Fig. 22.1
Title: Adiposopathy: Simplified relationship between pathogenic adipose tissue and cardiovascular disease [2]. Adiposopathy is promoted by unhealthy nutrition and a sedentary lifestyle in genetically and environmentally predisposed individuals. With impaired adipogenesis of peripheral, subcutaneous adipose tissue during positive caloric balance, existing fat cells may hypertrophy, circulating free fatty acids may increase, and lipids may be deposited in non-adipose tissue organs (e.g., liver, muscle, possibly pancreas) resulting in lipotoxicity. Adiposopathic endocrine and immune responses may be directly pathogenic to the cardiovascular system or otherwise interact with other body systems. If not mitigated by these other body organs, adiposopathy may indirectly cause or promote major atherosclerotic risk factors (type 2 diabetes mellitus, high blood pressure, or dyslipidemia)
Adiposopathy [2] is caused by positive caloric and sedentary lifestyle in genetically and environmentally susceptible patients [3]. Anatomically, adiposopathy is manifest by adipocyte hypertrophy, as well as increased visceral, pericardial, perivascular, and other periorgan adiposity, growth of adipose tissue beyond its vascular supply, increased number of adipose tissue immune cells, and “ectopic fat deposition” in other body organs [4]. Pathophysiological manifestations of adiposopathy include impaired adipogenesis, adipocyte organelle dysfunction, increased circulating free fatty acid levels, and adverse adipocyte and adipose tissue endocrine and immune responses [5]. As importantly, the pathogenic potential of adipose tissue is highly dependent upon interactions or cross talk with other body organs. If such actions and interactions are pathogenic, then the potential clinical manifestations of adiposopathy include hyperglycemia, high blood pressure, dyslipidemia, metabolic syndrome, atherosclerosis, fatty liver, and increased risk of cancer [2].
Hormonal abnormalities associated with adiposopathy include hyperandrogenemia in women and hypoandrogenemia in men. Thus, fat weight gain resulting in adiposopathy may “approximate the genders,” at least with respect to sex hormone status [6, 7]. Women may develop insulin resistance on the basis of inherited post-receptor defects in insulin signaling, irrespective of adiposity. Women who gain body fat may develop adiposopathic responses that also may contribute to insulin resistance. In either circumstance, the resultant compensatory hyperinsulinemia enhances ovarian androgen production and inhibits hepatic synthesis of sex hormone binding globulin (SHBG), which increases free testosterone. These mechanisms help account for the clinical presentations and common endocrinopathies associated with the polycystic ovarian syndrome (PCOS), which depending on the underlying pathophysiology, may occur with and without adiposity. The adverse clinical consequences of PCOS include infertility, increased risk of cardiovascular disease, and increased risk of T2DM [8]. Regarding men, adiposopathic responses may lead to hypoandrogenemia via various mechanisms [9]. Hyperinsulinemia may decrease SHBG, which decreases total circulating testosterone. Hyperleptinemia may suppress testicular androgen production. Increased adipose tissue aromatase activity may increase the conversion of testosterone to estradiol, which may decrease pulsatile luteinizing hormone secretion, which in turn, decreases testicular androgen production. The reduction in male androgens may promote even further adiposity, which may potentially worsen adiposopathy, creating a circular and interconnected pathologic process leading to a number of metabolic abnormalities. This is yet another example as to how adiposopathic responses lead to the clinical and laboratory findings described by the “metabolic syndrome,” as well as an increased risk of atherosclerotic coronary heart disease and T2DM [10].
Conceptually, the origins of the dyslipidemia associated with T2DM are shown in Figs. 22.2, 22.3, and 22.4. If during positive caloric balance, adipose tissue responds with pathogenic endocrine and immune responses, and if adipose tissue is unable to adequately store free fatty acids, then these pathogenic responses and energy overflow are directed to other body organs. If non-adipose tissue body organs (such as the liver and muscle) are not sufficiently “flexible” in their capacity to manage the adverse metabolic onslaught, then such organs may become dysfunctional, contributing to metabolic disease (Fig. 22.2). An illustrative example most applicable to this discussion is shown in Figs. 22.3 and 22.4. If due to genetic or environmental limitations, the liver is unable to oxidize the excessive free fatty acid load derived from visceral adipose tissue into the portal circulation, then this may clinically result in the common clinical findings among patients with T2DM and metabolic syndrome, such as fatty liver, increased VLDL secretion, as well as generation of small LDL and HDL particles, and increased triglyceride rich lipoprotein remnants [11].
