Unexpectedly, as high as 3.5% of otherwise normal individuals over age 40 may harbor a population of clonal (by light chain analysis) CD5+/19+/23+ B cells.9 These asymptomatic individuals do not have an absolute lymphocytosis, lymphadenopathy, or other clinical evidence of CLL and are referred to as having monoclonal B lymphocytosis (MBL). Furthermore, as many as 13% of family members of patients with familial CLL harbor a population of cells with an immunophenotype consistent with CLL, but do not fulfill CLL diagnostic criteria. Therefore, the prevalence of a monoclonal lymphoproliferative process is potentially much higher than previously appreciated. It is estimated that the rate of progression from MBL to CLL is 1 to 2% per year.10 Currently, there is no indication to perform screening for MBL.
Immunoglobulin Heavy Chain Variable Gene (IGHV) Mutation Status
Because CD5+ B cells are found in fetal spleen and because surface immunoglobulin D is present on cells that have not encountered antigen in the germinal center, it was long thought that CLL cells were derived from naïve B cells. Normal B-cell development involves an antigen-independent phase and an antigen-dependent phase. During the antigen-independent phase, B cells undergo rearrangement of the V, D, and J genes in the bone marrow. Somatic mutation of the heavy- and light-chain variable gene occurs after encounter with antigen in the germinal center. Somatic mutation has occurred when there is <98% sequence homology with the germline gene. The figure of 98% is used because polymorphisms may account for lesser degrees of disparity.11
In the 1990s, data emerged that a significant percentage of patients had mutation of the immunoglobulin heavy chain variable gene (IGHV) in their CLL cells. Subsequently, it was confirmed that approximately 50% of patients have a mutated IGHV and that this provided prognostic information; patients with an unmutated IGHV have significantly shorter survival.12,13 Characterization of the IGHV sequence is labor-intensive and has not been readily exportable to clinical laboratories. Thus, correlates with mutation status that may be more easily identified may be more accessible. A correlation between expression of CD38 and lack of somatic mutation was described.12 Although the correlation is significant and the presence of CD38, irrespective of mutation status, is associated with inferior survival, a significant minority of patients have mutated IGHV and yet express CD38, and vice versa. These patients may have an intermediate prognosis. Also, there is variation in CD38 expression over time and by disease site (e.g., blood versus bone marrow) in some patients.
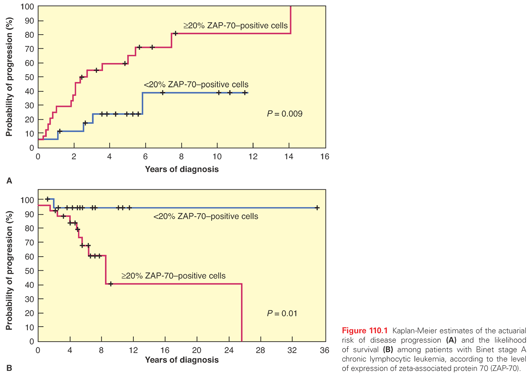
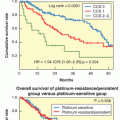
It was hypothesized that as patients with CLL can be segregated into two distinct prognostic categories based on IGHV mutation status, CLL may represent two separate disease entities, one derived from a naÏve B cell that expressed unmutated IGHV and the other derived from a memory B cell that had been exposed to antigen and displayed a mutated IGHV. This hypothesis has been examined using gene expression profiling.14 Investigators found that mutated and unmutated CLLs show a common gene expression pattern that is clearly distinguishable from that of other lymphomas, as well as normal B cells. Nevertheless, although the overall profile was similar, there were differentially expressed genes between the two groups. The gene that was most differentially expressed in one series was zeta-associated protein 70 (ZAP-70), with unmutated cases having significant expression of ZAP-70. Interestingly, ZAP-70 is normally found in T cells, where it functions as an intracellular signal–transduction molecule for the T-cell receptor. It was subsequently shown that ligation of the B-cell receptor (BCR) in CLL cells that expressed ZAP-70 produced greater tyrosine phosphorylation of cytosolic proteins than did stimulation of CLL cells that did not express ZAP-70; therefore, it may function in activating CLL cells. Expression of ZAP-70 was analyzed in 56 patients with CLL and was correlated with mutational status, disease progression, and survival in retrospective analyses (Fig. 110.1).15
