity to a tyrosine kinase inhibitor.
Acronyms and Abbreviations
ALL, acute lymphocytic leukemia; BCR, breakpoint cluster region; CCyR, complete cytogenetic response; CFU-GM, colony-forming unit–granulocyte-monocyte; CHR, complete hematologic response; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; CMML, chronic myelomonocytic leukemia; CMR, complete molecular response; DLI, donor lymphocyte infusion; FISH, fluorescence in situ hybridization; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-monocyte colony-stimulating factor; GRB2, growth factor receptor–bound protein-2; GTP, guanosine triphosphate; GTPase, guanosine triphosphatase; GVHD, graft-versus-host disease; HLA, human leukocyte antigen; HPRT, hypoxanthine phosphoribosyltransferase; hsp, heat shock protein; HUMARA, human androgen receptor assay; IFN, interferon; IL, interleukin; IRIS, International Randomized Study of Interferon; JAK, Janus-associated kinase; LTC-IC, long-term culture–initiating cell; MCP, monocyte chemotactic protein; MCyR, major cytogenetic response; MDS, myelodysplastic syndrome; MIP, macrophage inflammatory protein; MMR, major molecular response; NF-κB, nuclear factor-κB; NF1, neurofibromatosis tumor-suppressor gene; NK, natural killer; NOD, nonobese diabetic; OCT-1, organic cation transporter 1; PCR, polymerase chain reaction; PCyR, partial cytogenetic response; PDGFR, platelet-derived growth factor receptor; Ph, Philadelphia chromosome; PI3K, phosphatidylinositol 3′-kinase; Rb, retinoblastoma; RT-PCR, reverse transcriptase polymerase chain reaction; SCID, severe combined immunodeficiency; STAT, signal transducer and activator of transcription; TBI, total-body irradiation; TdT, terminal deoxynucleotidyl transferase; TGF, transforming growth factor; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; WT, Wilms tumor.
DEFINITION AND HISTORY
Chronic myelogenous leukemia (CML) is a multipotential hematopoietic stem cell disease characterized by anemia, extreme blood granulocytosis and granulocytic immaturity, basophilia, often thrombocytosis, and splenomegaly. The hematopoietic cells contain a reciprocal translocation between chromosomes 9 and 22 in more than 95 percent of patients with classic morphologic findings, which leads to an overtly foreshortened long arm of one of the pair of chromosome 22 (i.e., 22,22q−), referred to as the Philadelphia (Ph) chromosome. A rearrangement of the breakpoint cluster region gene (BCR) on the long arm of chromosome 22 defines this form of CML and is present even in the 10 percent of patients without an overt 22q abnormality by Giemsa chromosome banding. The natural history of the disease is to undergo clonal evolution into an accelerated phase and/or a rapidly progressive blast phase, an acute leukemia, highly refractory to therapy, which had been a frequent event prior to the introduction of tyrosine kinase inhibitors (TKIs) in 2001.
In 1845, Bennett1 in Scotland and Virchow2 in Germany described patients with splenic enlargement, severe anemia, and enormous concentrations of leukocytes in their blood at autopsy. Bennett initially favored an extreme pyemia as the explanation, but Virchow argued against suppuration as a cause. Additional cases were reported by Craige3 and others, and in 1847 Virchow4 introduced the designation weisses Blut and leukämie (leukemia). In 1878, Neumann5 proposed that the marrow not only was the site of normal blood cell production, but also was the site from which leukemia originated and used the term myelogene (myelogenous) leukemia. Subsequent observations amplified the clinical and laboratory features of the disease, but few fundamental insights were gained until the discovery by Nowell and Hungerford,6 who reported in 1960 that two patients with the disease had an apparent loss of the long arm of chromosome 21 or 22, an abnormality that was quickly confirmed7,8,9 and designated the Ph chromosome.7 Advanced cytogenetic techniques confirmed that it was chromosome 22. This observation led to a new approach to diagnosis, a marker to study the pathogenesis of the disease, and a focus for future studies of the molecular pathology of the disease. The availability of banding techniques to define the fine structure of chromosomes10,11 led to the discovery by Rowley12 that the apparent lost chromosomal material on chromosome 22 was part of a reciprocal translocation between chromosomes 9 and 22. The discovery that the cellular oncogene ABL1 on chromosome 9 and a segment of chromosome 22, the BCR, fuse as a result of the translocation provided a basis for the study of the molecular cause of the disease.13,14 The appreciation that the fusion gene encoded a constitutively active tyrosine kinase (BCR-ABL1) that was capable of inducing the disease in mice established the fusion gene product as the proximate cause of the malignant transformation. The search for, identification of, and clinical development of a small molecule inhibitor of the mutant tyrosine kinase provided a specific agent, imatinib mesylate, with which to inhibit the molecule that incites the disease.15 Several more potent congeners have also been synthesized (see “Etiology and Pathogenesis” below). Thomas and colleagues established that allogeneic hematopoietic stem cell transplantation could cure the disease.16 An engaging monograph on the discoveries and the scientists involved from the identification of the Ph chromosome to the development of imatinib has been published.17
EPIDEMIOLOGY
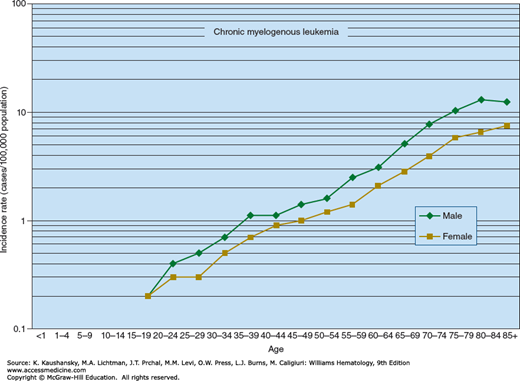
CML accounts for approximately 15 percent of all cases of leukemia, or approximately 6500 new cases in the United States in 2015. The age-adjusted incidence rate in the United States is approximately 2.3 per 100,000 persons for men and approximately 1.2 per 100,000 persons for women. The incidence around the world varies by a factor of approximately twofold. The lowest incidence is in Sweden and China (approximately 0.7 per 100,000 persons), and the highest incidence is in Switzerland and the United States (approximately 1.5 per 100,000 persons).18 The age-specific incidence rate for CML in the United States increases logarithmically with age, from approximately 0.2 per 100,000 per year in persons younger than 20 years to a rate of approximately 10.0 per 100,000 in octogenarians (Fig. 89–1). Although CML occurs in children and adolescents, less than 10 percent of all cases occur in persons between 1 and 20 years old. CML represents approximately 3 percent of all childhood leukemias. Multiple occurrences of CML in families are rare. There is no concordance of the disease between identical twins. There is no analytical epidemiologic evidence for a familial predisposition to CML in Swedish databases.19 There is some evidence that overweightness and obesity can increase the incidence of CML.20
ETIOLOGY AND PATHOGENESIS
Exposure to very high doses of ionizing radiation can increase the occurrence of CML above the expected frequency in comparable populations. Three major populations—the Japanese exposed to the radiation released by the atomic bomb detonations at Nagasaki and Hiroshima,21 British patients with ankylosing spondylitis treated with spine irradiation,22 and women with uterine cervical carcinoma who received radiation therapy23—had a frequency of CML (as well as acute leukemia) significantly above the frequency expected in comparable unexposed groups. The median latent period was approximately 4 years in irradiated spondylitics, among whom approximately 20 percent of the leukemia cases were CML; 9 years in the uterine cervical cancer patients, of whom approximately 30 percent had CML; and 11 years in the Japanese survivors of the atomic bombs, of whom approximately 30 percent of the leukemia patients had CML.24 Chemical leukemogens, such as benzene and alkylating agents, are not causative agents of CML, presumably because of their inability to induce the specific chromosome translocation required to cause the disease.25,26,27
CML results from the malignant transformation of a single hematopoietic stem cell. The disease is acquired (somatic mutation), given that the identical twin of patients with CML and the offspring of mothers with the disease neither carry the Ph chromosome nor develop the disease.28 The origin of CML from a single hematopoietic stem cell is supported by the following lines of evidence:
Involvement of erythropoiesis, neutrophilopoiesis, eosinophilopoiesis, basophilopoiesis, monocytopoiesis, and thrombopoiesis in chronic phase CML.29
Presence of the Ph chromosome (22q−) in erythroblasts; neutrophilic, eosinophilic, and basophilic granulocytes; macrophages; and megakaryocytes.30
Presence of a single glucose-6-phosphate dehydrogenase isoenzyme in red cells, neutrophils, eosinophils, basophils, monocytes, and platelets, but not in fibroblasts or other somatic cells in women with CML who are heterozygotes for isoenzymes A and B.31,32,33
Presence of the Ph translocation only on a structurally anomalous chromosome 9 or 22 of each chromosome pair in every cell analyzed in occasional patients with a structurally dissimilar 9 or 22 chromosome within the pair.34,35,36
Presence of the Ph chromosome in one, but not the other, cell lineage of patients who are a mosaic for sex chromosomes, as in Turner syndrome (45X/46XX)37 and Klinefelter syndrome (46XY/47XXY).38
Molecular studies showing variation in the breakpoint of chromosome 22 among different patients with CML but precisely the same breakpoint among cells within a single patient with CML.39,40
Combined DNA hybridization-methylation analysis of women who have restriction fragment length polymorphisms at the X-linked locus for hypoxanthine phosphoribosyltransferase (HPRT), which enables distinction of the two alleles of the HPRT gene in heterozygous females, coupled with methylation-sensitive restriction-enzyme cleavage patterns, which permits delineation of whether cells contain either the maternally derived or the paternally derived copy of the gene.41
The foregoing observations place the parent cell of the clone at least at the level of the hematopoietic multipotential cell.
Acquisition of the BCR-ABL1 fusion gene as a result of the t(9;22) (q34;q11.2) in a single primitive multipotential hematopoietic cell (possibly the pluripotential stem cell) results in the CML stem cell, necessary for the initiation and maintenance of the chronic phase of CML.42,43 The phenotype of the CML stem cell is not fully defined but they are among the CD34+CD33−Lin−Thy1+ KIT− fraction of CML cells.43 A proportion of CML stem cells is in the G0 phase of the cell cycle and is resistant to therapy with BCR-ABL1 inhibitors. These cells represent a pool for the regrowth of the tumor in most patients, if suppressive therapy is interrupted. The leukemia stem cell is resistant to TKI therapy, but a pan-BCL2 inhibitor has been found to sensitize marrow leukemia stem cells to tyrosine kinase inhibition.44 N-cadherin and WNT-β-catenin signaling are also thought to mediate microenvironmental protection of CML stem cells from TKIs.45 The acquisition of genetic and epigenetic events in a derivative BCR-ABL1–positive cell can result in evolution to accelerated phase and blastic transformation46 (see “Accelerated Phase and Blast Crisis of Chronic Myelogenous Leukemia” below).
Some patients in chronic phase CML have lymphocytes that are derived from the primordial malignant cell. Evidence for this finding includes the following: A single isoenzyme for glucose-6-phosphate dehydrogenase has been found in some T and B lymphocytes in women with CML who are heterozygous for isoenzymes A and B47; blood cells from patients with CML induced to proliferate with Epstein-Barr virus (presumptive B lymphocytes) are of the same glucose-6-phosphate dehydrogenase isoenzyme type, have cytoplasmic immunoglobulin heavy and light chains, and contain the Ph chromosome48; blood lymphocytes stimulated with B lymphocyte mitogens contain the Ph chromosome49,50; purified B lymphocytes from the blood in chronic phase CML contain an abnormal, elongated phosphoprotein coded for by the chimeric gene resulting from the t(9;22)51; and fluorescence in situ hybridization (FISH) has detected the BCR-ABL1 fusion gene in approximately 25 percent of B lymphocytes in some, but not all, patients in chronic phase.52,53 These findings suggest that B lymphocytes are derived from the malignant clone, placing the lesion closer to, if not in, the pluripotential lymphohematopoietic stem cell.47,48,49,50,51 Previous studies have found that the B lymphocyte pool is a mosaic, containing both Ph chromosome– and BCR-ABL1–positive cells and Ph chromosome– or BCR-ABL1–negative cells. Results of studies examining the derivation of T lymphocytes from the malignant clone are more ambiguous but indicate that T lymphocytes are derived from the malignant clone in some patients.47,49,54,55,56,57,58,59,60,61,62,63 Natural killer (NK) cells isolated from patients with chronic phase CML do not contain the BCR-ABL1 fusion gene.64 It is possible that myelopoiesis is invariably clonal and lymphopoiesis is an unpredictable mosaic derived largely from normal residual stem cells. This conclusion is supported by the finding that progenitors of T, B, and NK lymphocytes contain the Ph chromosome and BCR-ABL1, but most B-cell and all T-cell progenitors derived from the leukemic clone undergo apoptosis, leaving unaffected cells in the blood.65,66,67,68
The cell in which the mutation occurs may be even more primitive in that some endothelial cells generated in vitro express the BCR-ABL1 fusion gene, as do some cells in the patient’s vascular endothelium.69
Early studies indicated that the Ph chromosome may appear after the initial leukemogenic event.70,71,72,73 Patients with CML have developed the Ph chromosome during the course of the disease, have experienced periods of the disease when the Ph chromosome disappeared,74 or have had Ph chromosome–positive and Ph chromosome–negative cells concurrently.75,76,77,78,79
Nearly all, if not all, patients with CML have an abnormality of chromosome 22 at a molecular level (BCR rearrangement). Thus, earlier studies indicating an absence of a Ph chromosome was not a valid measure of the normality of chromosome 22. The molecular abnormality in CML involving the ABL1 gene on chromosome 9 and the BCR gene on chromosome 22 has been established as being the proximate cause of the chronic phase of the disease (see “Molecular Pathology” below).
Most, if not all, patients with CML have hematopoietic stem cells that, after treatment80,81,82 or culture in vitro,83,84,85 use of special cell isolation techniques,81,82 or use of cell transfer to nonobese diabetic (NOD)/severe combined immunodeficiency (SCID) mice88 do not have the Ph chromosome89,90 or the BCR-ABL1 fusion gene.91,92,93,94,95 The switch to Ph chromosome–negative cells in vitro is associated with a loss of monoclonal glucose-6-phosphate dehydrogenase isoenzyme patterns, indicating the persistence and reemergence of normal polyclonal hematopoiesis rather than reversion to a Ph chromosome–negative clone.96 In confirmation, BCR-ABL1+, CD34+, human leukocyte antigen (HLA)-DR− cells isolated from women with early phase CML are polyclonal using the human androgen receptor assay (HUMARA) to assess X chromosome inactivation patterns.97 Very primitive hematopoietic cells, the long-term culture–initiating cells (LTC-ICs), are present in Ph chromosome–negative cytapheresis samples collected during early recovery after chemotherapy for CML.98 These LTC-ICs are most commonly present when samples are collected within 3 months of diagnosis.99 Variable levels of BCR-ABL1–negative progenitors are found in the CD34+DR− population, but low levels are found in the CD34+CD38− population.95,100 Preprogenitors for the CD34+DR− cells are predominantly BCR-ABL1–negative in both marrow and blood at diagnosis.101 However, some cells with surface marker characteristics of very primitive normal hematopoietic cells do express the BCR-ABL1 gene.102 Both normal and leukemic SCID-repopulating cells coexist in the marrow and blood from CML patients in chronic phase, whereas only leukemic SCID-repopulating cells are detected in blast crisis.103,104
The leukemic transformation resulting from the BCR-ABL1 fusion oncogene is maintained by a relatively small number of BCR-ABL1 stem cells that favor differentiation over self-renewal.105 This predisposition to differentiation and progenitor cell expansion is mediated by an autocrine interleukin (IL)-3–granulocyte colony-stimulating factor (G-CSF) loop.105 The earliest progenitors have the capacity to undergo marked expansion of erythroid, granulocytic, and megakaryocytic cell populations, and have a decreased sensitivity to regulation.105,106,107 This expansion is especially dramatic in the more mature progenitor cell compartment.105,108 The proliferative capacity of individual granulocytic progenitors is decreased compared to normal cells. Thus, the progenitor cell population in marrow and blood expands proportionately more than the increase in granulopoiesis.109 BCR-ABL1 reduces growth factor dependence of progenitor cells.