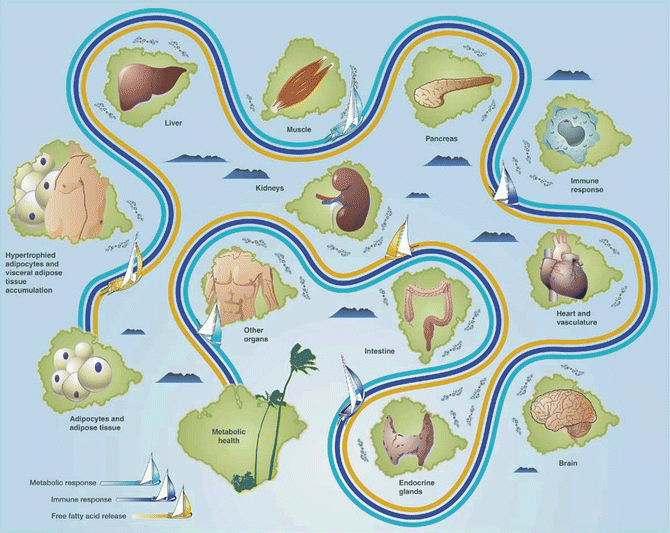
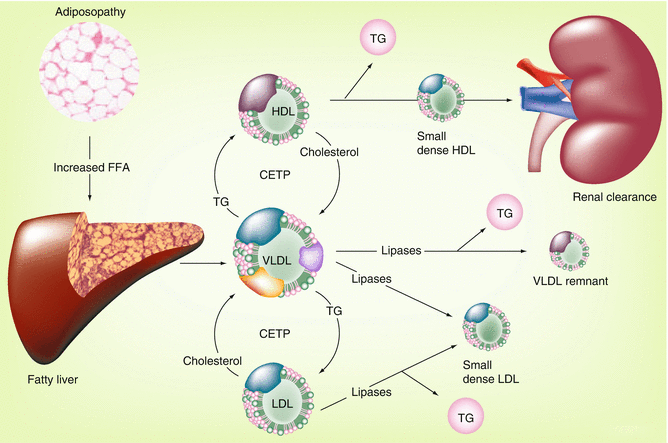
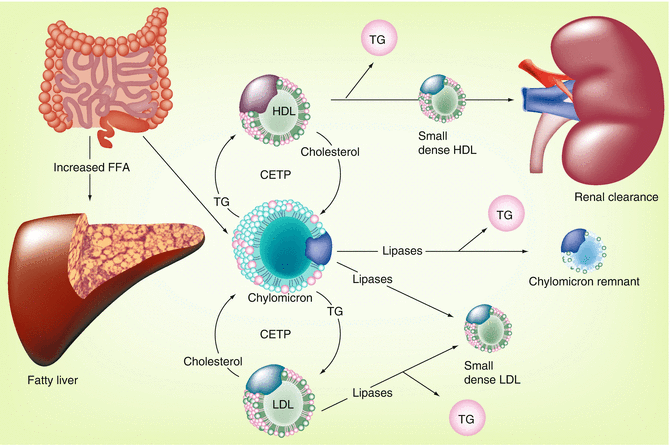
Fig. 22.2
Navigating the pathogenic potential of adiposopathy. Fat cell enlargement and accumulation of adipose tissue in the visceral area often result in pathogenic adipose tissue metabolic and immune responses, including the net release of free fatty acids, which may be lipotoxic to peripheral organs. The potential of pathogenic adipose tissue to cause metabolic disease is largely dependent on cross talk and interactions with, as well as responses of other body tissues
Fig. 22.3
Adiposopathy and the dyslipidemia associated with the metabolic syndrome [6]. Relation between pathogenic adipose tissue and the characteristic lipid pattern described by the metabolic syndrome: hypertriglyceridemia, low high-density lipoprotein (HDL) cholesterol levels, and small, dense low-density lipoprotein (LDL) particles. CETP cholesterol ester transfer protein, FFA free fatty acid, TG triglyceride, VLDL very low-density lipoprotein
Fig. 22.4
Postprandial contribution to the metabolic syndrome [6]. Adiposopathy may result in an inability of the adipose tissue organ to adequately clear postprandial FFA. This results in diversion (and potential lipotoxicity) of FFA to organs such as the liver. Such increases in FFA may also be lipotoxic to other organs, including muscle and pancreas (Fig. 22.2). The effect of excessive postprandial FFA upon lipid parameters is illustrated. CETP cholesteryl ester transfer protein, FFA free fatty acid, HDL high-density lipoprotein, LDL low-density lipoprotein, TG triglyceride
In addition to the increased VLDL secretion, as shown in Fig. 22.4, one of the endocrinopathies associated with adiposopathy is a relative decrease in lipoprotein lipase activity [4]. Thus, in addition to increased secretion of VLDL, adiposopathy may also result in a reduced capacity to hydrolyze triglycerides in VLDL particles, all resulting in the common clinical finding of hypertriglyceridemia. Subsequently, through the actions of various lipases, VLDL particles are converted to remnant lipoproteins (incompletely digested VLDL particles), which are thought to be atherogenic. Triglycerides carried by VLDL particles may also undergo a 1:1 stoichiometric exchange with LDL and HDL particles via cholesteryl ester transfer protein (CETP). Triglyceride-enriched LDL and HDL particles are better substrates for lipase activity, which results in the formation of smaller and more dense particles.