Molecular Abnormalities
Conventional chromosome banding identified cytogenetic abnormalities in 40% to 50% of CLL cases; trisomy 12 was most common. This technique is hampered by the low mitotic activity of CLL cells. Fluorescence in situ hybridization (FISH), using genomic DNA probes to detect aberrations in interphase cells, enhanced the ability to detect molecular abnormalities in CLL. FISH has shown molecular abnormalities in over 80% of CLL cases.7
Deletion 13q [del(13q)] is the most common genetic aberration in CLL; it is found by FISH as a sole abnormality in 55% of cases, followed by 11q deletion (18%) [del(11q)], 12q trisomy (16%), and 17p deletion (7%) [del(17p)].7 Prognosis in CLL has been correlated with the presence of these chromosomal abnormalities. When divided into five hierarchical prognostic categories in order of highest risk—del(17p), del(11q), 12q trisomy, no abnormalities, and del(13q) (sole abnormality)—the survival times were 32 months, 79 months, 114 months, 111 months, and 133 months, respectively. Patients with del(17p) or del(11q) had more advanced disease with extensive lymphadenopathy. With hierarchical categorization, patients are assigned according to their highest-risk abnormality, including when multiple abnormalities are present. Clonal evolution can occur over time, particularly in the setting of cytotoxic treatment; therefore, repeated FISH assessment is important with changes in clinical status, such as in patients needing retreatment.
The frequency of del(13q) led to a search for a potentially new tumor suppressor gene in that location. At least eight genes were identified and screened for alterations at the DNA or RNA level, or both, but studies failed to find consistent involvement of any of those genes. However, two potentially relevant microRNA (miR) genes, miR15 and miR16, were identified in the critical minimal deleted region of del(13q) and were noted to be deleted or downregulated in more than two-thirds of all CLL cases.16 MicroRNAs are nontranslated small RNAs that function to regulate gene expression. Both miR15 and miR16 negatively regulate BCL-2 transcript level; the absence of miR15 and miR16 in cases with del(13q) may lead to Bcl-2 overexpression and resultant resistance to apoptosis.
Whole-exome sequencing of CLL cases identified mutated genes that may contribute to the pathogenesis and biology of the disease.17–19 The frequency of mutated genes appears notably lower than the occurrence of cytogenetic abnormalities noted by FISH. Mutations are mostly private, and frequencies vary significantly by the patient population being characterized (e.g., untreated versus relapsed or refractory CLL). It is therefore unlikely that there is a single driver mutation that accounts for the disease. Indeed, the limited number of cases sequenced, the diversity in prior treatments among cases, and the relatively low frequency of mutations likely relate to the diversity in reported mutations. Consistently reported genes reported as mutated, albeit at low frequency, include NOTCH1, SF3B1, TP53, MYD88, XPO1, and ATM. Some of these mutations have been associated with clinical outcome such as TP53, SF3B1, NOTCH1, ATM, and BIRC3.18–20 Some mutations may result in decreased protein level or function, others may result in activation or increased protein levels, depending on the gene and mutation. Serial sampling of individual patients for whole-exome sequencing is leading to insights into diversities in clonal evolution and an appreciation for how different treatments may impact the emergence, frequency, and type of mutations and loss of others through the course of a patient’s disease.21–23
CLL cells disrupt immune function in patients with CLL. The most prominent manifestation of immune dysfunction is the increased risk and frequency of infections. Many patients with CLL succumb to infection or ineffectively treated autoimmunity. The treatments used for CLL, such as purine analogues, further immunosuppress patients and put them at increased risk for opportunistic infections and may exacerbate or unmask autoimmunity.
