FIGURE 32.1 Schematic representation of the t(9;22)(q34;q11) translocation that creates the Philadelphia (Ph) chromosome. The ABL and BCR genes reside on the long arms of chromosome 9 and 22, respectively. As a result of the (9;22) translocation, the BCR-ABL gene is formed on the derivative of chromosome 22 (22q–, Ph chromosome), while the reciprocal ABL-BCR resides on the derivative of 9q+.
Functional Domains of BCR-ABL and Kinase Activation
p210BCR-ABL contains several distinct domains (Fig. 32.2).13 The N-terminal coiled-coil domain of BCR allows BCR-ABL dimerization, which is critical for kinase activation. The p210BCR-ABL protein also retains the serine/threonine kinase and Rho guanine nucleotide exchange factor homology (Rho-GEF) domains of BCR, which are deleted in p190BCR-ABL, which may explain the differences in disease phenotype associated with the two variants. In contrast to BCR, the ABL sequence is almost completely retained, including SRC homology domains 2 and 3, the tyrosine kinase domain, a proline-rich sequence, and a large C terminus with nuclear localization signal, DNA-binding, and actin-binding domains. The N-terminal “cap” region of ABL negatively regulates kinase activity by binding to a hydrophobic pocket at the basis of the kinase domain, which in the Ib isoform is mediated by N-terminal myristoylation. It is believed that the replacement of the cap with BCR sequences contributes to constitutive kinase activation.
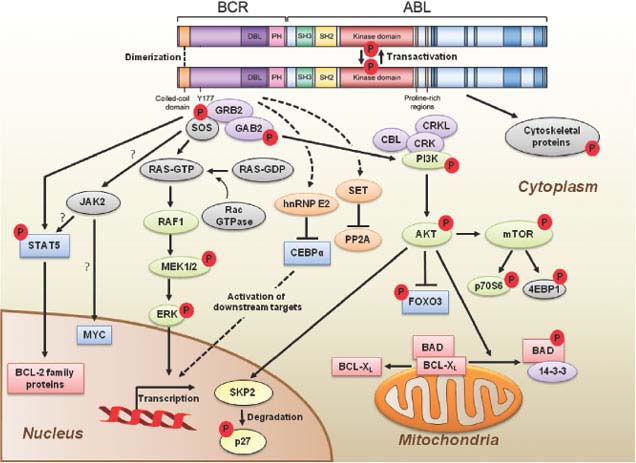
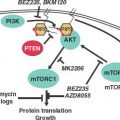
FIGURE 32.2 BCR-ABL domain structure and simplified representation of molecular signaling pathways activated in CML cells. Following dimerization of BCR-ABL, autophosphorylation generates docking sites on BCR-ABL that facilitate interaction with intermediary proteins such as GRB2. CrkL and CBL are also direct substrates of BCR-ABL that are part of a multimeric complex (purple). These BCR-ABL–dependent signaling complexes in turn lead to activation of multiple pathways whose net result is enhanced survival, inhibition of apoptosis, and perturbation of cell adhesion and migration. A subset of these pathways and their constituent transcription factors (blue), serine/threonine-specific kinases (green), cell-cycle regulatory protein (yellow), and apoptosis-related proteins (red) are shown. Several pathways were identified that are responsible for CML stem cell maintenance and/or BCR-ABL–mediated disease transformation (orange).
Signal Transduction
Numerous substrates and binding partners of BCR-ABL have been identified (Fig. 32.2). Current efforts are directed at linking these pathways to the specific phenotypic defects that characterize CML, such as increased proliferation, decreased apoptosis, defective adhesion to bone marrow stroma, and genetic instability.14 As comprehensive review of the multiple implicated pathways is beyond the scope of this chapter, we will focus on those for which strong evidence supports a rate-limiting role in disease pathogenesis.