Erythroid progenitors are expanded, erythroid precursor maturation is blocked at the basophilic erythroblast stage, and the extent of erythropoiesis is inversely proportional to the total white cell count.110
Phenotypic differences of stem and progenitor cells in CML patients compared to normal subjects have been identified.111 For example, a greater proportion of the circulating leukemic colony-forming unit–granulocyte-monocytes (CFU-GMs) express high levels of the adhesion receptor CD44112 and low levels of L-selectin113 in contrast to normal cells. Leukemic CD34+ cells overexpress the P glycoprotein that determines the multidrug resistance phenotype.114
BCR-ABL1–positive progenitors survive less well in long-term culture than do their normal counterparts. Leukemic CFU-GM colonies, unlike normal colonies, decrease in long-term cultures that are deficient in KIT ligand,115 whereas their proliferation is favored in the presence of KIT ligand.116 Macrophage inflammatory protein (MIP)-1α, renamed CCL3, does not inhibit growth factor-mediated proliferation of CD34+ cells from CML patients, as it does CD34+ cells from normal subjects, even though the CCL3 receptor is expressed.117 Another chemokine, monocyte chemotactic protein (MCP)-1 or CCL2, unlike CCL3, is an endogenous chemokine that cooperates with transforming growth factor beta (TGF-β) to inhibit the cycling of primitive normal, but not CML, progenitors in long-term human marrow cultures.118 Leukemic progenitors are less sensitive than normal progenitors to the antiproliferative effects of TGF-β.119
Primitive progenitors and blast colony-forming cells from patients with CML have decreased adherence to marrow stromal cells.120 This defect is normalized if stromal cells are treated with interferon (IFN)-α.121 As a result, BCR-ABL1–negative progenitors are enriched in the adherent fraction of circulating CD34+ cells in chronic phase CML patients. The most primitive BCR-ABL1–positive cells in the blood of patients with CML differ from their normal counterparts. They are increased in frequency and are activated, such that signals that block cell mitosis are bypassed.122
Ph chromosome–positive colony-forming cells adhere less to fibronectin (and to marrow stroma) than do their normal counterparts. Adhesion is fostered as a result of restoration of cooperation between activated β1 integrins and the altered epitopes of CD44.123,124 CML granulocytes have reduced and altered binding to P-selectin because of modification in the CD15 antigens.125 BCR-ABL1–induced defects in integrin function may underlie the abnormal circulation and proliferation of progenitors126,127 because growth signaling can occur through the fibronectin receptor.128 IFN-α restores normal integrin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment.129 There are conflicting data regarding the effects of TKI effects on adhesion of CML cells to stroma.130,131
BCR-ABL1–encoded fusion protein p210BCR-ABL binds to actin, and several cytoskeletal proteins are thereby phosphorylated. The p210BCR-ABL interacts with actin filaments through an actin-binding domain. BCR-ABL1 transfection is associated with increased spontaneous motility, membrane ruffling, formation of long actin extensions (filopodia), and accelerated rate of protrusion and retraction of pseudopodia on fibronectin-coated surfaces. IFN-α treatment slowly converts the abnormal motility phenotype of BCR-ABL1–transformed cells toward normal.132 Integrins regulate the c-ABL–encoded tyrosine kinase activity and its cytoplasmic nuclear transport.133 The p210BCR-ABL1 abrogates the anchorage requirement but not the growth factor requirement for proliferation.134
In normal cells exposed to IL-3, paxillin tyrosine residues are phosphorylated. In cells transformed by p210BCR-ABL1, the tyrosines of paxillin, vinculin, p125FAK, talin, and tensin are constitutively phosphorylated. Pseudopodia enriched in focal adhesion proteins134,135 are present in cells expressing p210BCR-ABL1.
The sum of evidence suggests that defects in adhesion (contact and anchoring) of CML primitive cells remove them from their controlling signals normally received from microenvironmental cells via cytokine messages. These signals retain the balance among cell survival, cell death, cell proliferation, and cell differentiation. Inappropriate phosphorylation of cytoskeletal proteins, possibly independent of the mutant tyrosine kinase, is thought to be the key factor in disturbed integrin function of CML cells.
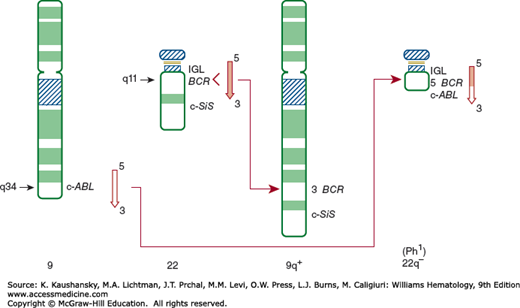
The genetic disturbance became evident with the knowledge that CML was derived from a primitive cell containing a 22q− abnormality.6,11 The abnormal chromosome contained only 60 percent of the DNA in other G-group chromosomes.136 Cytogenetic analysis indicated the G-group chromosome involved was different from the extra G-group chromosome in Down syndrome, which had been assigned number 21. Thus, the former was assigned number 22—even though it proved to be slightly longer than the chromosome involved in Down syndrome.11,137 The Paris Conference on Nomenclature decided not to undo the concept that Down syndrome is trisomy 21 and assigned the Ph chromosome and its normal counterpart, 22.138 Using quinacrine (Q) and Giemsa (G) banding, Rowley12 reported in 1973 that the material missing from chromosome 22 was not lost (deleted) from the cell, but was translocated to the distal portion of the long arm of chromosome 9. The amount of material translocated to chromosome 9 was approximately equivalent to that lost from chromosome 22, and the translocation was predicted to be balanced.12 Moreover, the breaks were localized to band 34 on the long arm of chromosome 9 and band 11 on the long arm of chromosome 22. Therefore, the classic Ph chromosome is t(9;22)(q34;q11), abbreviated t(Ph) (Fig. 89–2). The Ph chromosome can develop on either the maternal or the paternal member of the pair.139
Figure 89–2.
Schematic of normal chromosome 9 showing the ABL gene between bands q34 and qter of chromosome 22, which has the BCR and SIS genes between bands q11 and qter. The t(9;22) is shown on the right. The ABL from chromosome 9 is transposed to the chromosome 22 M-bcr sequences, and the terminal portion of chromosome 22 is transposed to the long arm of chromosome 9. The 22q− is the Ph chromosome. bcr, breakpoint cluster region; c-SiS, cellular homologue of the viral simian sarcoma virus-transforming gene; IGL, gene for immunoglobulin light chains. (Reproduced with permission from De Klein A: Oncogene activation by chromosomal rearrangement in chronic myelocytic leukemia. Mutat Res 1987 Sep;186(2):161–172.)
Mutations of the ABL1 gene on chromosome 9 and of the BCR gene on chromosome 22 are central to the development of CML (Fig. 89–3).140,141,142
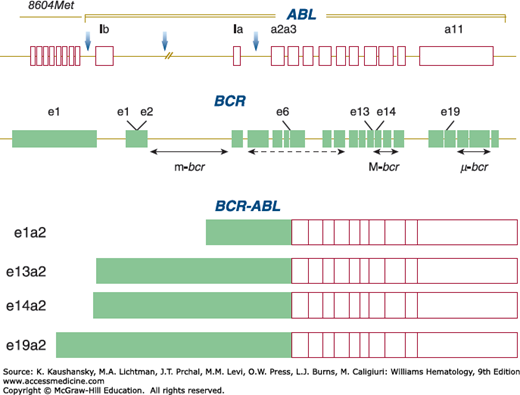
Figure 89–3.
Schematic of the normal ABL and BCR genes and of the BCR-ABL fusion transcripts. In the upper panel of the diagram, the possible breakpoint positions in ABL are marked by vertical arrows. Note the position immediately upstream of the ABL locus of the 8604Met gene. The BCR gene contains 25 exons, including first (e1) and second (e2) exons. The positions of the three breakpoint cluster regions, m-bcr, M-bcr, and μ–bcr, are shown. The lower panel of the figure shows the structure of the BCR-ABL messenger RNA fusion transcripts. Breakpoints in μ–bcr result in BCR-ABL transcripts with an e19a2 junction. The associated number designates the exon (location) at which the break occurs in each gene.
In 1982, the human cellular homologue ABL1 of the transforming sequence of the Abelson murine leukemia virus was localized to human chromosome 9.143 In 1983, ABL1 was shown to be on the segment of chromosome 9 that is translocated to chromosome 22144 by demonstrating reaction to hybridization probes for ABL1 only in somatic cell hybrids of human CML cells containing 22q− but not those containing 9q+. v-abl is the viral oncogenic homologue of the normal cellular ABL1 gene. This gene (v-abl) can induce malignant transformation of cells in culture and can induce leukemia in susceptible mice.145
The ABL1 gene is rearranged and amplified in cell lines from patients with CML.146 Cell lines and fresh isolates of CML cells contain an abnormal, elongated 8-kb RNA transcript,147,148,149,150 which is transcribed from the new chimeric gene produced by the fusion of the 5′ portion of the BCR gene left on chromosome 22 with the 3′ portion of the ABL1 gene translocated from chromosome 9 (Fig. 89–4).144 The fusion mRNA leads to the translation of a unique tyrosine phosphoprotein kinase of 210 kDa (p210BCR-ABL), which can phosphorylate tyrosine residues on cellular proteins similar to the action of the v-abl protein product.151,152,153,154,155 The ABL1 locus contains at least two alleles, one having a 500-bp deletion.157 In normal cells, the ABL1 protooncogene codes for a tyrosine kinase of molecular weight 145,000, which is translated only in trace quantities and lacks any in vitro kinase activity.152 The fusion product expressed by the BCR-ABL1 gene is hypothesized to lead to malignant transformation because of the abnormally regulated enzymatic activity of the chimeric tyrosine protein kinase.153,154,158,159 Construction of BCR-ABL1 fusion genes indicated that BCR sequences could also activate a microfilament-binding function, but the tyrosine kinase and microfilament-binding functions were not linked. Nevertheless, tyrosine kinase modification of actin filament function has been proposed as a step in leukemogenesis.160
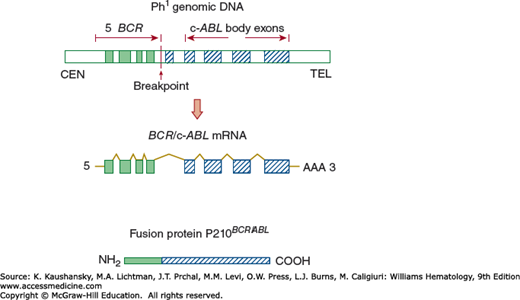
Figure 89–4.
Molecular effects of the Ph chromosome translocation t(9;22)(q34;q11). The upper panel shows the physically joined 5′ BCR and the 3′ ABL regions on chromosome 22. The exons are solid (from chromosome 22, BCR) and hatched (from chromosome 9, ABL). The middle panel depicts transcription of chimeric messenger RNA. The lower panel shows the translated fusion protein with the aminoterminus derived from the BCR of chromosome 22 and the carboxy-terminus from the ABL of chromosome 9. (Reproduced with permission from De Klein A: Oncogene activation by chromosomal rearrangement in chronic myelocytic leukemia. Mutat Res 1987 Sep;186(2):161–172.)