Early in the disease, in untreated CLL, the absolute number of T cells is increased with inversion of the T helper–T suppressor cell ratio.24,25 The CD4 to CD8 ratio continues to drop with disease progression or after therapy with nucleoside analogues or alemtuzumab. Qualitative functional assessment of T cells has been inconclusive. Normal and decreased CD4-cell function has been reported. Similarly, decreased, normal, or excessive CD8-cell function has been reported. Others have shown that T-cell functions may be impaired by immunosuppressive factors produced by CLL cells.24 Hypogammaglobulinemia is a common and progressive immune defect in patients with CLL and is another factor that increases the risk for infection. The pathogenesis of hypogammaglobulinemia in CLL is poorly understood. Impaired B-cell function and regulatory abnormalities of T cells, including the reversal of the normal helper–suppressor cell ratio, may play roles. In addition, CLL-derived natural killer (NK) cells have been shown to suppress immunoglobulin secretion by normal B cells in vitro.
The International Workshop on CLL (IWCLL) in 200826 updated the National Cancer Institute Working Group 1996 guidelines27 for diagnostic criteria and treatment for CLL.
International Workshop on Chronic Lymphocytic Leukemia Revised Diagnostic Criteria
1. A blood monoclonal B lymphocyte count >5 × 109/L, with <55% of the cells being atypical (prolymphocytes).
2. B-lymphocyte monoclonality should be demonstrated with cells expressing B-cell surface antigens (CD19, CD20, CD23), low-density surface immunoglobulin (M or D), and CD5.
A monoclonal B cell count >5 × 109/L was specified to distinguish CLL from small lymphocytic lymphoma in patients with palpable lymph nodes or splenomegaly. However, it is arguable as to whether that distinction is clinically relevant. Bone marrow aspirate typically shows >30% lymphocytes, with flow cytometry confirming monoclonality in the CD19/CD20/CD23/CD5+ population; however, diagnosis may be made solely on blood.
Other B-cell malignancies may also present with increased circulating lymphoid cells and should be differentiated from CLL. The diseases that may be confused with CLL are prolymphocytic leukemia (PLL), the leukemic phase of non-Hodgkin’s lymphoma (mantle cell lymphoma, follicular lymphoma, or splenic lymphoma with circulating villous lymphocytes), and hairy cell leukemia (HCL). Immunophenotyping is helpful in differentiating these disorders (Figs. 110.2, 110.3, and 110.4; see Table 110.1).



Clinical Manifestations
The majority of individuals diagnosed with CLL are asymptomatic and initially identified on routine blood count. Some patients remain asymptomatic for a long period of time. In patients presenting with symptoms, the most common is fatigue, which is generally mild. Sometimes, enlarged lymph nodes or the development of an infection is the initial manifestation of disease. Bacterial infections, such as pneumonia, are more common in patients who present with advanced-stage disease. Infections secondary to opportunistic organisms, particularly herpes zoster, may occur. Exaggerated skin reaction to a bee sting or an insect bite (Wells’ syndrome) is frequent in CLL. In contrast to the situation in lymphoma, fever in the absence of infection is rare in CLL. Lymph nodes, when enlarged, are usually discrete, freely movable, and nontender. Splenomegaly may occur, but massive splenomegaly is usually seen in patients with advanced disease. Splenic infarction is rare. Hepatomegaly occurs less frequently than splenomegaly. Skin involvement occurs in <5% of cases. Leptomeningeal leukemia is rare and, if present, is usually seen in patients with refractory disease. Malignant pleural effusions are also rare, and when present, are associated with aggressive disease and poor prognosis.
Laboratory Findings
Absolute lymphocyte counts range from 5 × 109/L to over 500 × 109/L. Leukostasis is uncommon in CLL, probably because of the small size and pliability of the leukemia cells. The lymphocyte count usually increases over time, but fluctuations in the absolute lymphocyte count of untreated patients may occur, particularly in the setting of infection. The lymphocytes are typically small and mature appearing, but there may be variations in cell morphology, with some lymphocytes being larger or atypical, whereas others may be plasmacytoid or cleaved or there may be prolymphocytes. Ruptured lymphocytes or “smudge” cells are commonly seen in the peripheral smear, reflecting fragility and distortion during preparation of the peripheral smear on the glass slide. Marrow infiltration by lymphocytes is universal, affecting from 30% to 100% of the cellularity, with overall increased marrow cellularity. The patterns of lymphoid infiltration of the marrow seen in biopsy specimens include nodular, interstitial, diffuse, or a combination. Patients with diffuse infiltration typically have advanced disease and a worse prognosis. Nodular and interstitial or “nondiffuse” patterns are associated with less advanced disease and better outcome.