Phosphatidylinositol-3 Kinase (PI3K)
PI3K is activated by autophosphorylation of tyrosine 177, which generates a high-affinity docking site for the SH2 domain of the GRB2 adapter, which in turn recruits GAB2 into a complex that activates PI3K.15,16 Consistent with a critical role of the Y177/GRB2/GAB2 axis, mutation of tryosine 177 to phenylalanine or lack of GAB2 abrogates myeloid leukemia.16 An alternative pathway of PI3K activation is complex formation between its p85 regulatory subunit, CBL, and CrkL, which bind to the SH2 and proline-rich domains of BCR-ABL.17 PI3K activates the serine-threonine kinase AKT, which suppresses the activity of the forkhead O transcription factors (FOXO), thereby promoting survival.18,19 Additionally PI3K enhances cell proliferation by promoting proteasomal degradation of p27 through up-regulation of SKP2, the F-Box recognition protein of the SCFSKP2 E3 ubiquitin ligase; absence of SKP2 from the leukemia cells prolongs survival in a murine CML model.20 Another important outlet of PI3K signaling is AKT-dependent activation of mTOR, which leads to the constitutive phosphorylation of ribosomal protein p70S6 kinase and 4E-BP1, which enhance protein translation and cell proliferation.21,22
RAS/Mitogen-activated Protein Kinase (MAPK) Pathways
GRB2-mediated recruitment and activation of SOS promotes exchange of GTP for GDP on RAS.15,23 GTP-RAS activates MAP kinase, promoting proliferation. Signaling from RAS to MAPK involves the serine-threonine kinase RAF-124,25 and RAC, another GTP–GDP exchange factor.26 A crucial role for the latter is supported by the fact that lack of RAC1/2 delays BCR-ABL leukemia in a murine model.27
Janus Kinase (JAK) Signal Transducer and Activator of Transcription (STAT) Pathway
BCR-ABL activates STAT5 through direct phosphorylation or indirectly through phosphorylation by HCK, a SRC family kinase, or JAK2.28,29 Active STAT5 induces the transcription of antiapoptotic proteins like MCL-1 and BCL-XL.30 Initial experiments in mice failed to demonstrate a critical role for STAT5.31 However, it was subsequently established that the mice used in this study were not null for STAT5, but expressed an N terminally deleted protein with partially retained function. In contrast, complete lack of STAT5 abrogates both myeloid and lymphoid leukemogenesis.32
Cytoskeletal Proteins
BCR-ABL phosphorylates several proteins involved in adhesion and migration, including FAK, paxillin, p130CAS and HEF1. This and activation of RAS15,33 are thought to impair integrin-mediated adhesion of CML progenitors to stroma and extracellular matrix, causing premature circulation as well as abnormal proliferation of Ph+ progenitors.34
DNA Repair
BCR-ABL impairs DNA damage surveillance by various mechanisms. For example, BCR-ABL has been shown to suppress checkpoint kinase 1 (CHK1) through inhibition of ATR35 or down-regulation of BRCA1, a substrate of ataxia telangiectasia mutated (ATM).36 Nonhomologous end-joining and homologous recombination, both critical double-strand break repair pathways, are defective in CML. BCR-ABL also up-regulates RAD51, inducing rapid but low-fidelity double-strand break repair on challenge with cytotoxic agents and induces reactive oxygen species that promote chronic oxidative DNA damage, double-strand breaks and point mutations. Lastly, telomere length decreases with disease progression from CP to BP.37
Although progress has been made to understand the extraordinary complexity of BCR-ABL signaling, a complete picture is still elusive. To overcome the limitations of investigating single pathway, quantitative proteomics is being used to establish a comprehensive picture of BCR-ABL signaling.38 These results suggest that cellular processes in CML, rather than relying on a single pathway, use integrated networks to fully realize their leukemogenic potential.