The breakpoints on chromosome 9 are not narrowly clustered, ranging from approximately 15 to more than 40 kb upstream from the most proximate region (first exon) of the ABL1 gene.143,144,161 The breakpoints on chromosome 22 occur over a very short, approximately 5 to 6 kb, stretch of DNA referred to as the breakpoint cluster region (M-bcr),162,163 which is part of a much longer BCR164,165 gene (see Fig. 89–4). Three main BCRs have been characterized on chromosome 22: major (M-bcr), minor (m-bcr), and micro (μ–bcr). The three different breakpoints result in a p210, p190, and p230 fusion protein, respectively (see Fig. 89–3). The overwhelming majority of CML patients have a BCR-ABL1 fusion gene that encodes a fusion protein of 210 kDa (p210BCR-ABL1), for which mRNA transcripts have e14a2 or a e13a2 fusion junction (see Fig. 89–3).166 The “e” represents the BCR exon and “a” the ABL1 exon sites involved in the translocation. A BCR-ABL1 with an e1a2 type of junction has been identified in approximately 50 percent of the Ph chromosome–positive acute lymphoblastic leukemia cases and results in the production of a BCR-ABL1 protein of 190 kDa (p190BCR-ABL). Almost all CML cases at diagnosis that encode a p210BCR-ABL also express BCR-ABL transcripts for p190.167 The biologic or clinical significance of these dual transcripts is not known. Transgenic mice expressing p210BCR-ABL develop acute lymphoblastic leukemia in the founder mice, but all transgenic progeny have a myeloproliferative disorder resembling CML.168
The BCR gene encodes a 160-kDa serine-threonine kinase, which, when it oligomerizes, autophosphorylates and transphosphorylates several protein substrates.169 Aberrant methylation of the M-bcr in CML occurs.166 The first exon sequences of the BCR gene potentiate the tyrosine kinase of ABL when they fuse as a result of the translocation.170 The central portion of BCR has homology to DBL, a gene involved in the control of cell division after the S phase of the cell cycle. The C-terminus of BCR has a guanosine triphosphatase (GTPase)-activating protein for p21rac, a member of the RAS family of guanosine triphosphate (GTP)-binding proteins.171 A reciprocal hybrid gene ABL-BCR1 is formed on chromosome 9q+ when BCR-ABL1 fuses on chromosome 22. The ABL-BCR1 fusion gene actively transcribes in most patients with CML.172
Variations in breakpoints involving smaller stretches of chromosome 9 and rearrangements outside the M-bcr of chromosome 22 can occur.37 In a few cases of CML with no evident elongation of chromosome 9, molecular probes have shown that ABL1 still is translocated to chromosome 22.173 In occasional patients with Ph chromosome–positive CML, the break in chromosome 22 is outside the M-bcr, and transcription of a fusion RNA of the usual type fails or a fusion RNA is transcribed that does not hybridize with the classic M-bcr complementary DNA (cDNA) probe.174
In cases in which the Ph chromosome is not found, BCR-ABL1 still may be located on chromosome 9 (a masked Ph chromosome).175 The BCR gene can recombine with genomically distinct sites on band 11q13 in complex translocations in a region rich in Alu repeat elements.176 ETV6/ABL1 fusion genes have also been found in BCR-ABL1–negative CML.177
The BCR breakpoint site has been examined as a factor in disease prognosis. Some studies have shown no correlation between CML chronicity and breakpoint site, although thrombocytosis may be more common with 3′ breakpoint sites and basophilia with 5′ breakpoint sites.178 No difference in response to IFN-α therapy was noted, and survival was not significantly different, although patients with 3′ deletions tended to have shorter survival.179 Others have observed a better response to IFN-α in patients with a 3′ rearrangement, which is being examined with imatinib mesylate therapy.180
CML patients with m-bcr breakpoints develop a blast crisis with monocytosis and an absence of splenomegaly and basophilia.181 The p230 (e19a2 RNA junction) encoded by μ–bcr is rarely expressed but has been associated with neutrophilic CML or thrombocytosis (see “Special Clinical Features” below). Other rare breakpoints have been described.182 For example, a case with a 12-bp insert between BCR and ABL1 resulted in a BCR-ABL1–negative (false-negative), Ph chromosome–positive CML with thrombocythemia.183 Another novel BCR-ABL1 fusion gene (e6a2) in a patient with Ph chromosome–negative CML encoded an oncoprotein of 185 kDa.184 Typical CML also has been associated with an e19a2 junction BCR-ABL1 transcript.185
Experimental support for the hypothesis that p210BCR-ABL1 tyrosine phosphoprotein kinase is transforming is provided by a retroviral gene transfer system that permits expression of the protein. Mouse marrow cells transfected with BCR-ABL1 develop clonal outgrowths of immature cells expressing the p210BCR-ABL1 tyrosine kinase. Some clones progress to a malignant phenotype, can be transplanted, and can induce tumors in syngeneic mice.186 Similar studies suggest that the p210BCR-ABL can transform 3T3 murine fibroblasts if the gag gene sequence from a helper virus cooperates.187 The BCR-ABL1 gene from a retroviral vector has been expressed in an IL-3–dependent cell line. Clones derived from the infected line transform over months to IL-3 independency, are capable of increased proliferation, and develop chromosomal abnormalities.188
A series of mouse models in which the BCR-ABL1 was used to induce leukemogenesis have been described.189,190,191,192,193,194,195,196,197 Lethally irradiated mice have been reconstituted with marrow enriched for cycling stem cells infected with a BCR-ABL1–bearing retrovirus. Fatal diseases with abnormal accumulations of macrophagic, erythroid, mast, and lymphoid cells develop.188 Classic CML did not occur, and complete transformation was not documented. The cell lines from spleen and marrow from mice with a BCR-ABL1 retrovirus infection were predominantly mast cells; however, in some cases these cell lines spontaneously switched to either erythroid and megakaryocytic, erythroid, or granulocytic lineages displaying maturation. They were transplantable (transformed) and contained the same proviral inserts as the original mast cell line.198 Murine marrow also has been infected with a retrovirus encoding p210BCR-ABL and transplanted into irradiated syngeneic recipients.189 Although several types of hematologic malignancies developed, a syndrome mimicking human CML also occurred. Mice transgenic for a p190BCR-ABL develop an acute lymphocytic leukemia (ALL) lymphoma syndrome190 that resembles human Ph chromosome–positive ALL. When a p210BCR-ABL transcript is introduced into a mouse germline (one-cell fertilized eggs), the p210 founder and progeny transgenic animals developed leukemia of B or T lymphoid or of myeloid origin after a relatively long latency period. In contrast, p190 transgenic mice exclusively developed leukemia of B-cell origin, with a relatively short period of latency. This finding was believed to be consistent with the apparent indolent nature of human CML during the chronic phase.191 When transgenic mice express p210BCR-ABL, the transgenes develop ALL, whereas the progeny develop a myeloproliferative disorder.192
Mouse models remain important for exploring the pathogenesis of the acute and chronic BCR-ABL1–mediated leukemias in vivo and in examining the potential effects of new drugs targeted at BCR-ABL1.199
BCR-ABL1 fusion genes can be found in the leukocytes of some normal individuals using a two-step reverse transcriptase polymerase chain reaction assay. Thus, although BCR-ABL1 may be expressed relatively frequently at very low levels in hematopoietic cells, only infrequently do the cells acquire the additional changes necessary to produce leukemia. This may be a dosage effect.200
The tyrosine phosphoprotein kinase activity of p210BCR-ABL1 has been causally linked to the development of Ph chromosome–positive leukemia in man.201–212 p210BCR-ABL1 is, unlike the ABL1 protein that is located principally in the nucleus, located in the cytoplasm making it accessible to a large number of interactions, especially components of signal transduction pathways.205,206,213 It binds and/or phosphorylates more than 20 cellular proteins in its role as an oncoprotein.206 A subunit of phosphatidylinositol 3′-kinase (PI3K) associates with p210BCR-ABL; this interaction is required for the proliferation of BCR-ABL1–dependent cell lines and primary CML cells. Wortmannin, a nonspecific inhibitor of the p110 subunit of the kinase, inhibits growth of these cells.207
The pathways and interactions invoked by BCR-ABL1 acting on mitogen-activated protein kinases are multiple and complex.214,215 A RAF-encoded serine-threonine kinase activity is regulated by p210BCR-ABL. Downregulation of RAF expression inhibits both BCR-ABL1–dependent growth of CML cells and growth factor–dependent proliferation of normal hematopoietic progenitors.208 The efficiency of cell transformation by BCR-ABL1 is affected by an adaptor protein that can relate tyrosine kinase signals to RAS. This involves growth factor receptor–bound protein-2 (GRB2). p210BCR-ABL also activates multiple alternative pathways of RAS.209 PI3K is constitutively activated by BCR-ABL1, generates inositol lipids, and is dysregulated through the downregulation by BCR-ABL1 of polyinositol phosphate tumor suppressors, such as PTEN and SHIP1.213 Figure 89–5 demonstrates interaction of p210BCR-ABL with various mediators of signal transduction.
Figure 89–5.
Major intracellular signaling events associated with BCR/ABL. Constitutive activation of ABL protein tyrosine kinase (PTK) induces phosphorylation of the tyrosine moiety of various substrates, including autophosphorylation of BCR/ABL and complex formation of BCR/ABL with adaptor proteins. This process subsequently activates multiple intracellular signaling pathways, including RAS activation and phosphatidylinositol 3′-kinase (PI3K) activation pathways. BCR/ABL also activates the c-MYC pathway, which involves ABL-SH2 domain. BCR/ABL inhibits apoptosis, possibly in part through upregulation of Bcl-2, and alters cellular adhesive properties, possibly by interacting with focal adhesion proteins and the actin cytomatrix. Broken lines indicate hypothetical pathways. ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; JNK, Jun N-terminal kinase; MEKK, MEK kinase; Sos, Son-of-sevenless; STAT, signal transducer and activator of transcription. (Reproduced with permission from Gotoh A, Broxmeyer HE: The function of BCR/ABL and related proto-oncogenes. Curr Opin Hematol 4(1):3–11, 1997.)
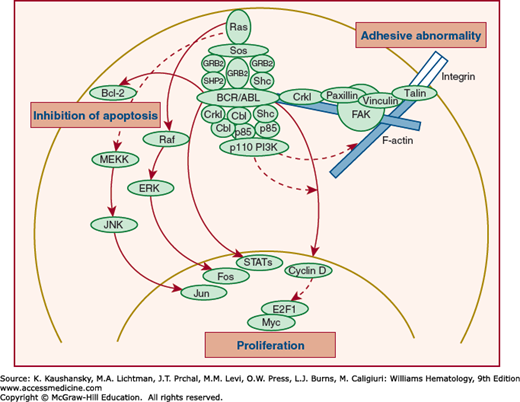
Reactive oxygen species are increased in BCR-ABL1–transformed cells and may act as a second messenger to modulate enzymes regulated by the reduction-oxidation (redox) equilibrium. An increase in these reactive oxygen products is postulated to play a role in the acquisition of additional mutations as a result of production of reactive oxygen species through the chronic phase, contributing to the progression to accelerated phase.213,216
The adaptor molecule CRKL is a major in vivo substrate for p210BCR-ABL, and it acts to relate p210BCR-ABL to downstream effectors. CRKL is a linker protein that has homology to the v–crk oncogene product. Antibodies to CRKL can immunoprecipitate paxillin. Paxillin is a focal adhesion protein210 that is phosphorylated by p210BCR-ABL. The p210BCR-ABL may be physically linked to paxillin by CRKL. CRKL binds to CBL, an oncogene product that induces B cell and myeloid leukemias in mice.211 The Src homology 3 domains of CRKL do not bind to CBL, but they do bind BCR-ABL. Therefore, CRKL mediates the oncogenic signal of BCR-ABL to CBL. The p120CBL and the adaptor proteins CRKL and c-CRK also link c-abl, p190BCR-ABL, and p210BCR-ABL to the PI3K pathway.212 The p120CBL also coprecipitates with the p85 subunit of PI3K, CRKL, and c-CRK. The p210BCR-ABL may, therefore, induce the formation of multimeric complexes of signaling proteins.217 These complexes contain paxillin and talin and may explain some of the adhesive defects of CML cells.218
Hef2 also binds to CRKL in leukemic tissues of p190BCR-ABL transgenic mice. Hef2 is involved in the integrin signaling pathway219 and encodes a protein that accelerates GTP hydrolysis of RAS-encoded proteins and neurofibromin. The latter negatively regulates granulocyte-monocyte colony-stimulating factor (GM-CSF) signaling through RAS in hematopoietic cells.220 p62DOK, a constitutively tyrosine-phosphorylated, p120RAS GAP-associated protein, which is rapidly tyrosine phosphorylated upon activation of the c-kit receptor,221 is also associated with ABL1.222
Nuclear factor (NF)-κB activation is also required for p210BCR-ABL-mediated transformation.223 Expression of p210BCR-ABL leads to activation of NF-κB–dependent transcription via nuclear translocation.224
Cell lines that express p210BCR-ABL also demonstrate constitutive activation of Janus kinases (JAKs) and signal transducers and activators of transcription (STATs), usually STAT5.225 STAT5 is also activated in primary mouse marrow cells acutely transformed by the BCR-ABL1226; p210BCR-ABL1 coimmunoprecipitates with and constitutively phosphorylates the common β subunit of the IL-3 and GM-CSF receptors and JAK2.227 Both ABL1 and BCR are also multifunctional regulators of the GTP-binding protein family Rho228,229 and the growth factor-binding protein GRB2, which links tyrosine kinases to RAS and forms a complex with BCR-ABL1 and the nucleotide exchange factor Sos that leads to activation of RAS.230
The p210BCR-ABL1 also activates Jun kinase and requires Jun for transformation.231 In some CML cell lines, p210BCR-ABL1 is associated with the retinoblastoma (Rb) protein.232 Loss of the neurofibromatosis (NF1) tumor-suppressor gene, a RAS GTPase-activating protein, also is sufficient to produce a myeloproliferative neoplasm in mice akin to human CML resulting from RAS-mediated hypersensitivity to GM-CSF.233
Whether p210BCR-ABL1 influences the expansion of the malignant clone in CML by inhibiting apoptosis is uncertain. In one study, the survival of normal and CML progenitors was the same after in vitro incubation in serum-deprived conditions and after treatment with X-irradiation or glucocorticoids.234 p210BCR-ABL1 inhibits apoptosis by delaying the G2/M transition of the cell cycle after DNA damage.235 The p210BCR-ABL1 also may exert an antiapoptotic effect in factor-dependent hematopoietic cells.236,237
p210BCR-ABL1 does not prevent apoptotic death induced by human NK or lymphokine-activated killer cells directed against CML or normal cells.238 In accelerated and blast phases, apoptosis rates were lower in CML neutrophils. G-CSF and GM-CSF considerably decreased the rate of apoptosis in CML neutrophils.239
Patients with CML present with a somewhat shortened mean telomere length in granulocytic cells but not blood T lymphocytes at diagnosis, but considerable overlap exists in the distribution of telomere length with healthy individuals.240,241,242 The rate of shortening of telomere length during the chronic phase is correlated with a more rapid onset of accelerated phase.240,242 Telomerase reverse transcriptase (TERT) is the catalytic subunit, expression of which is closely correlated with telomerase activity. In CML CD34+ cells containing BCR-ABL1, the expression of TERT is significantly lower than in normal CD34+ cells, consistent with accelerated shortening of telomeres in CML cells.243 A further significant decrease in telomere length occurs in the accelerated phase of CML. Telomerase activity is increased in the accelerated phase.244 When therapy permits restoration of Ph chromosome–negative cells in the blood, these cells have telomere length comparable to that in matched healthy controls.245
CLINICAL FEATURES
In the 70 percent of patients who are symptomatic at diagnosis, the most frequent complaints include easy fatigability, loss of sense of well-being, decreased tolerance to exertion, anorexia, abdominal discomfort, early satiety (related to splenic enlargement), weight loss, and excessive sweating.246,247,248 The symptoms are vague, nonspecific, and gradual in onset (weeks to months). A physical examination may detect pallor and splenomegaly. The latter was present in approximately 90 percent of patients at diagnosis, but with medical care being sought earlier, the presence of splenomegaly at the time of diagnosis is decreasing in frequency.247 Sternal tenderness, especially the lower portion, is common; occasionally, patients notice it themselves.
Uncommon presenting symptoms include those of dramatic hypermetabolism (night sweats, heat intolerance, weight loss) simulating thyrotoxicosis; acute gouty arthritis, presumably related in part to hyperuricemia; priapism, tinnitus, or stupor from the leukostasis associated with greatly exaggerated blood leukocyte count elevations249,250,251; left upper quadrant and left shoulder pain as a consequence of splenic infarction and perisplenitis; vasopressin-responsive diabetes insipidus252,253; and acne urticata associated with hyperhistaminemia.254 Acute febrile neutrophilic dermatosis (Sweet syndrome), a perivascular infiltrate of neutrophils in the dermis, can occur. In the latter situation, fever accompanied by painful maculonodular violaceous lesions on the trunk, arms, legs, and face are characteristic.255,256 Spontaneous rupture of the spleen is a rare event.257,258 Digital necrosis has been reported as a rare paraneoplastic event.259,260
In an increasing proportion of patients, the disease is discovered, coincidentally, when blood cell counts are measured at a periodic medical examination.
Hyperleukocytosis and symptoms or signs therefrom are a more common feature in patients who present with CML before the age of 20 years. The white cell counts at diagnosis are on average more than twice that in adults, the fraction of blood blasts, promyelocytes, and myelocytes is significantly higher, and clinical manifestations of hyperleukocytosis are far more frequent in children than adults.250
The presumptive diagnosis of CML can be made from the results of the blood cell counts and examination of the blood film.26,246,247 The blood hemoglobin concentration is decreased in most patients at the time of diagnosis. Red cells usually are only slightly altered, with an increase in variation from small to large size and only occasional misshapen (elliptical or irregular) erythrocytes. Small numbers of nucleated red cells are commonly present. The reticulocyte count is normal or slightly elevated, but clinically significant hemolysis is rare.246,261,262 Rare cases of mild erythrocytosis263,264 or erythroid aplasia265,266 have been documented.