Anemia (hemoglobin <11 g/dl) and thrombocytopenia (platelet count <100 × 109/L) are found in a minority of patients at diagnosis but develop with disease progression. A positive direct antiglobulin (Coombs’) test is seen in approximately 25% of cases, but overt autoimmune hemolytic anemia (AIHA) occurs less frequently. The incidence of a positive Coombs’ test increases significantly with clinical stage.28 Immune thrombocytopenia is usually diagnosed on the basis of a low platelet count in the presence of adequate numbers of megakaryocytes in the bone marrow. Neutropenia may also be encountered. These cytopenias may be the result of bone marrow failure due to “packed” marrow by CLL or occur as a result of an immune-mediated process or hypersplenism. Hypogammaglobulinemia occurs in approximately 50% of patients with CLL. At diagnosis, it may be noted in <10% of patients, but its incidence increases significantly with disease progression. Usually, all three immunoglobin classes (G, A, and M) are decreased, but in some patients, only one or two may be low. Significant hypogammaglobulinemia and neutropenia potentially result in increased susceptibility to bacterial infections.
Autoimmune Complications
When autoantibodies occur in CLL, they are usually targeted against hematopoietic cells, resulting in AIHA, immune thrombocytopenia, immune-mediated granulocytopenia, or pure red cell aplasia; AIHA is the most frequent.29 The autoantibodies are typically polyclonal and usually immunoglobulin G, indicating that they are not produced by the leukemic clone.30 The severity of the autoimmune phenomenon does not necessarily correlate with the severity of CLL, and such events may develop in patients whose disease is responding to therapy. Prednisone is the most commonly used treatment for autoimmune complications, with high initial response rates. It is usually given at a dose of 1 mg/kg orally and tapered once a response is noted. Relapses are not uncommon. Cyclosporin A is another effective therapy and can produce good results, even in steroid-refractory patients.31 CD20 and CD52 monoclonal antibodies (mAb) rituximab and alemtuzumab have been used alone or in combination with chemotherapy in some patients in whom standard therapy fails.32,33 Splenectomy is also a viable therapeutic option for refractory cases.28,34
Staging
The clinical course for individuals with CLL is variable; survival times range from 2 to over 20 years from diagnosis. In 1975, Rai et al.35 developed a staging system consisting of five stages (Rai 0 to IV) based on Dameshek’s model of orderly disease progression in CLL (Table 110.2). The Rai staging system was later modified into a three-stage system: low-risk (Rai 0), intermediate-risk (Rai I, II), and high-risk (Rai III, IV). A similar staging system was developed in Europe by Binet et al.36 Both classifications reflect bulk of disease and extent of marrow compromise (i.e., anemia, thrombocytopenia). Both staging systems have been recognized as simple and reliable predictors of survival (see Table 110.2). Although most patients in the high-risk group (Rai III, IV; Binet C) have a progressive clinical course and shortened survival, the course of the disease is not uniform. Patients in the low- and intermediate-risk groups may have an indolent disease course that spans years or even decades, or the course may be progressive and associated with a shortened survival. Thus, it is helpful to have prognostic factors associated with clinical outcomes particularly for the low-risk group. Several prognostic factors have been associated with shortened survival in CLL. These include a short lymphocyte doubling time (<6 months), a diffuse pattern of bone marrow infiltration, advanced age and male gender, abnormal karyotype, high serum levels of β2-microglobulin and soluble CD23, and a CLL-PLL category (11% to 54% prolymphocytes in the blood).37 Newer prognostic factors in CLL include IGHV mutation status, expression of CD38 and ZAP-70, and gene mutations. A prognostic model integrating cytogenetic abnormalities identified by FISH with mutated NOTCH1, SF3B1, BIRC3, and TP53 was proposed as a dynamic prognostic algorithm for overall survival (OS).20

TREATMENT AND RESPONSE CRITERIA
An unusual feature of CLL compared to other leukemias is that making the diagnosis is not necessarily an indication to initiate treatment. This is true for several reasons. CLL is a disease of the older population; it may be diagnosed in an asymptomatic patient and have a prolonged course; CLL is not curable with current standard treatment approaches; and a survival advantage was not demonstrated in clinical trials of early intervention. Given that the majority of patients are older than 70 years, may have serious comorbid conditions associated with aging, and may have indolent disease, a significant fraction of patients will die of other causes and may never require therapy for their CLL.