Murine Models of CML
The most commonly used murine model of CML is retroviral expression of BCR-ABL in bone marrow followed by transplantation into lethally irradiated syngeneic recipients, which develop a CML-like myeloproliferative neoplasm.39 Recently, an inducible transgenic mouse model has been developed, in which conditional BCR-ABL expression is under the control of the 3’ enhancer of the murine stem cell leukemia (SCL) gene. This model serves as a promising new tool for studying leukemogenic mechanisms in hematopoietic stem cells during disease initiation and progression.40 Lastly, xenograft models use various strains of immunodeficient mice for engraftment of primary CML cells.41 The problem with xenograft models is that the engraftment of CP cells is low, probably because of their compromised interactions with the microenvironment as a result of species differences in cytokines and adhesion molecules. More promising results have been obtained by injecting CML cells directly into the livers of newborn mice.42
CML Stem Cell
The origin of CML in a pluripotent hematopoietic stem cell (HSC) was elegantly demonstrated in the late 1970s in studies showing clonality of granulocytes, erythrocytes, and platelets of female patients heterozygous for glucose-6-phosphate dehydrogenase, a polymorphic X-chromosomal gene.43 BCR-ABL does not confer self-renewal, implying that Ph must be acquired by an HSC already endowed with this capacity.44 For unknown reasons the main cellular expansion occurs in the progenitor cell compartment, while at least initially the majority of HSC are Ph-negative.45 Serial xenograft studies have shown that CML leukemia stem cells (LSCs) reside within the quiescent CD34+38− fraction of bone marrow cells. Significant progress has recently been made by the identification of the IL-1 receptor-associated protein (IL-1RAP) as a surface marker specifically expressed on CD34+38− CML HSC.45a If confirmed, this will enable studies into the biology of these cells. Several genes were shown to have a critical role for leukemia-initiating cells maintenance in CML, including promyelocytic leukemia,46 the hematopoietic-specific Rac2 GTPase,47 smoothened, which is an essential component of the hedgehog pathway,48 and β-catenin. The fact that these genes are also critical for maintenance and self-renewal of normal HSCs may be an obstacle to exploiting them as therapeutic targets.
Progression to Blastic Phase
Disease progression is believed to be due to the accumulation of molecular abnormalities that lead to a loss of terminal differentiation capacity of the leukemic clone, which however continues to depend on BCR-ABL activity. BCR-ABL mRNA and protein levels are higher in BP than in CP cells, including CD34+ granulocyte macrophage progenitors (GMPs), which are expanded in BP.49 Another mechanism that enhances BCR-ABL activity in BP is inactivation of PP2A through up-regulation of SET, which in turn inhibits the PP2A phosphatase and its substrate protein tyrosine phosphatase I (SHP1). As SHP1 normally promotes BCR-ABL degradation through dephosphorylation, its reduced activity stabilizes BCR-ABL.50
The most striking feature of BP, the loss of differentiation capacity, suggests that the function of key myeloid transcription factors must be compromised. Occasionally, the differentiation block can be ascribed to mutations that result in the formation of dominant-negative transcription factors such as AML1-EVI or NUP98-HOXA9, which block differentiation or favor preferential growth of immature precursors.51–53 Isolated cases of myeloid transformation have been associated with the acquisition of core binding factor mutations typical of AML. A more universal mechanism appears to be the BCR-ABL–induced down-regulation of CCAAT/enhancer binding protein-α (CEBPα) through the stabilization of the translational regulator heterogeneous nuclear ribonucleoprotein E2 (hnRNP E2), which is low or undetectable in CP but readily detectable in BP CML.54 Interestingly, dominant-negative mutations of CEBPα are fairly common in AML,55 but rare in CML-BP.56
Aberrant activation of Wnt/β-catenin signaling is believed to contribute to CML progression by conferring self-renewal capacity to GMPs. Activated β-catenin regulates self-renewal by undergoing translocation to the nucleus where it interacts with lymphoid enhancer factor/T-cell factor and regulates the transcription of genes such as Myc and Cyclin D1.57 The acquisition of self-renewal by GMPs is expected to greatly increase the pool of LSCs in BP. Interestingly, expression microarray studies have implicated β-catenin activation not only in disease progression but also in resistance to tyrosine kinase inhibitors, supporting the view that drug resistance and disease progression share a common genetic basis.58,59 This has implications for prognostication as well as for the development of strategies to prevent progression and overcome resistance.
Conclusions
BCR-ABL orchestrates an integrated network of signaling pathways that upend the physiological control of proliferation, cell death, DNA repair and microenvironment interaction, and lead to the clinical phenotype of CML. Cooperation with additional genetic events that accumulate over time inevitably leads to BP and drug resistance. Although significant progress has been made toward understanding transformation and disease progression, much remains to be learned. The availability of genome-wide scanning tools will undoubtedly accelerate this process and hopefully lead to the discovery of new therapeutic targets to eliminate CML LSC, overcome drug resistance unless effective therapy is initiated early on, and improve the prognosis of patients whose disease has progressed to BP.