Cholesterol Flux and the Importance of Intestinal Cholesterol
Irrespective of cause, dyslipidemia in patients with T2DM is a modifiable risk factor, which is especially important given that patients with T2DM are at high risk for CHD [11]. Statins are the first treatment of choice to lower cholesterol levels in T2DM patients. However, when statins are not tolerated or if statin therapy alone is not sufficient in achieving LDL cholesterol treatment goals, then ezetimibe is another lipid-altering drug treatment option.
In both peripheral tissues and the liver, the major precursor for cholesterol synthesis is acetyl coenzyme A (acetyl-CoA), which gives rise to hydroxy-methylglutaryl coenzyme A (HMG-CoA). HMG-CoA reductase is the rate-limiting enzyme in cholesterol biosynthesis. This enzyme converts HMG-CoA to mevalonic acid. Statins inhibit HMG-CoA reductase. Clinically, statins are the most commonly used drug to treat high cholesterol and were originally termed HMG-CoA reductase inhibitors, reflecting their mechanism of action, which was to inhibit the rate-limiting step of cholesterol production.
Textbook descriptions differ when describing the origin of bodily cholesterol production. Strictly speaking, primate studies suggest the greatest amount of cholesterol produced in the body is derived from non-hepatic tissues, such as skin, muscle, and intestine, with the greatest amount of cholesterol produced per gram of tissue being endocrine organs, such as the adrenal gland and sex organs [12] (Fig. 22.5). This is because cholesterol is required for cell membranes, cellular functions, and especially for steroidogenesis. However, what is most clinically relevant regarding dyslipidemia and CHD is not where most total body cholesterol is produced but rather the origin of the cholesterol carried in the blood. Cholesterol is a waxy substance first described in gallstones. The Greek derivation of the term “cholesterol” refers to “chole” for bile and “stereos” for solid. Because cholesterol (a lipid) is insoluble in water, it must be packaged and carried in the blood by polar protein-containing biochemical particles, known as lipoproteins. Thus, most of the circulating cholesterol is hepatic/gastrointestinal in origin, in that most of the cholesterol carried by lipoproteins originates from the liver or intestine.
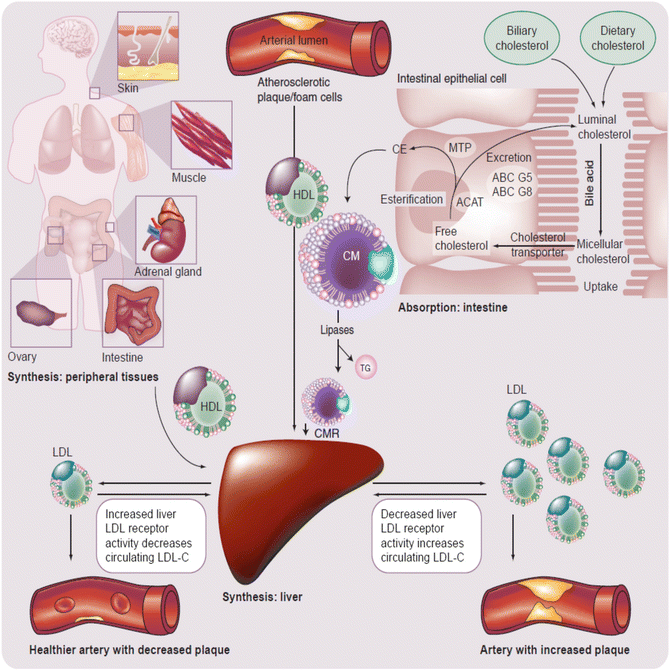
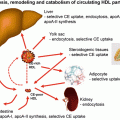
Fig. 22.5
Simplified, global overview of major organs involved in lipoprotein metabolism and subsequent risk for CHD [13]. The liver normally serves as the main regulatory organ determining LDL-C blood levels. Impaired hepatic cholesterol synthesis (such as through administration of “statins”) or impaired intestinal cholesterol delivery (such as through administration of cholesterol absorption inhibitors) result in increased hepatic LDL receptor activity with increased clearance of circulating LDL-C from the blood. Cholesterol from peripheral tissues, including macrophages associated with arterial cholesterol plaques, is transported to the liver via HDL particles. ABC adenosine triphosphate-binding cassette transporter, ACAT acyl-coenzyme A: cholesterol acyltransferase, CE cholesterol ester, CM chylomicron, CMR chylomicron remnant, HDL high-density lipoprotein, LDL low-density lipoprotein, MTP microsomal triglyceride transfer protein
Once released in the blood, different lipoproteins may undergo enzymatic exchanges of cholesterol (for TG) with other lipoproteins (e.g., via cholesteryl ester transfer protein) [13]. Furthermore, LDL particles may transfer cholesterol to peripheral tissues. In most cases peripheral tissues have the capacity to synthesize their own cholesterol. Conversely, HDL particles may transfer free cholesterol from peripheral tissues back to the liver via peripheral cholesterol transport (Fig. 22.5). The fact that cellular production of cholesterol is sufficient for cellular function and that lipoprotein delivery of cholesterol is not required for peripheral tissue function is supported by the lack of widespread tissue and organ failure among patients with homozygous familial hypercholesterolemia, a genetic disorder wherein tissue LDL receptors are lacking. Even when patients with homozygous familial hypercholesterolemia (with lack of LDL receptors) undergo steroidogenesis stress testing, endocrine glands (i.e., body tissues dependent upon cholesterol for steroidogenesis, and