The IWCLL revised criteria for active disease, an indication to initiate treatment,26 include constitutional symptoms attributable to CLL: weight loss (>10% of baseline weight within the preceding 6 months), extreme fatigue (Eastern Cooperative Oncology Group performance status 2 or higher), fever (temperature higher than 38°C or 100.5°F for at least 2 weeks) or night sweats without evidence of infection; evidence of progressive bone marrow failure characterized by the development of or worsening of anemia, thrombocytopenia, or both; AIHA or autoimmune thrombocytopenia, or both, poorly responsive to corticosteroid therapy; massive (>6 cm below the left costal margin) or progressive splenomegaly; massive (>10 cm in longest diameter) or progressive lymphadenopathy; and progressive lymphocytosis defined as an increase in the absolute lymphocyte count by >50% over a 2-month period, or a doubling time predicted to be <6 months. Hypogammaglobulinemia or monoclonal gammopathy alone are not sufficient criteria to initiate therapy.
Several European groups conducted trials in the 1980s to evaluate whether immediate treatment in patients with early stage disease could improve survival.38 These large randomized trials of immediate chlorambucil (CLB) therapy versus watch-and-wait were consistent in showing no survival benefit with early treatment. However, given significantly better current therapies, this question has been raised again. A limitation of randomizing all early stage patients is that approximately one-third of them may never require therapy for their disease, thus reducing the potential benefit of early treatment. Furthermore, the discovery of prognostic factors that identify early stage patients with a high likelihood of developing progressive disease may allow for randomized trials to more directly address this question of the benefit of early treatment.
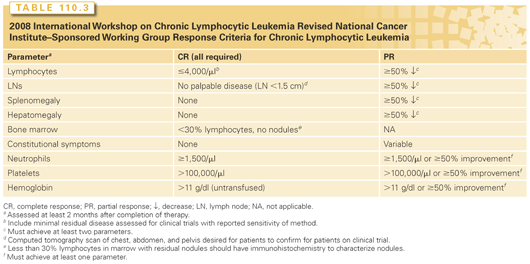
The response criteria published in 198839 by the National Cancer Institute Working Group on CLL were revised in 199627 and most recently updated by the IWCLL (Table 110.3).26 Updated recommendations made were that patients treated on clinical trial have evaluation of the blood or bone marrow by sensitive tests for minimal residual disease (MRD) such as multicolor flow cytometry or allele-specific polymerase chain reaction (PCR) for the IGHV gene and have confirmation of nodal response by computed tomography scan. In some patients who achieve complete remission by IWCLL criteria, one or both of these methods can demonstrate residual disease, referred to as MRD. Patients free of MRD following treatment have a longer remission duration and longer survival.40,41 Therefore, in addition to improving complete remission rates, investigators are focusing on eliminating MRD to improve treatment outcomes.
A sensitive four-color flow cytometry assay was developed to differentiate CLL cells (CD5/CD19 with CD20/CD38, CD81/CD22, and CD79b/CD43) from normal B cells.42 The assay can detect one CLL cell in 104 to 105 leukocytes. PCR techniques can also be used to assess MRD. Consensus primers for IGHV can be used in 70% to 80% of patients and may detect 1 in 104 residual cells. Allele-specific oligonucleotide primers generated for individual patients are more sensitive, detecting 1 in 105 CLL cells. Development of quantitative PCR techniques may aid in following patients over time but are technically complicated and therefore not available for routine clinical use.
Alkylating Agent-Based Treatments
For decades, the mainstay of therapy for CLL was alkylating agents, CLB and cyclophosphamide (CTX). These alkylating agents were given with or without corticosteroids. Various doses and schedules of oral CLB have been used. CLB is usually administered for several months, and the dose is adjusted to avoid its primary toxicity, myelosuppression. The overall response rate with either CLB or CTX monotherapy is approximately 40% to 60%, with 3% to 5% complete remission. Alkylating agents were combined with steroids to improve response rates.43 Alkylating agent–based combinations have also included an anthracycline. No superior alkylating-agent combination has been identified.