CHRONIC LYMPHOCYTIC LEUKEMIA
CLL is one of the most common types of leukemia in adults and has a relatively consistent immunophenotype including dim-surface immunoglobulin expression, CD19, CD20, CD23 along with the pan T-cell marker CD5.60 The impact on overall survival in young and elderly patients with CLL is quite substantial. Patients with a diagnosis under the age of 50 have a median expected lifespan of 12.3 years, which compares to 31.2 years in an age-matched control group.61 Although younger patients have poor outcome and shortened survival with CLL, several recent studies have also identified elderly patients as a high-risk group for poor survival following treatment.62–65 A small subset of CLL patients have indolent disease for many years and do not require therapy or intervention. Improving our understanding of the origin, biology, and progression of CLL will best help define both risk stratification of patients and also identify potential new treatments for this disease.
Origin of CLL
Attempts to identify the origin of CLL with respect to a normal B-cell counterpart has also occurred and remains a controversial area.66–68 Unlike most other B-cell lymphomas and leukemia with the exception of mantle cell lymphoma, CLL expresses typical mature B-cell markers with coexpression of CD5. This prompted many to hypothesize that CLL may be derived from CD5+ B cells whose immunoglobulin (Ig)VH is unmutated. However, the overall phenotype of CLL with expression of CD5, CD23, and CD19, and low levels of sIgM or IgD is not observed in any other type of normal B-cell counterpart. Additionally, investigators identified that approximately 40% of CLL cases are IgVH unmutated, whereas the remainder are IgVH mutated.69,70 These two groups also were shown to have distinct clinical features, prompting the hypothesis that CLL may represent two distinct diseases.69,70
Two sentinel articles examining mRNA gene expression profiling in CLL and normal B cells provided similar findings, suggesting CLL is in fact one disease with a common CLL gene signature.71,72 The first, by Klein et al.,71 examined cDNA gene expression profiles derived from IgVH unmutated, IgVH mutated, and normal B cells derived from different stages of differentiation. A nonsupervised analysis of gene expression profile demonstrated that IgVH mutated and unmutated CLL cases were not distinguished in any manner among a common profile typical of CLL. This CLL profile in the majority of samples had best resemblance to postgerminal center memory B cells and lacked any similarity to naive B cells, CD5+ B cells, and germinal center centroblasts. A supervised analysis of IgVH unmutated and mutated CLL did demonstrate distinct genes that could separate these two clinical subsets of CLL.
A second article, published concurrently by Rosenwald et al.,72 demonstrated similar findings of a common CLL phenotype described by Klein et al. as compared with other normal B cells and B-cell malignancies. In particular, the CLL gene phenotype was not shared by CD5+ normal B cells, thereby providing collaborating evidence that this is likely not the CLL cell of origin. Using a nonsupervised analysis of CLL samples, IgVH unmutated and mutated samples were intermingled. However, supervised analysis of IgVH unmutated and mutated CLL identified a number of genes differentially expressed in the former group related to B-cell receptor signaling and proliferation. In particular, ZAP-70 (zeta-chain associated protein kinase) overexpression was overexpressed in IgVH unmutated CLL as compared with IgVH mutated CLL.73–76
Subsequent studies have suggested that ZAP-70 expression may partly explain why IgVH unmutated CLL patients have more active evidence of B-cell receptor signaling on ligation of the B-cell receptor.77–79 Multiple studies confirming both the clinical prognostic significance of IgVH mutational status and/or ZAP70 expression have subsequently been reported. IgVH and/or ZAP70 represent very strong independent variables in predicting early disease progression, treatment remission duration, and survival of CLL patients. Unfortunately, the extreme variability in measurement of ZAP-70 expression among different investigators has limited full application of this biomarker clinically.