The total leukocyte count is always elevated at the time of diagnosis and is nearly always greater than 25 × 109/L; at least half the patients have total white counts greater than 100 × 109/L (Fig. 89–6).26,246,247 The total leukocyte count rises progressively in untreated patients. Rare patients may have dramatic cyclic variations in white cell counts as much as an order of magnitude with cycle intervals of approximately 60 days.267,268 Granulocytes at all stages of development are present in the blood and are generally normal in appearance (Fig. 89–7). In this series, the mean blast cell prevalence was approximately 3 percent but can range from 0 to 10 percent; progranulocyte prevalence was approximately 4 percent; myelocytes, metamyelocytes, and bands accounted for approximately 40 percent; and segmented neutrophils accounted for approximately 35 percent of total leukocytes (Table 89–1). Often, there is a “myelocyte bulge” in which the differential count shows an exaggerated proportion of myelocytes compared to the proportion observed in normal persons. Hypersegmented neutrophils are commonly present.
| Percent of Total Leukocytes (Mean Values) | |
|---|---|
| Myeloblasts | 3 |
| Promyelocytes | 4 |
| Myelocytes | 12 |
| Metamyelocytes | 7 |
| Band forms | 14 |
| Segmented forms | 38 |
| Basophils | 3 |
| Eosinophils | 2 |
| Nucleated red cells | 0.5 |
| Monocytes | 8 |
| Lymphocytes | 8 |
Figure 89–6.
Total white cell count and platelet count of 90 patients with CML at the time of diagnosis. The cumulative percent of patients is on the ordinate, and the cell count is on the abscissa. Fifty percent of patients had a white cell count greater than 100 × 109/L and a platelet count greater than approximately 300 × 109/L at the time of diagnosis.
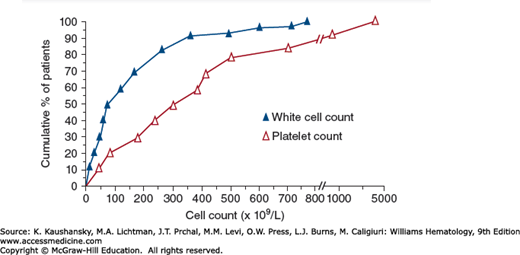
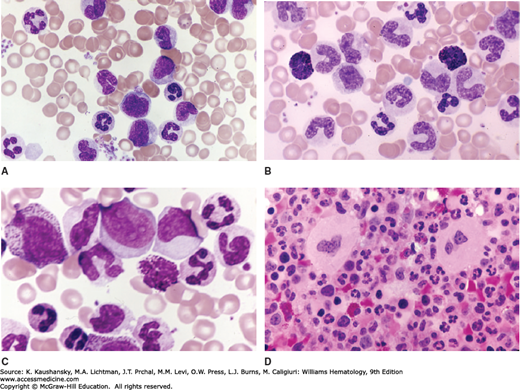
Figure 89–7.
Blood and marrow cells characteristic of chronic myelogenous leukemia. A. Blood film. Elevated leukocyte count. Elevated platelet count (aggregates). Characteristic array of immature (myelocytes, metamyelocytes, band forms) and mature neutrophils. B. Blood film. Elevated leukocyte count. Characteristic array of immature (myelocytes, metamyelocytes, band forms) and mature neutrophils. Two basophils in the field. Absolute basophilia is a constant finding in CML. C. Blood film. Elevated leukocyte count. Characteristic array of immature (promyelocytes, myelocytes, metamyelocytes, band forms) and mature neutrophils. Basophil in the field. Two myeloblasts in upper center. Note multiple nucleoli (abnormal) and agranular cytoplasm. D. Marrow section. Hypercellular. Replacement of fatty tissue (normally approximately 60 percent of marrow volume in adults of this patient’s age) with hematopoietic cells. Intense granulopoiesis and evident megakaryocytopoiesis. Decreased erythropoiesis. (Reproduced with permission Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Neutrophil alkaline phosphatase activity is low or absent in more than 90 percent of patients with CML.269,270,271 The mRNA for alkaline phosphatase is undetectable in neutrophils of patients with CML.272 The activity increases toward or to normal in the presence of intense inflammation or infection and when the total leukocytic count is decreased to or near normal with treatment.271,273 CML neutrophils regain alkaline phosphatase activity after infusion into leukopenic recipients, suggesting the effect of regulators or factors extrinsic to the neutrophils. With the availability of specific markers, BCR-ABL1 in CML and JAK2 mutations in polycythemia, leukocyte alkaline phosphatase is no longer used for diagnostic purposes.
The proportion of eosinophils usually is not increased, but the absolute eosinophil count nearly always is increased. Rarely, eosinophils are so prominent that they dominate the granulocytic cells and lead to the designation Ph chromosome–positive eosinophilic CML. An absolute increase in the basophil concentration is present in almost all patients, and this finding can be useful in preliminary consideration of the differential diagnosis.26,278 Basophilic progenitor cells are increased in the blood.279 The proportion of basophils usually is not greater than 10 to 15 percent during the chronic phase but may, in rare patients, represent 30 to 80 percent of the total leukocyte count during chronic phase and lead to the designation of Ph chromosome–positive basophilic CML.280 Flow cytometry using anti-CD203c provides very accurate assessment of the basophil frequency. Basophils may be hypogranulated or have an immature phenotype and may be left uncounted in an optical differential white cell count. Anti-CD203c recognizes these cells as basophils.281 Granules of basophils in patients with CML, unlike normal basophils, contain mast cell α-tryptase.281,282 Granulocytes containing both eosinophilic and basophilic granules (mixed granulation) are commonly present.283
The total absolute lymphocyte count is increased (mean: approximately 15 × 109/L) in patients with CML at the time of diagnosis284 as a result of the balanced increase in T-helper and T-suppressor cells.285 B lymphocytes are not increased.288 T lymphocytes also are increased in the spleen.286 NK cell activity is defective in CML patients as a result of decreased maturation of these cells in vivo287,288 and a decrease in the absolute number of circulating NK cells in patients with CML. The latter change can perhaps be related to increased apoptosis.289 The CD56 bright subset of NK cells is particularly decreased. These cells are reduced more as CML progresses, and they respond less to stimuli that recruit clonogenic NK cells compared to NK cells from normal subjects.290
The platelet count is elevated in approximately 50 percent of patients at the time of diagnosis and is normal in most of the rest.291 The median value in patients at diagnosis is approximately 400 × 109 cells/L. The platelet count may increase during the course of the chronic phase. Platelet counts greater than 1000 × 109/L are not unusual, and platelet counts as high as 5000 to 7000 × 109/L have occurred. Thrombohemorrhagic complications of thrombocytosis are infrequent. Occasionally, the platelet count may be below normal at the time of diagnosis, but this finding usually signals an impending progression to the accelerated phase of the disease (see “Accelerated Phase and Blast Crisis of Chronic Myelogenous Leukemia” below) and may also occur in with massive splenomegaly.
Functional abnormalities of neutrophils (adhesion, emigration, phagocytosis) are mild; are compensated for by high neutrophil concentrations; and do not predispose patients in chronic phase to infections by either usual or opportunistic organisms.292,293,294 Platelet dysfunction can occur but is not associated with spontaneous or exaggerated bleeding. A decrease in the second wave of epinephrine-induced platelet aggregation is the most common abnormality and is associated with a deficiency of adenine nucleotides in the storage pool.295,296
Morphology The marrow is markedly hypercellular, and hematopoietic tissue takes up 75 to 90 percent of the marrow volume, with fat markedly reduced (see Fig. 89–7).297,298 Granulopoiesis is dominant, with a granulocytic-to-erythroid ratio between 10:1 and 30:1, rather than the normal 2:1 to 4:1. Erythropoiesis usually is decreased, and megakaryocytes are normal or increased in number. Eosinophils and basophils may be increased, usually in proportion to their increase in the blood. Mitotic figures are increased in number. Mast cells are often seen, and uncommonly a juxtamembrane domain mutant of KIT coincides with BCR-ABL1 in CML.299 Rare reports of marrow mastocytosis have been explained by a KIT mutation as an additional genetic abnormality or by dual clones in the marrow.300,301 Macrophages that mimic Gaucher cells in appearance are sometimes seen. This finding is a result of the inability of normal cellular glucocerebrosidase activity to degrade the increased glucocerebroside load associated with markedly increased cell turnover.302 Macrophages also can become engorged with lipids, which, when oxidized and polymerized, yield ceroid pigment. This pigment imparts a granular and bluish cast to the cells after polychrome staining; such cells have been referred to as sea-blue histiocytes.302
Collagen type III (reticulin fibrosis), which takes the silver impregnation stain, is commonly increased at the time of diagnosis in nearly half the patients,303 and is correlated with the proportion of megakaryocytes in the marrow.304,305 Increased fibrosis also is correlated with larger spleen size, more severe anemia, and a higher proportion of marrow and blood blast cells.
The marrows of CML patients have a mean doubling of microvessel density compared to healthy controls and have more angiogenesis in marrow than other forms of leukemia.306,307,308 This increased marrow vascularity decreases to normal after treatment.309
Progenitor Cell Growth Cells that form colonies of neutrophils and macrophages or eosinophils (CFUs) are increased in the marrow and blood. The increase in CFUs in marrow is approximately 20-fold normal and in blood approximately 500-fold normal. The CFUs are of lighter buoyant density than those in normal marrow.100 More primitive progenitors that can initiate long-term cultures of hematopoiesis also are markedly increased.311 Spontaneous blood-derived granulocyte-macrophage colony growth is common, although CFUs also respond to growth factor stimulation.
Cytogenetics The marrow and nucleated blood cells of more than 90 percent of patients with clinical and laboratory signs that fall within the criteria for the diagnosis of CML contain the Ph chromosome (22q−) as measured by G-banding, and virtually all patients have the t(9;22)(q34;q11)(BCR-ABL1) by FISH. The Ph chromosome is present in all blood cell lineages (erythroblasts, granulocytes, monocytes, megakaryocytes, T- and B-cell progenitors) but is not present in the majority of blood B lymphocytes or in most T lymphocytes.54,56 Approximately 70 percent of patients in the chronic phase have the classic Ph chromosome in their cells.312 The remaining 20 percent also have a missing Y chromosome [t(Ph),−Y]; an additional C-group chromosome, usually number 8 [t(Ph),+8]; an additional chromosome 22q− but without the 9q+ [t(Ph), 22q−]; or t(Ph) plus either another stable translocation or another minor clone. These variations have not been shown to affect the duration of the chronic phase. Deletion of the Y chromosome occurs in approximately 10 percent of healthy men older than 60 years.313,314
Variant Ph chromosome translocations occur in approximately 5 percent of subjects with CML and involve complex rearrangements (three chromosomes), and every chromosome except the Y chromosome can be involved.315,316,317,318,319 The Ph chromosome, that is, 22q−, is present, but the gross exchange of chromosomal material involves a chromosome other than 9 (simple variant) or involves exchange of material among chromosomes 9 and 22 and a third or more chromosomes (complex variant; Fig. 89–8). High-resolution techniques have indicated that 9q34-qter is transposed to 22q11 in simple and in complex translocations.320,321 Thus, the fusion of 9q34 with 22q11 seems to occur in the cells of most patients with CML.323 Complex translocations involving chromosome 3 have been notable.322,323,324 In rare cases, a reciprocal translocation with a chromosome other than 9 to chromosome 22 is larger than usual, and the posttranslocation shortening of the long arms of 22 is not apparent. This circumstance has been referred to as a masked Ph chromosome or masked translocation because the 22q− is not evident by microscopic examination,325,326 although t(9;22) may occur as judged by banding techniques or molecular probes.327
Figure 89–8.
Translocations involved in chronic myelogenous leukemia. The positions of the ABL gene in each of the chromosomes before and after the translocation are noted. The origin of the chromosomal segments in each of the translocated chromosomes is indicated by a bracket on the side of the chromosome. (Reproduced with permission from Rosson D, Reddy EP: Activation of the abl oncogene and its involvement in chromosomal translocations in human leukemia. Mutat Res 1988 May;195(3):231–243.)
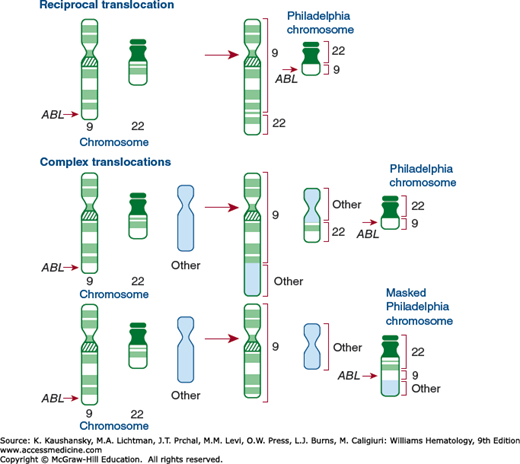
Approximately 10 percent of patients have a deletion of the derivative 9 chromosome adjacent to the chromosome breakpoint. Although this deletion is thought to be an important factor in resistance to drug effects with IFN therapy, it does not appear to be significant with the use of imatinib.214
Molecular Probes In a small proportion of patients with a clinical disease analogous to CML, cytogenetic studies do not disclose a classic, variant, or masked Ph chromosome. In these cases, use of a panel of restriction enzymes and Southern blot analyses with a molecular probe for the breakpoint cluster region on chromosome 22 nearly always detects rearrangement of fragments. This finding has led to the conclusion that almost all cases of CML have an abnormality of the long arm of chromosome number 22 (BCR rearrangement).328,329,330,331,332 Ph chromosome–negative CML cells with BCR rearrangement can express p210BCR-ABL1, and such patients have a clinical course similar to Ph chromosome–positive CML.328,333,334,335,336
The ability to identify the molecular consequences of the t(9;22), that is, BCR rearrangement, mRNA transcripts of the mutant fusion gene, and p210BCR-ABL1, has resulted in diagnostic tests supplementary to cytogenetic analysis.332 These tests include Southern blot analysis of BCR rearrangement,334,335,336,337,338 polymerase chain reaction (PCR) amplification of the abnormal mRNA,339 and a less complex variation on the latter, a hybridization protection assay.340
PCR can achieve a sensitivity of one positive cell in approximately 500,000 to one million cells. This extreme sensitivity requires special care in analysis and the inclusion of negative controls.341,342,343,344 Fusions e13a3, e14a3, and e19a2 are not detectable with standard PCR primers.344A
A multicolor FISH method to detect the BCR-ABL1 fusion in patients with CML is a rapid and sensitive alternative to Southern blot and PCR-dependent methods.345 For diagnostic purposes, FISH is simple, accurate, and sensitive, and can detect the various molecular fusions (e.g., e13a2, e14a2, e1a2).346,347,348,349,350 Interphase FISH is faster and more sensitive than cytogenetics in identifying the Ph chromosome. If the concentration of CML cells is very low, interphase FISH may not detect BCR-ABL1, so it has limited use for detecting minimal residual disease.351 Hypermetaphase FISH allows analysis of up to 500 metaphases per sample in 1 day. Several factors influence the false-positive and false-negative rates of FISH identification of BCR-ABL1, including definition of a fusion signal, nuclear size, and the genomic position of the ABL1 breakpoint.352 Double BCR-ABL fusion signals (double-fusion [D]-FISH) have been proposed as being more accurate than the fusion signal used in dual color (single-fusion) S-FISH, because in the latter case a small percentage of the normal BCR and ABL1 signals overlap.353
The frequency of cytogenetic analysis can be reduced if patients are monitored by molecular methods such as competitive reverse transcriptase (RT)-PCR. Molecular analyses can be performed on blood samples and therefore are much easier to use than cytogenetic analysis of marrow cell metaphases. Quantitative RT-PCR is the method of choice for monitoring patients for residual disease or reappearance of disease after marrow transplantation and for following response to TKIs once routine cytogenetics and FISH are negative for the Ph chromosome. Competitive PCR can detect reappearance of or increasing levels of BCR-ABL1 RNA transcripts prior to clinical relapse in patients after transplantation.354,355,356
Uric Acid An increased production of uric acid with hyperuricemia and hyperuricosuria occurs in untreated CML.357 Uric acid excretion often is two to three times normal in patients with CML. If aggressive therapy leads to rapid cell lysis, excretion of the additional purine load may produce urinary tract blockage from uric acid precipitates. Formation of urinary urate stones is common in patients with CML, and some patients with latent gout may develop acute gouty arthritis or uric acid nephropathy.358 The likelihood of complications from urate overproduction is greatly increased by starvation, acidosis, renal disease, or diuretic drug therapy.