Purine Analogues
Purine analogues, including fludarabine monophosphate, 2-chlorodeoxyadenosine (2-CdA), and pentostatin (deoxycoformycin), all have activity in treating patients with CLL.43
In a phase 2 trial conducted at MD Anderson Cancer Center, fludarabine was given at a dose of 30 mg/m2 per day for 5 days every 4 weeks. A response rate of 59% was observed in 68 previously treated patients, with 15% achieving complete remission. A subsequent study explored the combination of fludarabine and prednisone. Response rates were identical to those seen with fludarabine monotherapy, but the addition of prednisone was associated with increased incidence of Pneumocystis jiroveci and Listeria monocytogenes infections. The major side effects associated with fludarabine were myelosuppression and immunosuppression, with low CD4 counts lasting for many months to years after completion of treatment.43
Single-arm studies evaluated fludarabine in previously untreated patients. Overall response rates were higher at 70% to 80%, and complete remission was seen in 10% to 25%.43 An oral formulation of fludarabine was evaluated in relapsed patients with CLL. Seventy-eight patients received oral fludarabine 40 mg/m2 per day for 5 days every 4 weeks for six to eight courses. The overall response rate was 51%, which was almost identical to prior trials using the intravenous formulation as a salvage regimen. Furthermore, oral fludarabine was used in the first-line Leukemia Research Foundation CLL4 trial44 comparing fludarabine plus CTX versus fludarabine versus CLB, demonstrating efficacy and tolerability with the oral formulation, which is now approved by the US Food and Drug Administration (FDA).
Comparative Studies
A randomized European trial compared six courses of fludarabine versus six courses of CTX, doxorubicin, and prednisone (CAP) in 196 patients with Binet stage B or C CLL (Table 110.4).45 In previously treated patients, a significantly higher overall response rate was observed with fludarabine compared to CAP. In previously untreated patients (see Table 110.4), the response rates with fludarabine were similar to those with CAP, but the duration of response was significantly longer with fludarabine. The French Cooperative Group on CLL randomized nearly 1,000 previously untreated patients to one of three treatment regimens: fludarabine, CTX, doxorubicin, prednisone, and vincristine, or CAP.46 Higher overall response rate and longer time to progression were seen with fludarabine. Infection rates were similar, but extramedullary toxicity was less with fludarabine.
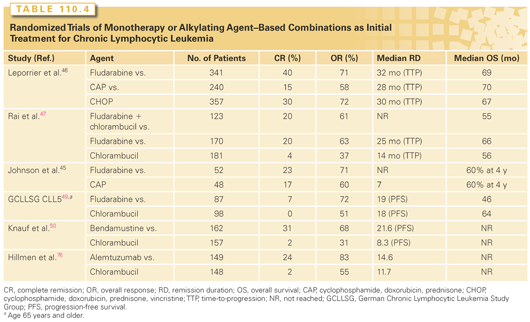
Results from an Intergroup trial with 509 previously untreated patients with CLL showed significantly higher complete and overall remission rates in patients treated with fludarabine versus those given CLB (see Table 110.4).47 A third arm, fludarabine plus CLB, was closed early because of infection-related toxicity. Crossover was allowed for patients with no response or relapse. Half the patients who failed to respond to CLB responded to fludarabine, including a 14% complete remission rate. In contrast, only 7% of patients who failed to respond to fludarabine achieved partial response with CLB. Although longer response duration and improved progression-free survival (PFS) were noted in patients treated with fludarabine, no difference in OS was found between the two groups in the initial report. However, updated data, with significantly longer follow-up, reported improved OS for patients treated initially with fludarabine; this difference in the survival curves emerged after 6 years follow-up.48 The German CLL Study Group (GCLLSG) CLL5 evaluated fludarabine versus CLB monotherapy as initial treatment for patients older than 65 years (see Table 110.4).49 Surprisingly, while treatment with fludarabine was associated with superior complete (7% versus 0%) and overall (72% versus 51%) response rates, there was no associated improvement in PFS or OS for these “elderly” patients. Thus, standard first-line treatment of the elderly does not require fludarabine.