Chromosomal Abnormalities in the Pathogenesis of CLL
In CLL, conventional metaphase cytogenetics can identify chromosomal aberrations in only 20% to 50% of cases because of the low in vitro mitotic activity of CLL tumor cells.80 Early nonstimulated metaphase karyotype studies of CLL demonstrated abnormalities in descending frequency of occurrence, including trisomy 12, deletions at 13q14, structural aberrations of 14q32, and deletions of 11q, 17p, and 6q.81 In addition, complex karyotype (three or more abnormalities) occur in approximately 15% of patients and were noted in these early studies to predict for rapid disease progression, Richter’s transformation, and inferior survival.82–84 The use of CD40L or combination of interleukin (IL)-2 and cytosine-phosphate-guanosine (CpG) stimulation prior to metaphase analysis has also been reported with identification of translocations in 33 of 96 patients (34%) that were both balanced and unbalanced, which is associated with significantly shorter median time from diagnosis to requiring therapy and overall survival.85 Subsequent comparative genomic hybridization (CGH) and global single nucleotide polymorphism (SNP) array studies in CLL have confirmed these deletions and also other chromosomal deletions in CLL.86–88 Increasing aberrations in these same studies of CGH or SNP arrays have been associated with more aggressive disease.
Given the limitation of standard or stimulated karyotype analysis and inability to feasibly analyze patients by CGH or SNP arrays, interphase cytogenetics of known abnormalities are used to identify common, clinically significant aberrations in CLL. The largest study of interphase cytogenetics resulted in improved sensitivity to detect partial trisomies (12q12, 3q27, 8q24), deletions (13q14, 11q22-23, 6q21, 6q27, 17p13), and translocations (band 14q32) in more than 80% of all cases. In a large study of 325 patients by Dohner et al.,89 a hierarchical model consisting of five genetic subgroups was constructed on the basis of regression analysis of CLL patients with chromosomal aberrations. The patients with a 17p deletion had the median survival time of 32 months and shortest treatment-free interval (TFI) of 9 months, whereas patients with 11q deletion followed closely with 79 months and 13 months, respectively.89 The favorable 13q14 deletion group had a long TFI of 92 months and a median survival of 133 months, while the group without detectable chromosomal anomalies and those with trisomy 12 fell into the intermediate group with median survival of 111 and 114 months. Their TFI was 33 and 49 months, respectively. Based on this pivotal study, CLL patients are prioritized in a hierarchical order (deletion 17p13 > deletion 11q22-q23 > trisomy 12 > no aberration > deletion 13q14).89 Of interest, patients with high-risk interphase cytogenetic abnormalities or other complex abnormalities are almost always found to have IgVH unmutated or ZAP-70–positive CLL.90 The impact of high-risk interphase cytogenetics relative to disease progression, outside its association with IgVH unmutated CLL, remains uncertain.
Gene Mutation or Deletion Versus Epigenetic Silencing in the Pathogenesis of CLL
In many types of cancer including leukemia and lymphoma, the presence of activating mutations of specific oncogenes and also inactivating mutations and/or deletions of tumor suppressor genes directly contribute to the pathogenesis of the disease. In CLL only a few known tumor suppressor genes have been noted to be mutated or deleted in tumor cells. Of these, the p53 tumor suppressor gene has been the most rigorously studied based on it being one of the most commonly silenced genes in cancer. Deletions of the p53 gene are generally noted to occur in 3% to 10% of patients at diagnosis and generally occur as part of loss of large regions in del(17p13).89,91 Mutations of p53 are also relatively uncommon and occur in a similar low proportion of CLL patients. The presence of deletion and mutation of p53 is more common but not uniform, with a proportion of patients having only one of the aberrations. Outcome in all studies is negatively influenced by the presence of a del(17p13.1), whereas only a subset of studies have shown that loss of p53 predicts aggressive disease course with rapid progression to time requiring therapy and inferior survival.92–97 Both del(17p) and p53 mutations become more frequent as the disease progresses.96 Overall, this suggests that loss of p53 function by mutation or deletion may be an important contributor to disease progression, but it likely does not contribute to the early pathogenesis of the disease. Indirect inactivation of p53 by mutation of ATM also occurs in CLL and correlates with more rapid disease progression.93,98–100 Similar to p53, loss of ATM becomes more frequent with disease progression and also is likely a late event in the pathogenesis of this disease.