Serum Vitamin B12–Binding Proteins and Vitamin B12 Neutrophils contain vitamin B12–binding proteins, including transcobalamins I and III (synonym: R-type B12-binding protein or cobalophilin).359,360,361,362 Patients with myeloproliferative neoplasms have an increased serum level of vitamin B12–binding capacity, and the source of the protein is principally mature neutrophilic granulocytes.359,360 The increase in transcobalamin level and the resultant increase in vitamin B12 concentration are particularly notable in CML, although any increase in the number of neutrophilic granulocytes, as in leukemoid reactions, can be accompanied by an increase in serum vitamin B12–binding protein levels and vitamin B12 concentration.362 The serum vitamin B12 level in CML patients is increased on average to more than 10 times normal.363 The increase is proportional to the total leukocyte count in untreated patients and falls toward normal levels with treatment, although increased vitamin B12 levels commonly persist even after the white cell count is lowered to near normal with therapy.
Pernicious anemia and CML may rarely coexist. In this situation, the tissues are vitamin B12 deficient, but the serum vitamin B12 level may be normal because of the elevated level of transcobalamin I, a binder with a very high affinity for vitamin B12.363
Whole Blood Histamine Mean histamine levels are markedly increased in patients in chronic phase (median: approximately 5000 ng/mL) compared to healthy individuals (median: approximately 50 ng/mL); and, this elevation is correlated with the blood basophil count.364 Cases of exaggerated basophilia and disabling pruritus, urticaria, and gastric hyperacidity have occurred, associated with enormous increases (several hundredfold) of blood histamine concentration.365,366
Serum Lactic Dehydrogenase, Potassium, Calcium, and Cholesterol The level of serum lactic acid dehydrogenase (LDH) is elevated in CML.367 Pseudohyperkalemia resulting from the release of potassium from white cells during clotting368 and spurious hypoxemia or pseudohypoglycemia from in vitro utilization of oxygen or glucose by granulocytes can occur. Hypercalcemia369 or hypokalemia370 has occurred during the chronic phase of the disease, but such complications are very rare until the disorder transforms to acute leukemia. Elevated serum and urinary lysozyme levels are features of leukemia with greater monocytic components and are not features of CML.371 Serum cholesterol is decreased in patients with CML.372,373
Serum Angiogenic Factors Angiogenin, endoglin (CD105), vascular endothelial growth factor (VEGF), β-fibroblast growth factor, and hepatocyte growth factor are increased strikingly in the serum of CML patients.307,308,374,375
SPECIAL CLINICAL FEATURES
Either of two syndromes—thrombocythemia with the Ph chromosome and BCR-ABL1 rearrangement or thrombocythemia without a Ph chromosome but with the BCR-ABL1 rearrangement—may precede the overt signs of CML or its accelerated phase.376,377,378,379 In general, the disease closely mimics classic essential thrombocythemia initially: marked platelet elevation, extreme megakaryocytic hyperplasia, normal or mildly elevated white cell count, no or very slight myeloid immaturity in the blood, and minimal anemia. Minor bleeding, such as epistaxis, erythromelalgia, or signs of thrombosis, such as cerebral or limb ischemia, are occasionally present. In some cases, the absolute basophil count is mildly elevated. Approximately 5 percent of patients with apparent essential thrombocythemia have a Ph chromosome.376 In another study, two of 121 patients with essential thrombocythemia had BCR-ABL1 transcripts, and one of these patients also had a Ph chromosome in the marrow cells, whereas in a different study, four of 32 patients with thrombocythemia had low levels of BCR-ABL1 transcripts in blood cells. Approximately one in 20 patients with CML present with the features of essential thrombocythemia.377,378 Evolution to blast crisis may occur.380,381
A rare variant of BCR-ABL1–positive CML has been described in which the elevated white cell count is composed principally of mature neutrophils.382,383 The white cell count is lower (on average: 30 to 50 × 109/L) at the time of diagnosis than is the case with classic CML (median: 100 to 150 × 109/L). Moreover, patients with neutrophilic CML usually do not have basophilia, notable myeloid immaturity in the blood, prominent splenomegaly, or low leukocyte alkaline phosphatase scores. The cells of these patients have the Ph chromosome but have an unusual BCR-ABL1 fusion gene in that the breakpoint in the BCR gene is between exons 19 and 20. This breakpoint location results in fusion of most of the BCR gene with ABL1 (e19a2 type BCR-ABL1), which leads to a larger fusion protein (230 kDa) compared to the fusion protein in classic CML (210 kDa; see Fig. 89–3). This correlation between genotype and phenotype has not been observed in all cases.384 This variant usually has an indolent course, which may be the result of very low levels of mRNA for p230 and the undetectable or barely detectable p230 protein in cells.385
A small portion of patients with BCR-ABL1–positive CML have the breakpoint on the BCR gene in the first intron (m-bcr), resulting in a 190-kDa fusion protein instead of the classic 210-kDa protein observed in most patients with CML (see Fig. 89–3). The m-bcr molecular lesion is similar to that observed in approximately 60 percent of patients with BCR rearrangement-positive ALL. In patients with m-bcr CML, monocytes are more prominent, the white cell count is lower on average, and basophilia and splenomegaly are less prominent than in disease with classic BCR breakpoint (M-bcr). The few reported cases had a short interval before either myeloid or lymphoid blast transformation developed.386,387
Approximately 15 percent of patients present with symptoms or signs referable to leukostasis as a result of the intravascular flow-impeding effects of white cell counts greater than 300 × 109/L.249 Hyperleukocytosis is more prevalent in children with Ph chromosome–positive CML.250 The effects of total leukocyte counts from 300 to 800 × 109/L include impaired circulation of the lung, central nervous system, special sensory organs, and penis, resulting in some combination of tachypnea, dyspnea, cyanosis, dizziness, slurred speech, delirium, stupor, visual blurring, diplopia, retinal vein distention, retinal hemorrhages, papilledema, tinnitus, impaired hearing, and priapism.251 In asymptomatic patients with hyperleukocytosis, initial treatment with hydration and hydroxyurea usually can be used to decrease the white cell count. Hydroxyurea treatment should be designed to accomplish a gradual decrease in white cell count over a few days so as to avoid the tumor lysis syndrome. If signs of hyperleukocytosis are present, hydration, leukapheresis, and hydroxyurea can be used simultaneously; hydroxyurea dose should be selected to avoid exaggerated tumor lysis.
CML may emerge in patients with established chronic lymphocytic leukemia (CLL).388,389,390 A few patients have presented with simultaneous occurrence of the two diseases.391,392 A single case of lymphocytic leukemoid reaction simulating CLL that regressed as CML emerged has been reported.393 In some cases, the CLL lymphocytes did not contain the Ph chromosome, whereas the CML cells did, suggesting the presence of two independent clonal disorders.388,389,393,394 In other cases, the Ph chromosome was present in the myeloid and lymphoid cells, indicating a common origin.392 Patients may present with Ph chromosome–positive acute lymphoblastic leukemia and, following chemotherapy-induced remission, develop the features of typical CML.395
DIFFERENTIAL DIAGNOSIS
The diagnosis of CML is made based on the characteristic granulocytosis, white cell differential count, increased absolute basophil count, and splenomegaly coupled with the presence of the Ph chromosome or its variants (90 percent of patients) or a BCR rearrangement on chromosome 22 (>95 percent of patients).
Patients with other chronic hematopoietic stem cell diseases, such as polycythemia vera, essential thrombocythemia, or primary myelofibrosis, only occasionally have closely overlapping features. For example, the total white cell count is greater than 30 × 109/L in more than 90 percent of patients with CML and increases inexorably over weeks or months of observation, whereas the total white cell count is less than 30 × 109/L in more than 90 percent of patients with the three other classic chronic clonal myeloid diseases and usually does not change significantly over months to years. Polycythemia vera is associated with increased red cell mass and hemoglobin concentration and displays clinical signs of plethora; CML does not have these features. Patients with primary myelofibrosis invariably have marked teardrop poikilocytes and other severe red cell shape, size, and chromicity changes, as well as prominent nucleated red cells in the blood; CML rarely has these features. Patients with essential thrombocythemia have a platelet count greater than 450 × 109/L and usually only mild neutrophilia (<20 × 109/L); the slight neutrophilia distinguishes it from the proportion (approximately 25 percent) of CML patients with platelet counts greater than 450 × 109/L, who at the time of diagnosis have white cell counts above 25 × 109/L. In addition, patients with the clinical features of polycythemia vera or primary myelofibrosis do not have the Ph chromosome or BCR rearrangement in their blood and marrow cells, except in extremely rare cases. A very small proportion of patients with apparent essential thrombocythemia has BCR-ABL1 transcripts in their marrow and blood cells, and occasionally a Ph chromosome and may represent an atypical initial phase of CML (see “BCR-ABL1–Positive Thrombocythemia” above). The presence of a mutation in the JAK2 gene in more than 95 percent of patients with polycythemia vera is an important distinguishing feature (Chap. 84). The blood cells of approximately 50 percent of patients with primary myelofibrosis or essential thrombocythemia carry the JAK2 gene mutation and in those with primary myelofibrosis who do not, a significant proportion have a mutation in the calreticulin or the c–MPL gene (Chap. 86).
Increased awareness of the features of related disorders, such as chronic myelomonocytic leukemia (CMML) and chronic neutrophilic leukemia, and an appreciation that older patients are prone to atypical clonal myeloid diseases, have minimized the inappropriate diagnosis of Ph chromosome–negative CML, which should be avoided unless the clinical features are characteristic of classic CML and a masked Ph chromosome or BCR rearrangement is not found.
Reactive leukocytosis can occur with absolute neutrophil counts of 30 to 100 × 109/L. Usually these leukemoid reactions occur in the setting of an overt inflammatory disease (e.g., pancreatitis), cancer (e.g., lung), or infection (e.g., pneumococcal pneumonia). If the incitant is not apparent, the absence of granulocytic immaturity, basophilia, or splenomegaly, and the absence of BCR/ABL1 in blood cells virtually eliminates classic CML as a consideration.
The precise diagnosis of CML is helpful in estimating the patient’s prognosis, identifying the utility of TKIs, and assessing the timing of special therapies, such as allogeneic hematopoietic stem cell transplantation.
The Ph chromosome has been found rarely in patients with apparent polycythemia vera,396 polycythemia vera that later evolves into Ph chromosome–positive CML,397,398,399 primary myelofibrosis,400,401 and myelodysplastic syndrome (MDS).402,403 Molecular studies to determine the presence of the BCR-ABL1 were not performed in cases reported before 1985. Essential thrombocythemia with a Ph chromosome and/or BCR-ABL1 rearrangement in blood cells was discussed earlier (see “Special Clinical Features” above). Rare cases of aplastic anemia have presented with BCR-ABL1–positive cells or have evolved into BCR-ABL1 CML.404,405
THERAPY
Hyperuricemia and hyperuricosuria are frequent features of CML at diagnosis or in relapse.406 The need for treatment of hyperuricemia is a function of the elevated pretreatment serum uric acid concentration, blood white cell concentration, spleen size, and dose of cytolytic therapy planned. If these variables suggest a high risk for a significant amount of cell lysis, allopurinol 300 mg/day orally and adequate hydration to maintain a good urine flow should be instituted prior to therapy. Allopurinol is associated with a high frequency of allergic skin reactions and should be discontinued after the blood leukocyte count and spleen size have decreased and the risk of exaggerated cell lysis has passed. If hyperuricemia is extreme, usually over 9 mg/dL, rasburicase can be administered.407 Rasburicase is a recombinant urate oxidase that converts uric acid to allantoin. Rasburicase, unlike allopurinol, reduces the uric acid pool very rapidly, does not result in the accumulation of xanthine or hypoxanthine, and does not require alkalinization of urine facilitating phosphate excretion.408 Although the manufacturer recommends a dose every day for 5 days, several reports have indicated that one injection will produce a rapid and sustained decrease in serum uric acid, significantly decreasing the cost of therapy.409 Another alternative is to use allopurinol for a few days after one injection of rasburicase. A dose of 0.2 mg/kg of ideal body weight of rasburicase intravenously has been used.410
A TKI is now used as initial therapy in patients with CML. In cases where the white cell count is markedly elevated, hydroxyurea can be used prior to or in conjunction with a TKI. If rapid cytoreduction is required because of signs of the hyperleukocytic syndrome, leukapheresis and hydroxyurea often are combined.
Leukapheresis can control CML only temporarily. For this reason, it is rarely used in chronic phase CML and is useful in only two types of patients: the hyperleukocytic patient in whom rapid cytoreduction can reverse symptoms and signs of leukostasis (e.g., stupor, hypoxia, tinnitus, papilledema, priapism),249,250,251 and in the pregnant patient with CML who can be controlled by leukapheresis treatment without other therapy either during the early months of pregnancy when therapy poses a higher risk to the fetus or, in some cases, throughout the pregnancy.411,412 Because of the large body burden of leukocytes in marrow, blood, and spleen, and the high proliferative rate in CML, leukocyte reduction by apheresis is less efficient than in other types of leukemia.249,251 Leukapheresis reduces the burden of tumor cells subject to chemotherapeutically induced cytolysis and thus the production and the excretion of uric acid. In hyperleukocytic nonpregnant patients, leukapheresis is best used in conjunction with hydroxyurea to ensure rapid and optimal reduction in white cell count.
Hydroxyurea 1 to 6 g/day orally, depending on the height of the white cell count, can be used to initiate elective therapy.413 Urgent treatment of extraordinary total white cell counts may require higher doses. The dose of hydroxyurea should be decreased as the total white cell count decreases and usually is given at 1 to 2 g/day when the total white cell count reaches 20 × 109/L. The drug should be temporarily discontinued if the white cell count drops below 5 × 109/L. If hydroxyurea is being used in combination with a TKI, it is usually tapered and discontinued once a hematologic response to the TKI is observed.