Bendamustine has a benzimidazole (purinelike) ring structure with an alkylating group and has potent alkylating agent activity, inducing intra- and interstrand DNA crosslinks. Bendamustine was compared to CLB in a randomized phase 3 trial for previously untreated patients with CLL (see Table 110.4).50 Treatment with bendamustine was associated with superior PFS (21.6 months versus 8.3 months), and complete (31% versus 2%) and overall (68% versus 31%) response rates; this was the basis for FDA approval of this agent. Myelosuppression was mild but more frequent with bendamustine; this did not result in an increased infection rate.
Fludarabine inhibits excision repair of DNA interstrand crosslinks induced by CTX, thereby potentiating activity and providing a rationale for combining these agents.51 Phase 2 trials, which combined fludarabine and CTX (FC), suggested increased efficacy compared to that seen in historical patients treated with fludarabine monotherapy.52–54 Three large randomized trials evaluated the efficacy of FC versus fludarabine monotherapy in previously untreated patients (Table 110.5). In the GCLLSG CLL4 trial, previously untreated patients younger than 65 with indications for treatment were randomized to receive six courses of FC or fludarabine.55 The U.S. Intergroup E2997 trial56 randomized previously untreated patients to receive FC or fludarabine and the UK Leukemia Research Foundation CLL4 trial44 randomized patients to FC, fludarabine, or CLB in a 1:1:2 randomization. All three trials demonstrated superior PFS with FC treatment over fludarabine or CLB, and this was associated with superior complete and overall response rates. There was more myelosuppression with the combination, yet there was no difference in the incidence of infections in any of the trials. None of these trials showed a difference in OS with the follow-up available. Patients with del(17p) were confirmed to be high-risk with lower response rates and shorter survival compared to patients with other chromosome abnormalities, regardless of treatment. Patients with del(11q) were high-risk, but there appeared to be a large benefit for those patients treated with an alkylating agent combined with fludarabine. Patients with an unmutated IGHV gene had similar response rates to those with a mutated IGHV; however, their PFS and OS was shorter for those with an unmutated IGHV gene.
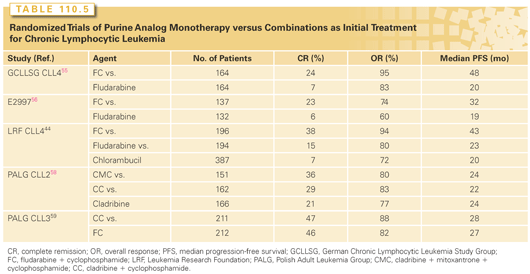
Cladribine (2-CdA) and pentostatin (2-deoxycoformyin) have activity in treating CLL.57 There are no head-to-head comparisons available for purine analogue monotherapy. The large randomized Polish Adult Leukemia Group (PALG)-CLL2 trial compared cladribine monotherapy, cladribine with CTX, versus cladribine with CTX and mitoxantrone (CMC) as initial therapy and demonstrated a higher complete response rate for CMC compared to the other treatments (see Table 110.5).58 Neutropenia was more common with CMC, as was infection. There were no significant differences in PFS or OS rates among the three arms. The PALG-CLL3 phase 3 trial demonstrated equivalent efficacy with cladribine with CTX and FC for previously untreated patients with CLL (see Table 110.5).59 Patients with del(17p) had poor outcomes with either treatment.
Monoclonal Antibodies
Alemtuzumab is a humanized mAb targeting CD52, an antigen that is highly expressed on CLL cells and normal T and B lymphocytes. Alemtuzumab was originally approved by the FDA based on the pivotal trial that demonstrated monotherapy activity in fludarabine-refractory patients with CLL (Table 110.6).60–74 Complete and partial remissions were noted in 2% and 31% of 93 treated patients, respectively; 55 (59%) had stable disease. Similar results were reported in a trial with a similar patient population treated with the same dose and duration of alemtuzumab administered subcutaneously.75 Subcutaneous administration eliminates the infusion-related side effects, although local injection site reactions occur. Alemtuzumab was very effective at eliminating disease in the peripheral blood and bone marrow; bulky lymphadenopathy was less effectively treated, a pattern observed in other studies with alemtuzumab. The T-cell suppression that occurred with this agent has been concerning. As a consequence, a significant incidence of infections (including cytomegalovirus reactivation) is associated with therapy. Antibacterial and antiviral prophylaxis should always be used with alemtuzumab. Other studies confirmed the activity of alemtuzumab in less heavily pretreated patients (see Table 110.6). A randomized first-line trial demonstrated superior PFS associated with alemtuzumab monotherapy compared to CLB, as well as higher complete and overall response rates (see Table 110.4).76

The mechanism of action for alemtuzumab is independent of p53. TP53 is a gene deleted in patients who have loss of chromosome 17p. Loss of p53 function by deletion or mutation confers resistance to treatment with standard chemotherapy such as CLB and purine analogues. Alemtuzumab was reported to have activity in patients with leukemia cells that lack p53 function.77 Despite treatment with alemtuzumab, patients with del(17p) have short remission duration and OS. Alemtuzumab was also combined with high-dose methylprednisolone for previously treated and treatment-naïve patients with better results, although there is concern for immunosuppression and risk for infection (see Table 110.6).