Whereas enhanced B-cell receptor signaling and ZAP-70 expression has been shown to contribute to the pathogenesis of CLL, efforts to identify mutations in this pathway have been limited. A screen of the tyrosine kinase kinome in selected exons that encode the kinase domain was reported from 95 CLL patients in whom 65 different kinases were examined based on relevance to B-cell biology.101 No somatic tumor-associated mutations were found. The estimated frequency of mutations in the CLL genome based on this study was less than one mutation per 6.21 Mb DNA. The low proportion of mutations fall well under that observed in most solid tumors and also supports that gene mutations may not be a major contributor to the pathogenesis of this disease.
Finally, efforts to identify mutations in commonly deleted gene regions in CLL have been undertaken. Greatest attention has been placed on the most common del(13q14), where no protein coding gene mutation has been observed. However, the Croce group in 2002 identified that the microRNAs miR15 and miR16 resided in the minimally deleted region that was lost in approximately 70% of CLL patients.102 MicroRNAs are small noncoding RNAs that effectively inhibit translation or induce degradation of numerous genes based on partial complementarity to site at the 3’ untranslated region of mRNA. Further support for the role of miR16.1 in the pathogenesis of CLL came from identification of both germ line mutations in a small number of CLL patients with a history of familial CLL.103 Additionally, it was noted that New Zealand Black mice that develop both autoimmune disease and an indolent murine CLL and also have an acquired point mutation in the 3’ flanking sequence of microRNA mir-16-1.104 The linkage of the mir-15a/16-1 complex to the development of murine CLL in a spontaneous mouse model suggests that mir-15a/16-1 is at least one of the molecular aberrations responsible for CLL. Klein et al.,105 in a recent article, have demonstrated that a mouse with deletion of the miR15/miR16 region also develops a disease similar to human CLL, confirming the ultimate importance of this miR cluster in the development of CLL. These findings related to miRs and absent mutations suggest that miRs and other forms of epigenetic silencing, as opposed to mutational silencing of genes, are likely much more important in the pathogenesis and progression of CLL.
The role of microRNA and other noncoding genes in transcriptional and translational silencing of genes in CLL is now widely expanding. A subsequent study has demonstrated miR15 and miR16 target genes involved in disrupting apoptosis (bcl-2).106 Multiple additional studies have demonstrated several other known important genes in the pathogenesis of CLL. Similarly, other forms of epigenetic silencing are also being shown to be relevant. Examples of alternative forms of epigenetic silencing can occur via promoter CpG methylation, recruitment of specific corepressor complexes, or recruitment of specific chromatin remodeling ATPase screening of CLL patients demonstrate distinct patterns of methylation by several groups.107–110 Multiple oncogenes (bcl-2, TCL1, ZAP70)111–113 have been shown to be aberrantly hypomethylated (hence expressed), whereas tumor suppressor genes (DAPK1, SFRP1, ID4, and PTPRO)114–117 are hypermethylated (hence silenced), suggesting promoter methylation is one important key to gene regulation as part of malignant transformation. The current model for gene silencing likely involves loss of gene transcriptional activity followed by methylation.118,119
CLL and Proliferation
For many decades, CLL was viewed as a nonproliferating leukemia whose pathogenesis was driven solely by disrupted apoptosis and extended tumor cell survival. This paradigm was in part perpetuated based on the nonproliferating blood compartment. However, as with normal B cells, it has come to be recognized that CLL cell proliferation likely occurs in sites where microenvironment stimulation can occur, such as the lymph node and bone marrow. In such sites, proliferation centers are observed with a high proportion of dividing CLL cells that are often surrounded by either T cells or accessory stromal cells capable of providing cytokine costimulation.120,121 Advances in technology including oral intake of heavy water have come forward to accurately measure all body compartments of CLL and assess the birth rate of CLL tumor cells in vivo in patients.122 These studies have demonstrated a broad range of proliferation of CLL cells that varies based on disease state and also IgVH mutational status.123,124 As one might expect, this same proliferation rate identified through heavy water studies in CLL was also shown to be predictive of disease progression. Collectively, these studies have at least partially discredited the theory that CLL is purely an accumulative disease, and rather focused study on specific compartments of the body that have very different biologic features of proliferation.