Anagrelide can be used for platelet reduction in patients who present with elevated platelet counts. This agent acts directly to decrease megakaryocyte mass, and it can lead to a precipitous fall in platelet counts. In occasional patients who still have significant thrombocythemia after a TKI is initiated, combination with anagrelide is associated with a normalization of platelet counts.414
Imatinib mesylate (imatinib) was the first TKI developed, and it was approved by the FDA for initial therapy of CML in 2002. Subsequently, two second-generation TKIs, nilotinib and dasatinib, were approved for initial therapy in 2010. This approval was based upon superior cytogenetic and molecular response rates with nilotinib and dasatinib at benchmark time points and lower rates of conversion to accelerated or blast phase as compared with imatinib. Thus far, an overall survival advantage of dasatinib or nilotinib compared with imatinib has not been shown.415 Third-generation TKIs, bosutinib and ponatinib, are under study. Table 89–2 compares these inhibitors.
| Imatinib (Gleevec) | Nilotinib (Tasigna) | Dasatinib (Sprycel) | Bosutinib (Bosulif) | Ponatinib (Iclusig) | |
|---|---|---|---|---|---|
| Indications | First-line therapy (CP, AP, BP); relapsed/refractory Ph+ ALL | First-line therapy (CP), resistance or intolerance to imatinib (CP and AP) | First-line therapy (CP), resistance or intolerance to other TKIs (CP, AP, or BP); Ph+ ALL with resistance or intolerance to prior therapy | Second-line therapy (CP, AP, BP with resistance or intolerance) | Resistance or intolerance to prior TKI or Ph+ ALL resistant or intolerant to all other TKIs; all T315I + casesI |
| Usual dosing | CP 400 mg/day AP/BP/progression 600–800 mg/day | CP 300 mg BID AP/BP 400 mg BID | CP 100 mg/day AP/BP 140 mg/day | 500 mg/day | 45 mg/day |
| Common toxicities (nonhematologic) | GI disturbance, edema (including periorbital), muscle cramps, arthralgias, Hypophosphatemia, rash | Rash, GI disturbances, elevated lipase, hyperglycemia, low phosphorus, increased LFTs | Edema, pleural effusions, GI symptoms, rash, low phosphorus | GI (diarrhea), rash, edema, fatigue, low phosphorus, elevated LFTs | HBP, rash, GI, fatigue, headache |
| Other significant toxicities | Elevated LFTs (usually appear in first month); rare cardiac toxicity reported | Peripheral vascular disease, PT prolongation, pancreatitis | Pulmonary arterial hypertension, QTc prolongation | Arterial and venous thrombosis, pancreatitis, liver failure, ocular toxicity, cardiac failure | |
| Drug–drug interactions | CYP3A4 inducers decrease levels CYP3A4 inhibitors may increase levels It is an inhibitor of CYP3A4 and CYP2D6 Pgp substrate | CYP3A4 inhibitors increase levels CYP3A4 inducers may decrease levels Inhibitor of CYP3A4, CYP2C8, CYP2C9, CYP2D6 Induces CYP2B6, CYP2C8, and CYP2C9 | CYP3A4 inhibitors increase levels CYP3A4 inducer decrease levels Antacids decrease levels H2 antagonists/proton pump inhibitors decrease levels | CYP3A inhibitors and inducers may alter levels Acid-reducing medication may lower levels | Strong CYP3A inhibitors increased serum levels |
| Administration considerations | Taken with food | Taken on empty stomach; avoid food 2 hours before and 2 hours after dose | Can be taken with or without a meal | Taken with food | Taken with and without food |
| Black box warnings | None | QT prolongation and sudden death | None | None | Arterial thrombosis; hepatotoxicity |
| Other considerations | Approved in pediatric patients (340 mg/m2/day) in CP | Keep potassium, Mg, calcium, phosphorus repleted | Ascites and pericardial effusion can also occur; has CSF penetration | Has activity with T315I mutations; Available in U.S. through ARIAD PASS program |
Imatinib Patients with newly diagnosed, chronic phase CML can be started on imatinib, 400 mg/day by mouth. The goal of imatinib therapy is to decrease the cells bearing the t(9;22) translocation (leukemic cells) to the lowest levels possible, during which process normal (polyclonal) hematopoiesis is restored. The efficacy of imatinib is judged by measuring three benchmarks: hematologic response, cytogenetic response, and molecular response as defined in Table 89–3.416,417 These benchmarks are used to determine its maximal therapeutic effect. The time to achieve a maximal effect is variable, but as long as a patient is having a continued reduction in the size of the leukemic clone as judged by cytogenetic or PCR measurements, and has met response benchmarks, the drug is continued at 400 mg/day. If the patient stops responding before a complete cytogenetic remission or complete molecular remission is achieved, the dose can be increased to 600 mg/day or to 800 mg/day (400 mg every 12 hours), if tolerated. Alternatively, another TKI can be used. About two-thirds of patients who do not have a significant hematologic response or who relapse while receiving imatinib at a dose of 400 mg/day achieve a complete or partial hematologic response with higher doses, but few cytogenetic responses occur.418 Some patients without a cytogenetic response can enter a partial or complete cytogenetic response(CCyR) with higher doses of imatinib. Unfortunately, the responses to higher doses of imatinib in patients lacking a hematologic or cytogenetic response at 400 mg/day usually are transient.419,420
| Complete hematologic response (CHR) | White cell count <10 × 109/L, platelet count <450 × 109/L, no immature myeloid cells in the blood, and disappearance of all signs and symptoms related to leukemia (including palpable splenomegaly) lasting for at least 4 weeks. |
| Minor cytogenetic response (mCyR) | >35% of cell metaphases are Philadelphia (Ph) chromosome–positive by cytogenetic analysis of marrow cells. |
| Partial cytogenetic response (pCyR) | 1–35% of cell metaphases are Ph chromosome–positive by cytogenetic analysis of marrow cells. |
| Major cytogenetic response (MCyR) | <35% of cell metaphases contain the Ph chromosome by cytogenetic analysis of marrow cells. |
| Complete cytogenetic response (CCyR) | No cells containing the Ph chromosome by cytogenetic analysis of marrow cells. |
| Major molecular response (MMR) | BCR-ABL1/ABL1 ratio <0.1% or a 3-log reduction in quantitative polymerase chain reaction (qPCR) signal from mean pretreatment baseline value, if International Standard (IS)-based PCR not available. |
| Complete molecular response (CMR) | BCR-ABL1 mRNA levels undetectable by qPCR with assay sensitivity at least 4.5 logs below baseline (IS). |
Several studies have examined use of imatinib at doses higher than 400 mg/day. Patients with newly diagnosed chronic phase CML treated with imatinib, 800 mg/day, administered in two 400-mg doses, every 12 hours, had a frequency of 90 percent CCyR, and 96 percent had at least a major cytogenetic response (MCyR). At a median of 15 months, no patients had progressed and 63 percent showed blood BCR-ABL1/ABL1 percentage ratios of less than 0.05 percent. Twenty-eight percent of patients had undetectable BCR-ABL1 blood levels.421 In one trial, major molecular remission (MMR) at 12 and 24 months was higher in those receiving doses of imatinib greater than 600 mg/day.422 In another trial, patients receiving 400 mg twice per day had a MCyR of 90 and 96 percent at 12 and 18 months, respectively. MMR rates were 48 percent and 54 percent at 6 and 12 months, respectively. These results compared favorably to historical data in the IRIS (International Randomized Study of Interferon) trial studying 400 mg/day of imatinib, but myelosuppression, rash, fatigue, and musculoskeletal symptoms were greater with these higher doses. Responses were, also, more rapid with the higher doses. More edema, gastrointestinal symptoms, rash, and myelosuppression occurred at the higher doses.423 Despite these reports the current starting dose is customarily 400 mg/day, balancing both effectiveness and tolerability in newly diagnosed patients. Moreover, the more rapid response with higher doses of imatinib may not translate into better long-term results.424,425 For example, in another trial, MMR and CCyR at 12 months were not significantly different between standard and high-dose patients, although patients in higher-risk categories based on Sokal scores, fared better with high-dose imatinib.424 (See “Course and Prognosis” below for an explanation of the Sokal Score.)
Doses of imatinib lower than 400 mg/day result in fewer CCyR and a shorter duration of that response. Patients who are older and who have lower body weight may only tolerate a lower dose, but they are less likely to achieve a CCyR.426 If however, a patient is on a lower dose (e.g., 300 mg/day) for a special reason (body size or tolerance level) and achieves a complete hematologic response (CHR) and CCyR within 12 months of onset of therapy, acceptable outcomes without excess toxicity can result.427
Some patients have been followed for up to 8 years on imatinib in the IRIS trial (IFN vs. STI571).428 With median followup of 60 months, the best observed MCyR and CCyR rates were 89 percent and 82 percent, respectively. Only 7 percent had progressed to accelerated or blast phase, and overall survival rate was 89 percent. The best MMR rate was 86 percent during the 8-year followup. No patient with an MMR at 12 months progressed to accelerated or blast phase. By 8 years of follow up on the IRIS trial, 22 percent of patients had discontinued imatinib treatment because of an unsatisfactory response or toxicity, and only 55 percent remained on study. The 5-year probability of remaining in MCyR while receiving imatinib was approximately 60 percent. Achieving a CCyR correlated with progression-free survival, but achieving a MMR conferred no further survival benefit.429
Patients with variant Ph chromosome translocations have a similar prognosis to that of patients with classic Ph chromosome translocations who are treated with imatinib.430 (See Fig. 89–3 for a diagram of breakpoints.) Patients with the e13a2, p210BCR-ABL translocation respond well to imatinib, with similar rates of complete cytogenetic remission.431 The e13a2 transcript may be more sensitive to imatinib than the e14a2 transcript.432 In a patient with both e1a2 and e14a2 fusion transcripts, only the p210 e14a2 transcript disappeared, whereas the e1a2 transcript persisted during progression to blast phase. No mutation in the kinase domain of ABL1 was found.433 Thus, different clones in a patient may have a different sensitivity to imatinib. Deletions of the derivative chromosome 9 do not influence the response and outcomes in CML chronic phase when using imatinib.434
More than 80 percent of children with chronic phase CML who are treated with imatinib, 260 to 570 mg/m2, enter a complete cytogenetic remission. Imatinib is now approved for use in pediatric patients. Weight gain is the most common side effect of imatinib.435 In patients who were older than age 60 years, similar cytogenetic response rates and survival rates were noted as in younger patients in the late chronic phase who were treated concurrently, suggesting that age is not usually a factor in response.436,437
Imatinib is usually tolerated. Most adverse effects are manageable and seldom require permanent cessation of therapy. Reduction to subtherapeutic doses is not recommended; it is better to interrupt therapy for a time.438
Myelosuppression is common, especially at treatment onset when the CML clone accounts for most of the blood cells. Dose reduction to less than 300 mg/day is not advisable for myelosuppression. The drug should be stopped until blood counts recover. G-CSF or GM-CSF can prevent or treat neutropenia.439,440 Platelet transfusion may be used for severe thrombocytopenia. Patients with imatinib-induced chronic cytopenias have inferior responses.441 Myelosuppression is an independent adverse factor for achieving cytogenetic responses with imatinib.442 Erythropoiesis-stimulating agents may be used to raise hemoglobin levels, and their use does not appear to affect CML outcomes, but may increase the risk of thrombosis.443 Severe irreversible marrow aplasias after imatinib exposure can occur.444
The main side effects noted with imatinib include fatigue, edema, nausea, diarrhea, muscle cramps, and rash.445 Elevated hepatic transaminases can occur. Mild transaminase elevations often respond to glucocorticoid use.446 Hepatotoxicity is uncommon, occurring in approximately 3 percent of patients, usually within 6 months of onset of imatinib use. Acute liver failure has been described.447 The severe periorbital edema occasionally observed is postulated to be a drug effect on the function of platelet-derived growth factor receptor (PDGFR) and KIT expressed by dermal dendrocytes. Surgical decompression of severe edema rarely has been required.448 Although no effects on spermatogenesis have been reported, women of childbearing age are at risk of teratogenic effects on their fetus.448
Weight gain is associated with imatinib use.449 Patients with renal impairment require lower doses of imatinib.450 Hypophosphatemia451 and altered bone and mineral metabolism have occurred.452,453 Cutaneous reactions with imatinib therapy occur in approximately 15 percent of patients.454 Except for severe reactions (approximately 5 percent of patients), such as Stevens-Johnson syndrome, exfoliative dermatitis, and erythema multiforme, cutaneous reactions rarely require permanent discontinuation of therapy. With milder reactions, concomitant glucocorticoid therapy or brief discontinuation of imatinib with gradual reintroduction at a lower dose and then a gradual increase in dose can be accomplished.456,457 With very mild cases, concurrent treatment with antihistamine or other symptomatic therapy may be successful. Oral desensitization regimens have been described that allow some patients to continue imatinib therapy. Hair depigmentation458 and hypopigmentation of the skin,459 probably related to the inhibition of the KIT receptor tyrosine kinase by imatinib, have been reported.
Imatinib has been found to cause regression of marrow fibrosis.460 One study found that the extent of marrow fibrosis in CML is not a prognostic factor with imatinib therapy,461 whereas another study observed that although imatinib reverses marrow fibrosis in patients with CML, it does not change the unfavorable prognosis associated with fibrosis.462
Imatinib reverses exaggerated VEGF secretion in patients with CML,463 and it may reverse exaggerated marrow angiogenesis.464 It can reduce marrow cellularity and normalize morphologic features regardless of cytogenetic response. BCR-ABL1–positive cells persist in patients despite prolonged treatment responses with imatinib.465
Mean plasma trough concentration of imatinib and its metabolite, CGP74588, obtained at about 1 month (presumptive steady-state) was 979 ± 530 ng/mL. The rate of CCyR and MMR was higher within the highest quartiles of imatinib trough levels.466 Some therapists suggest that imatinib plasma levels be checked in cases of suboptimal response in order to adjust the dose, but access to this monitoring is not routinely available.467 Comedications and population covariates, such as body weight and white cell count, had no, or minimal, effect on imatinib clearance.468 Patients with CML on hemodialysis have been successfully treated with imatinib.469 Therapy interruptions and nonadherence with oral imatinib usage are common, and patient education and close monitoring are important to ensure compliance.470
Dasatinib Dasatinib is a second-generation oral BCR-ABL1 inhibitor with dual inhibition of ABL1 and SRC.471 It can bind to both the active and inactive conformation of the ABL1 kinase domain, so it may be affected by mutations resulting in resistance.471
Dasatinib was first studied for use in initial therapy in a phase II trial that accrued 62 patients. Ninety-eight percent achieved a CCyR, and the median time to CCyR was 3 months. The MMR rate was 82 percent. Responses were durable, and the recommended treatment schedule based on a safety profile was 100 mg, once daily.472 A randomized, phase III trial compared the efficacy of dasatinib to imatinib.473 In this study, 259 patients received dasatinib, 100 mg/day, and 260 received imatinib, 400 mg/day. After 12 months of followup, the rates of CCyR by 3, 6, and 9 months were 54, 73, and 78 percent, respectively, for patients on dasatinib as compared to 31, 59, and 67 percent, respectively, for those on imatinib. The 24-month CCyR was 80 percent for patients using dasatinib, as compared to 74 percent for those on imatinib. The MMR showed a similar trend, and the median time to MMR was 15 months for those using dasatinib, compared to 36 months for those using imatinib.