Rituximab is a chimeric immunoglobulin G1 CD20 mAb; CD20 is expressed on malignant and normal B cells.78 Relatively low levels of CD20 are expressed on CLL cells compared to normal B or neoplastic B cells of other lymphomas. In addition, circulating CD20 was demonstrated in the plasma of patients with CLL; this may inhibit the capacity of rituximab to bind to CLL cells, resulting in rapid clearance and negatively affecting pharmacokinetics.79 Rituximab binds to the large-loop domain of CD20 and mediates antileukemic activity predominantly through antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity (CDC) (type I CD20 mAb). Standard-dose rituximab monotherapy has limited activity in treating CLL (see Table 110.6). Dose-intense80 and dose-dense81 rituximab monotherapy increased efficacy (see Table 110.6). In addition, greater efficacy was seen when rituximab was used as first-line therapy (see Table 110.6).82 Maintenance rituximab is not routine practice for patients with CLL.
The primary toxicity seen with rituximab is usually with the initial infusion and is predominantly fever and chills. These symptoms are generally mild to moderate, subside with completion or discontinuation of the infusion, and abate with subsequent infusions. Although normal B cells are also targeted by rituximab, trials to date have shown no significant decrease in immunoglobulin levels, and infection rates are low.
Rituximab was combined with alemtuzumab based on the rationale of targeting two distinct antigens expressed on CLL cells as well as the differential effectiveness by disease site; rituximab has activity in treating lymph node disease, and alemtuzumab is highly effective at clearing blood and bone marrow. Efficacy and tolerability were demonstrated in both untreated and previously treated CLL (see Table 110.6). In addition, rituximab was combined with high-dose methylprednisolone in an active regimen for untreated and previously treated patients (see Table 110.6). Immunosuppression was seen with this combination, owing to the use of high-dose steroids, and was effectively managed with prophylactic antibiotics.
Ofatumumab is a fully human immunoglobulin G1 CD20 mAb that binds to an epitope encompassing both large- and small-loop domains of CD20 and is highly effective at CDC (type I CD20 mAb). Ofatumumab monotherapy was first evaluated in phase 1 and 2 trials of escalating doses of four weekly infusions (see Table 110.6).83 The pivotal trial that led to FDA approval of ofatumumab enrolled patients who were refractory to fludarabine and alemtuzumab (FA-ref) as well as fludarabine-refractory patients with bulky (>5 cm) lymph nodes (and therefore poor candidates for treatment with alemtuzumab) (BF-ref); regulatory approval was based on outcome for the FA-ref group. Patients received ofatumumab 2,000 mg intravenously weekly for 8 weeks, then monthly for 4 months. The overall response rate was 58% and 47% for the FA-ref and BF-ref group, respectively. The median PFS and OS were 5.7 months and 13.7 months, respectively, in the FA-ref and 5.9 months and 15.4 months, respectively, in the BF-ref group, representing clinical benefit for these patients compared to historic outcomes with available treatment (see Table 110.6).84 First infusion-associated toxicity was most common and effectively managed with premedication. Infection was seen, but expected in these highly refractory, heavily pretreated patients. Response rates, median survival, and adverse effects were similar between rituximab-treated, rituximab-refractory, and rituximab-naïve patients.85
Chemoimmunotherapy
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