CLL and Disrupted Apoptosis
As the normal cell from which the disease CLL is derived is uncertain, it is quite difficult to equivalently compare to know if a difference in spontaneous apoptosis exists. However, several studies derived from CLL do provide evidence that apoptosis at least in vivo is disrupted. Despite the rarity of Bcl-2 gene rearrangement in B-CLL, overexpression of Bcl-2 mRNA and bcl-2 protein expression is common and has been shown to contribute to both disrupted spontaneous apoptosis and also ex vivo drug resistance.112,125–128 Similarly, other antiapoptotic bcl-2 family member proteins including MCL-1, A1, and BCL-XL have also been shown to be elevated either in resting CLL or in CLL cells exposed to soluble and contact factors present in the microenvironment and they also contribute to drug resistance.129–131
Finally, a host of transcription factors involving the NFkB,132 WNT,133 hedgehog,134 and JAK/STAT135 signaling pathway have been shown to be constitutively active and also to contribute to disrupted apoptosis and drug resistance in CLL. In particular, differential activation of NF-kappa B activation in CLL119,136–139 as compared with normal resting B cells and its relevant prognostic significance140–142 with respect to predicting outcome and also its positive role in regulating many of the antiapoptotic genes up-regulated in CLL has generated particular interest.
B-cell Receptor Signaling in CLL
Identification of the divergent natural history of CLL based on IgVH mutational status, ZAP-70 expression, and associated enhanced B-cell receptor signaling has raised interest in the role of this in the pathogenesis of CLL.77–79,143 Activation of the proximal lyn kinase and also downstream syk has been demonstrated in CLL.144–146 Additionally, increased activity of the PI3-kinase pathway as measured by both lipid kinase activity and also baseline phosphorylation of p-AKT has been preliminarily reported.147,148 Complementing this finding, a recent study demonstrated that mature memory B-cell development was in great part dependent on the PI3-kinase pathway.149 A recent study using the isoform-specific inhibitor of PI3-kinase-δ demonstrated that much of the survival protection generated by the microenvironment from stromal cells, cytokines (CD40L, IL-6, TNF-α), and fibronectin contact is mediated via PI3-Kinase δ isoform signaling.150 Of great interest has been the transition of these B-cell receptor kinase pathway inhibitors to clinical trials. Here, with syk, PI3-kinase δ isoform inhibitors, and BTK inhibitors they have demonstrated dramatic and often rapid clinical responses with relatively favorable toxicity profile in CLL patients. The success of such therapeutics further emphasizes the importance of BCR signaling in the pathogenesis of CLL.
Progression of CLL: Role of Genomic Instability and Clonal Evolution
Two small studies of previously untreated patients have been examined for features associated with clonal evolution and have noted this to be more frequent in patients with IgVH unmutated status151 or those expressing the surrogate marker for IgVH mutational status, ZAP70.91 The smaller of these studies noted clonal evolution to occur more commonly among those patients with progression.151 One recently published study in which patients with long telomere length were more likely to have IgVH mutated disease and del(13q14), whereas those with del(11q22.3), del(17p13.1), complex karyotype (more than abnormalities) and IgVH unmutated disease were likely to have extended telomeres.152 Furthermore, one small study suggested long telomere length among patients with IgVH unmutated disease could identify patients with an expected extended progression-free survival.153 The contribution of clonal evolution, telomere length, and global hypomethylation in CLL progression will require further study.
Conclusion
A significant amount of data has been produced concerning the pathogenesis of CLL. Emerging from such work is the importance of epigenetics in the progression of CLL from normal B cells and also enhanced B-cell receptor signaling. Murine mouse models have demonstrated the importance of NF-kB, Bcl-2, TCL1, and loss of miR 15, MiR 16, and Leu2 in the pathogenesis of CLL. It is likely that current lines of investigation, along with emerging technologies related to whole-genome signaling, global proteomic assessment, and miR profiling, will lead to further advances in risk stratification and improvement in CLL therapy.
Selected References
The full list of references for this chapter appears in the online version.
1. Deininger MW. Chronic myeloid leukemia: an historical perspective. American Society of Hematology Education Program Book, 50th Anniversary Review. 2008:418.
2. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst 1960;25:85.
3. Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290.
5. Groffen J, Stephenson JR, Heisterkamp N, et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984;36:93–99.
6. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247:824–830.
7. Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996;88:2375–2384.