Long-term followup data have confirmed faster and deeper responses to dasatinib as compared to imatinib.474 At 4 years, 76 percent of dasatinib-treated patients had attained a MMR (BCR/ABL1 <0.1 percent) as compared to 63 percent using imatinib. At 3, 6, and 12 months, more patients achieved molecular responses using dasatinib than those using imatinib. There were fewer patients who progressed to accelerated or blast phase using dasatinib as compared to imatinib. To date, there is no difference in progression-free survival or overall survival between the two groups.474 In a randomized study, with a minimal followup of 3 years, the proportion of patients with BCR-ABL1 transcript levels less than 10 percent was higher in those using dasatinib as compared to those using imatinib.475 Better responses were observed at 3, 6, and 12 months in the patients using dasatinib. The achievement of an early molecular response was predictive of improved progression-free and overall survival.476
Toxicity of Dasatinib Most grade 3 or 4 adverse events with dasatinib are hematologic and all cell lines can be affected. Some patients may have bleeding from inhibition of platelet aggregation.477 Adverse events noted less frequently with dasatinib than imatinib include nausea, vomiting, myalgia, rash, and fluid retention, including superficial edema. Pleural effusions can be seen with dasatinib use. In one trial, 14 percent of patients had grade 1 or 2 pleural effusions at 24 months, but grade 3 or 4 effusions occurred in only 2 patients. This toxicity did not affect drug efficacy. Dasatinib may also increase the risk of pulmonary arterial hypertension at any time, and this is an indication to discontinue dasatinib.478 Dasatinib may prolong the QTc interval, and it should be used with caution in those who have long QT syndrome or those taking drugs that may lengthen the QT interval.474 Hypophosphatemia was found in 7 percent of cases.474
Dasatinib is metabolized primarily by hepatic cytochrome P450 (CYP) 3A4 enzymes, so inducers of this enzyme may decrease the effective dose, and inhibitors may increase the effective dose. Increases or decreases in administered dose may be needed to compensate for these effects. Antacids can also reduce dasatinib effects.479 Lymphocytosis from the clonal expansion of NK/T cells has occurred during dasatinib treatment.480 In a population of dasatinib-treated patients with large granular lymphocyte expansion, 90 percent had T-cell receptor delta rearrangements, the functional significance of which is unknown.481 Lymph node follicular hyperplasia has been noted on dasatinib therapy.482 Unlike the case with imatinib, dasatinib cellular uptake is not affected by octamer-binding protein-1 (OCT-1) activity, which is a substrate of the efflux proteins, ABCB1 and ABCG2. Resistance to dasatinib is often found with point mutations in ABL1 at residue 315 or 317.
Nilotinib Unlike dasatinib, nilotinib is a selective, orally bioavailable, ATP-competitive inhibitor of BCR-ABL1 which is 20 to 50 times more potent than imatinib in vitro.483 Like imatinib, it does not induce apoptosis in CD34+ CML cells.484 As with dasatinib, nilotinib was first tested as initial therapy in phase II trials,485 which were followed by a randomized phase III trial. In one phase II trial, 51 patients received nilotinib 400 mg, twice a day, and 98 percent entered CCyR and 76 percent had a MMR by 6 months.486 The phase III trial compared nilotinib, 300 mg twice daily, 400 mg twice daily, and imatinib 400 mg daily.487 At 12 months, the MMR which had been chosen as the primary end point, was 44 percent (nilotinib 300 mg dose), 43 percent (nilotinib 400 mg dose), and 22 percent (imatinib 400 mg daily). The CCyR rates were 15 percent higher with nilotinib than imatinib. The rate of progression to accelerated or blast phase was 4 percent at 1 year with imatinib and less than 1 percent with nilotinib. These improvements were observed in each prognostic group based on Sokal risk groups. The patients using either 300 or 400 mg doses had minimal differences, so in 2010, nilotinib was approved at a dose of 300 mg twice daily for initial CML therapy. At 4 years of followup, more patients using either dose of nilotinib achieved a MMR than those using imatinib (73 and 70 percent vs. 53 percent).488 The rates of progression to accelerated and blast phase were also lower for nilotinib than imatinib. The 4-year freedom-from-progression and overall-survival rates were not different between the two groups. MMR rates at 3 years were higher for those patients using nilotinib, 300 mg, twice per day, in the three risk groups of Sokal (low risk, 79 percent; intermediate risk, 76 percent; and high risk, 52 percent progression as compared to those using imatinib (65, 55, and 30 percent progression, respectively).488 There was a reduced incidence of BCR-ABL1 mutations in in patients using nilotinib compared to those using imatinib. Nilotinib use led to fewer (less than half as many) treatment-emergent BCR-ABL1 mutations than did imatinib treatment, and to reduced rates of progression to accelerated phase and blast crisis in patients with these mutations.489
Toxicity of Nilotinib Nilotinib is rarely associated with edema or muscle cramps. Grades 3 and 4 cytopenias were seen in 29 percent of cases in one trial.487 Grade 3 or 4 elevations in lipase, bilirubin, and hyperglycemia were observed in 17, 8, and 12 percent of patients, and hypophosphatemia was seen in 16 percent. QTc prolongation can occur, so one should monitor the electrocardiogram (EKG) readings for 7 days after starting therapy and with dose changes.483 Electrolyte abnormalities should be corrected at outset of treatment. Nilotinib may be associated with an increased risk of peripheral vascular disease, which may be arterial or venous.490 If thrombosis occurs, it should no longer be used for therapy.
Bosutinib and Ponatinib These TKIs are not approved for use in initial therapy. In the one trial, which compared efficacy for bosutinib, 500 mg once daily, with imatinib, 400 mg daily in newly diagnosed chronic phase patients, the primary end point of CCyR at 12 months was 70 percent for bosutinib and 63 percent for imatinib.491,492 Results from followup of MMR rates, transformation rates, and durability of remission are not yet available.
Summary of Tyrosine Kinase Inhibitor Selection for Initial Therapy of Chronic Phase Chronic Myelogenous Leukemia
The goal of initial TKI therapy is to achieve a CCyR within 12 months or no later than 18 months of therapy, and to prevent progression to the accelerated or blast phase. How best to achieve these goals remains controversial. Hence, the National Comprehensive Cancer Network (NCCN) guidelines list imatinib, nilotinib, and dasatinib as all being acceptable TKIs for initial treatment of CML.492 Many clinicians would choose a second-generation TKI, given the rapidity and depth of response and the lower rates of transformation to advanced phases of the disease, but others use imatinib because of the lack of proof of prolongation of survival with nilotinib or dasatinib. In those with intermediate- or high-risk disease as assessed by the Sokal and Hasford models (see “Course and Prognosis” below for details of these scores), nilotinib or dasatinib may be preferred over imatinib to achieve rapid, better responses—the “hit hard, hit early” approach.493 Thus, until survival data are available, any one of the three approved TKIs may be used for initial therapy. Choice of agent may be dependent upon cost considerations, ease of administration, patient risk scores or perceived risk,494 and the drug’s side-effect profile (see Table 89–2).
Table 89–3 contains definitions of hematologic, cytogenetic, and molecular responses. The guidelines for periodic monitoring patients who are in chronic phase and receiving TKI therapy are shown in Table 89–4. The median BCR-ABL1 levels for imatinib-treated patients can decrease over at least 5 years. Table 89–5495 lists the milestones at 3, 6, 12, and 18 months expected of patients as indicators of an appropriate response in patients treated initially with a TKI.434 There is variation in an individual patient’s time to maximal response. Consequently, if a patient has not met those precise milestones but shows a continued decrease in the proportion of Ph chromosome–positive cells on cytogenetic examination of marrow, or if in a CCyR, a continued decrease in the level of the PCR signal for BCR-ABL1 and is near the benchmark, the treatment can be continued. Only (1) failure to meet the benchmarks at 3, 6, or 12 months or (2) loss of response as defined as loss of a CHR or CCyR, (3) development of new cytogenetic abnormalities, (4) acquisition of a BCR-ABL1 mutation, or (5) an increase in the BCR-ABL1/ABL1 ratio of 1-log or more on serial RT-PCR testing or into the range associated with reappearance of the Ph chromosome on G-banding should generate a change in treatment to limit the risk of progression of the disease. Because of the variability in PCR testing, those changes should be confirmed within 1 month. Patients who have 100 percent Ph chromosome–positive cells after 6 months of therapy have a minimal chance of achieving a MCyR or CCyR and may be offered allogeneic stem cell transplantation, if applicable.417,496
1. At diagnosis, before starting therapy, obtain Giemsa-banding cytogenetics and measure BCR-ABL1 transcript numbers by qPCR using marrow cells. If marrow cannot be obtained, use FISH on a blood specimen to confirm the diagnosis. 2. At 3, 6, 9, and 12 months after initiating therapy, measure qPCR for BCR-ABL1 transcripts. (If qPCR using the International Standard is not available, perform marrow cytogenetics.) If there is a rising level of BCR-ABL1 transcript or 1 log increase after MMR achieved, qPCR should be repeated in 1 to 3 months. 3. At 12 months obtain marrow cytogenetics for cells with Ph chromosome if no CCyR or MMR. 4. Once CCyR is obtained, monitor qPCR on blood cells every 3 months for 3 years and then every 4 to 6 months, thereafter. If there is a rising level of BCR/ABL1 transcripts (1 log increase after MMR achieved), repeat quantitative PCR in 1 to 2 months for confirmation. 5. These guidelines presume continued response to a TKI until CCyR achieved. If this does not occur see text for approach. 6. Mutation analysis should be performed with loss of chronic phase, loss of any previous level of response, inadequate initial response (BCR/ABL1 transcripts >10%) at 3 or 6 months or no CCyR at 12 or 18 months, and a 1-log increase in BCR/ABL after MMR once achieved. |
| Time of Observation (months) | Disease Response | ||
|---|---|---|---|
| Unsatisfactory | Suboptimal Response/Warning | Optimal Response | |
| 3 | No CHR and/or Ph+ >95% | BCR/ABL1 >10% and/or Ph+ 36–95% | BCR/ABL1 ≤10% and/or MCyR |
| 6 | BCR/ABL1 >10% and/or no MCyR | BCR–ABL1 1–10% and/or MCyR | BCR/ABL1 <1% and/or CCyR |
| 12 | BCR/ABL1 >1% and/or no CCyR | BCR–ABL1 <0.1–1% | BCR/ABL1 <0.1% |
| 18 | No CCyR | CCyR if no MMR | CCyR or MMR |
Achieving a cytogenetic response is associated with progression-free survival in patients treated with imatinib and is an important goal of therapy (97 percent in those with a CCyR vs. 81 percent in those without a MCyR).497 At 5 years of followup, of those patients who achieved CCyR on imatinib in the IRIS study, only 3 percent had progressed to accelerated or blast phase during treatment.428 A CCyR at 1 year may be the major predictor for overall survival and progression-free survival.498 In patients in chronic phase treated with imatinib, nilotinib, or dasatinib, early responses (at 3 months) in BCR-ABL1 transcript reduction or decrease of Ph chromosome frequency predicted for better outcomes as measured by event-free or overall survival. For example, patients with less than 10 percent BCR-ABL1 transcripts at 3 months had a 3-year event-free survival of 95 percent or greater, whereas those with greater than 10 percent transcript level at 3 months had a 61 percent event-free survival.499
Molecular response is determined by the decrease in BCR-ABL1 mRNA by PCR. This is the only means to measure the depth of the response once a CCyR is attained. The achievement of MMR after treatment with imatinib is associated with durable long-term CCyR and a lower rate of disease progression. Only 5 percent of patients achieving a MMR with imatinib lost a CCyR compared to 37 percent who did not achieve that degree of response.500 Also, the 5-year followup of the IRIS trial showed that no patient with a CCyR and MMR at 12 months had progressed to a more advanced phase of the disease.497 The IRIS study 7-year followup also showed that in those with a MMR at that point in time, progression was rare. The estimated event-free survival was 95 percent for those with MMR at 18 months compared with 86 percent in those without a MMR.501 To date, there is no evidence that a change of therapy would improve survival in those with CCyR but not a MMR. In those with stable CCyR after treatment with a second-generation TKI, achievement of MMR may not have significance as a predictor of survival.499
The time taken to achieve MMR is also thought to have prognostic significance. In the IRIS study, the chance of disease progression was higher in those who failed to achieve a 1-log reduction in BCR/ABL1 transcripts by 3 months or a 2-log reduction by 6 months.502 A BCR-ABL1 transcript level greater than 10 percent after 3 months is a significant predictor for long-term outcomes.503 In another analysis, chronic phase patients treated with imatinib 400 mg/day who had BCR-ABL1 transcripts of 9.84 percent or less at 3 months had better overall, progression-free, and event-free survival at 8 years than did those with values of 9.84 percent or greater.503 Also, there is evidence that early molecular response to initial therapy with dasatinib or nilotinib in patients with CML is a predictor of overall response. In one trial with dasatinib, patients with BCR-ABL1 transcripts of 10 percent or less at 3 months had significantly better 5-year progression-free (92 vs. 67 percent) and 4-year overall survival than did those with greater than 10 percent transcripts (95 vs. 83 percent).504 In another study with nilotinib, patients with BCR-ABL1 of 10 percent or less at 3 months also had improved 4-year progression-free survival compared to those with BCR-ABL1 greater than 10 percent at 3 months (95 vs. 85 percent).505 Progression was defined as transformation to accelerated or blast phase.
Rising BCR-ABL1 Transcript Levels Rising BCR-ABL1 transcripts can indicate a new mutation or cytogenetic relapse. A significant change has been defined as either a twofold increase, serially increasing levels, or a 1-log increase.492,495 There are no current recommendations for therapy changes based on an increase in BCR-ABL1 transcripts. Serial increases or increases of 1-log or more should trigger ABL mutational analysis and frequent monitoring of BCR-ABL1 transcripts.
Suboptimal Therapeutic Responses The initial European Leukemia Network guidelines defined suboptimal response as no cytogenetic response at 3 months, less than a partial cytogenetic response (PCyR) at 6 months, a PCyR at 12 months, and less than a MMR at 18 months.495,506 The significance of a suboptimal response is dependent on the cause; for example, this may be insignificant if the result of drug intolerance or noncompliance as opposed to drug resistance (see “Acquired Resistance” below). The significance also depends on time of suboptimal response with earlier time points indicating a worse prognosis. Currently, CCyR and PCyR at 3 months are considered optimal and suboptimal responses. Suboptimal responses are labeled as a “warning” response in the Network guidelines.495 These response levels may trigger ABL mutation analysis or closer monitoring.
Because of its earlier development, most of these data are obtained with imatinib treatment. Clonal abnormalities in cells lacking a detectable Ph chromosome or BCR-ABL1 rearrangements have been detected in patients undergoing imatinib therapy who previously were treated with IFN-α.507,508 These cytogenetic changes were noted in seven patients at a median of 13 months of imatinib therapy, and trisomy 8 was the most frequent abnormality. All of these patients had MCyRs to imatinib.507 The presence of additional chromosomal abnormalities is considered to be a feature of the accelerated phase of CML. In some patients, clonal evolution may be related to imatinib resistance.509 Clonal abnormalities may be present in up to 10 percent of patients taking imatinib.510 Some of these cases may be associated with a MDS, especially in those patients with previous exposure to cytarabine and idarubicin. The antiproliferative effect of imatinib allows restoration of polyclonal hematopoiesis in CCyR, which could permit the manifestation of a Ph chromosome–negative disorder.511 Some investigators have found that, with the possible exception of +8, +Ph, and i(17), additional chromosomal abnormalities at diagnosis are not associated with an inferior outcome to imatinib therapy.512,513 In contrast, another group found that development of trisomy 8 in patients taking imatinib, while associated with pancytopenia, did not result in signs of disease progression. In a series of 34 CML patients who developed Ph chromosome–negative clones while taking imatinib, the most common abnormalities were trisomy 8 and monosomy 7. In 11 of these patients, no archival evidence of these clones was present before imatinib therapy was initiated, and none of the patients developed myelodysplasia.514 In patients treated at diagnosis with imatinib, 9 percent developed chromosomal abnormalities in Ph chromosome-negative metaphases. These appeared at a median of 18 months, and the most common abnormalities were −Y and +8. Most were temporary and disappeared within 5 months. Only one patient with −7 progressed to acute myelogenous leukemia (AML).515 Cytogenetic clonal evolution may not be an important impediment to achieving a MCyR or CCyR with imatinib, but it is an independent poor prognostic factor for survival of patients in chronic and accelerated phases of CML.516 Imatinib therapy may overcome the poor prognostic significance of derivative chromosome 9 in CML.517
Noncompliance with therapy is associated with poorer outcomes. In one trial, patients with a suboptimal response had higher nonadherence (23 percent) than did those with optimal responses (7 percent).518 In another study, adherence was the only independent predictor for achieving a complete molecular response (CMR) on imatinib. Patients with an adherence rate of 85 percent or less had a greater chance of losing their CCyR at 2 years (27 percent) than did those with better adherence (1.5 percent). They, also, had a lower chance of remaining on imatinib.519 Adherence has also been correlated with level of molecular response. In patients using a TKI for about 5 years, median adherence was 98 percent (range: 24 to 100 percent). If adherence was greater than 90 percent, there was a higher probability for a 3-log reduction in BCR/ABL1 transcripts and a CMR. If adherence was less than 80 percent, no MMRs occurred.520 The poor adherence to second-generation TKIs has not been studied for a sufficient duration to determine its impact. Management of side effects is of importance in maintaining a high rate of adherence.