9. Huntly BJ, Guilhot F, Reid AG, et al. Imatinib improves but may not fully reverse the poor prognosis of patients with CML with derivative chromosome 9 deletions. Blood 2003;102:2205.
12. Melo JV, Gordon DE, Cross NC, et al. The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood 1993;81:158–165.
16. Sattler M, Mohi MG, Pride YB, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell 2002;1:479–492.
18. Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010;463:676.
20. Agarwal A, Bumm TG, Corbin AS, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood 2008;112:1960.
23. Kardinal C, Konkol B, Lin H, et al. Chronic myelogenous leukemia blast cell proliferation is inhibited by peptides that disrupt Grb2-SoS complexes. Blood 2001;98:1773–1781.
29. Ilaria RL, Jr., Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem 1996;271:31704.
32. Hoelbl A, Schuster C, Kovacic B, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med 2010;2:98–110.
33. Verfaillie CM, Hurley R, Zhao RC, et al. Pathophysiology of CML: do defects in integrin function contribute to the premature circulation and massive expansion of the BCR/ABL positive clone? J Lab Clin Med 1997;129:584–591.
34. Ramaraj P, Singh H, Niu N, et al. Effect of mutational inactivation of tyrosine kinase activity on BCR/ABL-induced abnormalities in cell growth and adhesion in human hematopoietic progenitors. Cancer Res 2004;64:5322–5331.
35. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007;7:441–453.
38. Brehme M, Hantschel O, Colinge J, et al. Charting the molecular network of the drug target Bcr-Abl. Proc Natl Acad Sci U S A 2009;106:7414.
39. Pear WS, Miller JP, Xu L, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 1998;92:3780–3792.
40. Koschmieder S, Gottgens B, Zhang P, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 2005;105:324–334.
41. Agliano A, Martin-Padura I, Mancuso P, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int J Cancer 2008;123:2222–2227.
43. Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med 1977;63:125–130.
44. Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6:587–596.
46. Ito K, Bernardi R, Morotti A, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008;453:1072.
48. Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009;458:776–779.
50. Perrotti D, Neviani P. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br J Cancer 2006;95:775–781.
57. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004;351:657–667.
58. McWeeney SK, Pemberton LC, LoWWriaux MM, et al. A gene expression signature of CD34+ cells to predict major cytogenetic response in chronic-phase chronic myeloid leukemia patients treated with imatinib. Blood 2010;115:315–325.
59. Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A 2006;103:2794–2799.
60. Matutes E, Wotherspoon A, Catovsky D. Differential diagnosis in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol 2007;20:367–384.
69. Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–1854.
70. Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999;94:1840–1847.
71. Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 2001;194:1625–1638.
72. Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001;194:1639.
75. Rassenti LZ, Huynh L, Toy TL, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 2004;351:893–901.
77. Chen L, Apgar J, Huynh L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood 2005;105:2036–2041.
85. Mayr C, Speicher MR, Kofler DM, et al. Chromosomal translocations are associated with poor prognosis in chronic lymphocytic leukemia. Blood 2006;107:742–751.
89. Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–1916.
91. Shanafelt TD, Witzig TE, Fink SR, et al. Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 2006;24:4634.
101. Brown JR, Levine RL, Thompson C, et al. Systematic genomic screen for tyrosine kinase mutations in CLL. Leukemia 2008;22:1966–1969.
102. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002;99:15524–15529.
103. Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005;353:1793–1801.
104. Raveche ES, Salerno E, Scaglione BJ, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007;109:5079–5086.
105. Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010;17:28–40.
116. Raval A, Tanner SM, Byrd JC, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007;129:879.
119. Chen SS, Raval A, Johnson AJ, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2009;106:13433–13438.
124. Messmer BT, Messmer D, Allen SL, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 2005;115:755-764.
132. Furman RR, Asgary Z, Mascarenhas JO, et al. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J Immunol 2000;164:2200–2206.
150. Herman SE, Gordon AL, Wagner AJ, et al. The phosphatidylinositol 3-kinase-{delta} inhibitor CAL-101 demonstrates promising pre-clinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010;116:2078–2088.
151. Stilgenbauer S, Sander S, Bullinger L, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 2007;92:1242.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