The development of resistance to imatinib is not surprising.521,522 Its specificity and “snug fit” into the ABL1-kinase pocket provide the ideal circumstance for resistance.523 Some cases demonstrate primary resistance to imatinib, and gene profiling has demonstrated differential expression of about 46 genes in responders compared to nonresponders.524 Even in patients with CCyR, malignant progenitors at the LTC-IC stage persist. Chronic phase CML stem cells are resistant to imatinib and are genetically unstable.525 These cells have a high level of BCR-ABL1 transcription, and they are thought to express transporter proteins that result in abnormal imatinib flux.526 Mathematical models suggest that imatinib rapidly eliminates leukemic progenitors, but does not deplete CML stem cells. Such models predict the probability of developing resistant mutations and can estimate the time that resistance will emerge.527 Several potential mechanisms of resistance include BCR-ABL1 amplification in the presence of imatinib, P-glycoprotein–mediated drug efflux, altered drug metabolism, acquisition of BCR-ABL1–independent signaling characteristics, and point mutations in the ABL1 kinase domain that decrease imatinib binding. Each of these mechanisms of resistance may have clinical relevance.
Primary Resistance Primary resistance to imatinib is defined as lack of CHR at 6 months or failure to achieve any level of cytogenetic response at 6 months, a MCyR at 12 months, or a CCyR at 18 months. This may occur in 15 to 25 percent of patients. Primary resistance may often be the result of inadequate plasma concentration because of binding of the drug to proteins, such as albumin or α1-acid glycoprotein. In an analysis of the IRIS study, plasma levels of imatinib following the first month of treatment proved to be a significant predictor for clinical response. Plasma levels are not available for clinical use, however, so these have minimal influence on treatment decisions when responses are not as expected. Only one gene, prostaglandin-endoperoxide synthase 1/cyclooxygenase1 (PTGS1/COX1) was found to differentiate primary imatinib resistance. Eleven genes were associated with secondary resistance after imatinib therapy in those without an ABL1 kinase domain mutation.528 Expression of the OCT-1, which mediates drug influx, is thought to be important for imatinib but not dasatinib effectiveness.529,530 Many CML patients who have a suboptimal response to imatinib have low OCT-1 activity, but this can be overcome with higher doses of imatinib or use of dasatinib, which uptake is not dependent on OCT-1 expression.530 OCT-1 expression is associated with MMR at 12 and 24 months, and it is a predictor of the long-term risk of resistance and of transformation in patients treated with imatinib.531 CML CD34+ cells overexpress the drug transporter ABCG2, and imatinib, dasatinib, and nilotinib are substrates for ABCB1 and ABCG2. Overexpression of MDR1 has been associated with decreased intracellular concentration of imatinib.532
Acquired Resistance Acquired resistance is that which occurs after exposure to TKIs or other treatments. Amplified gene expression and increased BCR-ABL1 protein expression are often reported in resistant patients. Duplication of the Ph chromosome and isodicentric chromosomes are a possible mechanism of resistance to imatinib.533,534
Mutations in the ABL1 kinase domain are a frequent mechanism of resistance. Kinase domain mutations were the only independent predictor for the loss of CCyR and progression when compared to those without a mutation.535 Mutations in the ABL1 kinase domain may predate imatinib treatment,536 and several BCR-ABL1 kinase domain mutants associated with imatinib resistance remain sensitive to the drug, suggesting a need for characterization before a resistant phenotype can be attributed to the given mutation.537 The mutant clone does not always have a proliferative advantage.538 Some of these mutations may lie outside the kinase domain, and more than 40 such mutations have been described. Screening early phase CML patients for mutations before the start of imatinib therapy is not cost-effective because of their low incidence, but in patients with evidence of an increase in CML cells while on imatinib, mutation searches are indicated.539 BCR-ABL1 kinase domain point mutations are rare in those who have had good cytogenetic responses to imatinib, and when detected in that setting, their presence does not always predict relapse.540 Mutations in the ABL1 portion of the BCR-ABL1 oncogene are present in approximately 40 percent of patients who do not achieve a CHR or CCyR to imatinib. ABL1 mutations were found in those patients with both primary and acquired resistance. Amino acid substitutions in seven residues accounted for 85 percent of all mutations associated with resistance.541 The mutations most associated with resistance are Thr315ILe, Gly250Glu, Glu255Lys, and Thr253His substitutions. Few of the described mutations directly affect imatinib binding.542 Mutations in the ABL1–ATP phosphate-binding loop (P-loop) are most closely associated with a poor prognosis,543 and these P-loop mutations predict for disease progression. Overall survival is worse for P-loop and for T315I mutations, but not significantly different when other mutations are present.544
Ultra-deep sequencing approaches have shown that routine Sanger sequencing underestimates BCR-ABL1 mutation in 55 percent of samples where the missed mutations had low abundance.545 Mass spectrometry can detect a 0.05 to 0.5 percent level of mutations, as well.546 For many mutations, the concentration that inhibits 50 percent (IC50) of various TKIs and response have not been documented.547
Second-generation BCR-ABL1 inhibitors (see “Dasatinib and Nilotinib” below) are able to overcome imatinib-resistant mutants, with the exception of the T315I mutations (Table 89–6). Mutations F317L and V299L are resistant to dasatinib and mutations Y253H, E255K, and F359I are resistant to nilotinib. Ponatinib was active against T315I and against other BCR-ABL1 mutations resistant to dasatinib or nilotinib.548 Ponatinib may be effective against individual ABL1 point mutations but may not overcome some compound mutations, which are two or more mutations in the same BCR/ABL1 molecule. Some mutations may be polyclonal as well.549 The T315I mutation results in steric hindrance, which precludes access of some TKIs to the ATP-binding pocket of the ABL1 kinase domain.550 In a series of 27 patients with T315I mutation, survival was dependent on stage of disease, with many of the chronic phase patients described as having an indolent course.551 In addition to ponatinib, agents such as IFN-α and homoharringtonine have also been proposed as therapy for those with the T315I mutation.552
| Mutation | Treatment Recommendation |
|---|---|
| T315I | Ponatinib or stem cell transplant |
| V299L, T315A, F317L/V/I/C | Consider nilotinib over dasatinib; bosutinib and ponatinib may be effective |
| F359V/C/I | Consider dasatinib rather than imatinib; bosutinib and ponatinib may be effective |
| Others | Any tyrosine kinase inhibitor not previously used or high-dose imatinib |
In some cases of resistance associated with imatinib, other signal pathways independent of BCR-ABL1 may become important in cell proliferation.553 These include heat shock protein (hsp) 70,554 survivin,555 LYN kinase,556 SRC,557 and GRB2, among others.558
Dose escalation, combination therapy, and treatment interruption have been proposed as means to overcome drug resistance.559 Combination therapy from the outset560 also has been proposed to prevent development of resistance. Treatment interruption to stop clonal selection of resistant cells has been proposed.557 Gene-expression profiles may be useful to predict the clinical effectiveness of imatinib for CML treatment, thereby allowing individualized therapy from the outset.560 In patients with relapse or resistance, alternative approaches include increasing the dose of imatinib or switching to dasatinib or nilotinib.561 Allogeneic hematopoietic stem cell transplantation is not recommended unless patients have inadequate response or intolerance to multiple TKIs or have the T315I mutation. Mutational analysis is not recommended at diagnosis but is of help in selecting TKI therapy for patients with an inadequate initial response or loss of response to TKI therapy. Mutation type may dictate choice of the next TKI.562
Management of Resistance
Dose Escalation If the patient is on imatinib, 400 mg/day, dose escalation to 800 mg/day can be tried. This may be most efficacious in patients with cytogenetic relapse who had achieved a cytogenetic response with the initial dose of imatinib, but is not likely to benefit those who have not had a cytogenetic response with imatinib at 400 mg/day.
Second-Generation Tyrosine Kinase Inhibitor Therapy Several TKIs are approved for use in the case of imatinib resistance or intolerance. In general, outcome with each is comparable, so the choice of agent often depends on its side-effect profile or in some cases on mutation type (see Tables 89–2 and 89–6).562
The 3-month molecular response after initiation of a second TKI is also a predictor of overall and event-free survival for those patients in chronic phase when switched from imatinib to another TKI.505 A BCR-ABL1 level of 10 percent or less at 3 months is desirable. In patients treated with nilotinib after imatinib resistance or intolerance, the 4-year progression-free and overall survival was 85 and 95 percent, respectively, if BCR-ABL1 transcripts were 10 percent or less as compared to 42 and 71 percent for those with BCR-ABL1 transcripts greater than 10 percent at 3 months.563
Either dasatinib or nilotinib can be used in cases of imatinib resistance or intolerance. Dasatinib is 325-fold more potent than imatinib; and, as a dual inhibitor of SRC and ABL1 kinases, dasatinib is able to bind to BCR-ABL1 with less-stringent conformational requirements.564 In patients resistant to imatinib, dasatinib, 140 mg/day (70 mg q12h), resulted in a higher proportion of MCyR, CCyR, and MMR than did 800 mg/day (400 mg q12h) of imatinib. Treatment failure was decreased and progression-free survival was improved with dasatinib.565 Unlike imatinib, dasatinib penetrates the blood–brain barrier.566 In long-term followup of 670 patients with imatinib-resistant or intolerant CML, treated with various doses of dasatinib, 28 percent of patients remained on study treatment at 6 years. Survival was in the range of 76 to 83 percent and a MMR was achieved in 45 percent on a dose of 100 mg daily. Molecular and cytogenetic responses at 3 and 6 months were predictive of survival. For those with BCR-ABL1 transcripts less than 10 percent at 3 months, progression-free survival was 68 percent, whereas it was only 26 percent for those with BCR-ABL1 transcripts greater than 10 percent.567 Nilotinib in a dose of 400 mg every 12 hours induced approximately 40 percent of patients who were resistant to or intolerant of imatinib into a MCyR, and approximately 30 percent into a CCyR. Nilotinib, 400 mg BID, is the recommended dose for those intolerant or resistant to imatinib.
Bosutinib and Ponatinib In addition to dasatinib and nilotinib, bosutinib and ponatinib, third-generation TKIs, are active against many of the imatinib-resistant BCR-ABL1 kinase domain mutations and are effective treatment options for CML resistant to standard-dose imatinib. Both were FDA approved for this indication in 2012. Bosutinib is active with F317L, Y253H, and F359C/I/V mutations. Ponatinib is also effective against many mutations resistant to nilotinib or dasatinib and has activity in cases with a T315I mutation (see Table 89–6).
Bosutinib is a dual SRC-ABL1 TKI that resulted in a CCyR in 24 percent and CHR in 73 percent of patients who had used two prior TKIs.568 All mutations except T315I were responsive. It is approved for chronic phase, accelerated phase, or blast phase in those resistant or intolerant to prior TKI therapy. The dose is 500 mg/day, orally, with food. Diarrhea, nausea, decreased platelet count, other gastrointestinal complaints, rash, and anemia were the most common adverse events, and most of these were of low-grade. Bosutinib is not yet approved for use as an initial agent in CML, as it did not demonstrate significant improvement in CCyR rates at 1 year compared to imatinib. It was, however, associated with higher 1-year MMR rates, faster time to response, and less disease progression.491
Ponatinib blocks native and mutated BCR-ABL1 including the gatekeeper mutation T315I. In a phase I dose escalation study, pancreatitis, rash, and myelosuppression were major toxicities. In 12 patients with T315I mutations, 92 percent had a MCyR. In those with accelerated or blast phase, or Ph chromosome–positive ALL, 36 percent had a major hematologic response and 32 percent had a MCyRs. Arterial thrombotic events occurred, however.548 Retrospective analyses of these incidents led to temporary withdrawal of this medication, which is now used primarily in cases of T315I mutation or failure of several other TKIs. If vascular occlusion occurs, therapy should be halted immediately.569 Ponatinib has a black box warning for vascular occlusion, heart failure, and hepatotoxicity.
A third-line TKI should be considered in patients who have failed two prior generations of TKI therapy, but responses tend to be infrequent and are usually not durable, so clinical trials, other agents, or allogeneic hematopoietic stem cell transplantation should be considered. A prior CCyR on either imatinib or subsequent nilotinib or dasatinib therapy was the only predictor of a cytogenetic remission on a third-line therapy.570
Omacetaxine Omacetaxine (homoharringtonine) is a Cephalotaxus alkaloid that has activity against the T315I mutation. Omacetaxine was approved on the basis of a study that recruited patients who had already been on two or more TKIs. In those enrolled in chronic phase, 67 percent had a CHR; a MCyR or a CCyR was achieved in 22 and 4 percent, respectively. Median overall survival was 30 months.570 In those with T315I mutations enrolled on a separate study, MMR was achieved in 17 percent of patients and the T315I clone was reduced in 61 percent of patients.571 The most common side effects with this medication are cytopenias. This agent also has activity in accelerated phase CML, but little in blast phase. It was approved by the FDA in October 2012 for chronic and accelerated phase patients intolerant of other therapy or in cases not responding to two or more TKIs.
Several inhibitors of signal transduction mediators involved in the downstream effects of BCR-ABL have been proposed for use in imatinib-resistant CML. These inhibitors include the JAK2 inhibitor AG490,572 SRC kinase inhibitors, mTOR (mammalian target of rapamycin) inhibitors, such as rapamycin,573 the proteasome inhibitor bortezomib,574,575 histone deacetylators,576,577 PI3K or MEK (mitogen-activated kinase) inhibitors and inhibitors of the WNT/β-catenin pathway thought to be active in CML stem cells. Imatinib resistance often is associated with restored activation of the BCR-ABL1 signal transduction pathway, suggesting that BCR-ABL1 remains a valid target to overcome resistance in these cases.578 BCR-ABL1 point mutations isolated from patients with imatinib-resistant CML are sensitive to inhibitors of the BCR-ABL1 chaperone hsp90, such as geldanamycin.579 Many of these agents have not yet entered clinical trials, but some are in early phase trials, such as inhibitors of the hedgehog pathway, which is activated in CML but not normal hematopoietic stem cells.580 Some are being used in conjunction with imatinib in resistant cases.
Combined Therapy Agents that have been proposed for use in combination to improve response rates or to overcome resistance to imatinib have included IFN-α, cytarabine, omacetaxine multiagent chemotherapy, arsenic trioxide, and decitabine, with some supporting in vitro data.581,582,583,584,585,586 Combining imatinib with chemotherapeutic agents is more myelosuppressive, and final effects on response rates and survival have yet to be determined. This approach is rarely used in chronic phase CML, but is used in accelerated or blast phase or in Ph chromosome–positive acute lymphoblastic leukemia.587
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