thus among the most potentially vulnerable to deficiency of cholesterol transport) hormone production is not diminished, again, due to sufficient intracellular cholesterol production. In fact, it is only after administration of statins in patients with homozygous familial hypercholesterolemia that minor, nonclinically significant reductions in steroidogenesis are observed [14]. The modest, if any clinical effect upon peripheral tissues with statins is likely because statin concentrations are effectually higher in the liver than peripheral tissues, with greater inhibition of cholesterol synthesis in the liver versus extrahepatic tissues by HMG-CoA reductace inhibitors [15].
Excessive delivery and accumulation of cholesterol in arterial subendothelia, as derived from LDL particles, can result in atherosclerotic lesions. The transfer of cholesterol from vascular subendothelial to the liver by HDL particles is often described as “reverse cholesterol transport,” which technically, is not an adequate term to describe the totality of HDL peripheral cholesterol transport function. Quantitatively, cholesterol transport by HDL particles is not restricted to “reversing” cholesterol flux. As noted before, cholesterol is substantially produced from cellular synthesis in peripheral tissues, without the need for cholesterol transport to peripheral tissues for normal cellular function. Thus, the term “reverse cholesterol transport” is best considered a subset of HDL transport function, restricted to describing HDL’s function in specifically retrieving cholesterol from arterial plaques.
If the net transfer of cholesterol is such that cholesterol accumulates in the subendothelial space of arterial vessels, then this is an important step in the progression of atherosclerosis and an important promoter of CHD events. Conversely, if the net transfer dynamics are such that no cholesterol accumulation occurs in the vascular subendothelium, then this reduces the risk of CHD. Overall, the production, exchange, and transfer of cholesterol is in constant flux, and the predisposition to CHD is highly dependent upon the net effect of this flux.
Beyond tissue production, another important contributor to the cholesterol pool, and thus contributor to cholesterol flux, is intestinal cholesterol. Dietary sterols may not substantially contribute to circulating cholesterol and other sterols, except during times of very high cholesterol consumption, because up to three-quarters of the cholesterol delivered to the intestine is derived from biliary cholesterol excretion from the liver, not dietary consumption [12]. Once in the intestinal lumen, both biliary and dietary cholesterol (and other lipids) interact with bile acids allowing for micelle formation, which enhances transport of cholesterol through the jejunal brush border membranes into intestinal epithelial cells. Once in intestinal cells, free cholesterol may be returned to the intestinal lumen through a heterodimer of adenosine triphosphate (ATP)-binding cassette (ABC) transporters G5 and G8 or esterified and then eventually packaged into chylomicron particles which then deliver intestinal cholesterol to peripheral tissues and the liver. Intestinal cholesterol absorption, cholesterol synthesis, and blood cholesterol are thus all interrelated. If intestinal absorption of cholesterol is reduced, then less cholesterol is delivered to the liver, hepatic cholesterol synthesis is increased, the number and activity of hepatic LDL surface receptors are increased, more LDL cholesterol is cleared from the blood, and LDL cholesterol levels are decreased.
Ezetimibe: Mechanism of Action
Ezetimibe is a lipid-altering drug approved to lower cholesterol. Its “mibe” suffix reflects that its discovery was during the evaluation of the clinical utility of various acyl-CoA cholesterol acyltransferase (ACAT) inhibitors, in that “mibe” is a designated name for this group of agents (e.g., avasimibe, pactimibe). Curiously, most ACAT inhibitors do not have clinically meaningful effects upon intestinal cholesterol absorption. Conversely, at approved doses, ezetimibe has no clinically significant ACAT activity. When ezetimibe was approved for clinical use in 2002, the precise molecular target was unknown and was classified as a cholesterol absorption inhibitor. Subsequently, ezetimibe was found to competitively inhibit the sterol transporter, Niemann-Pick C1-like 1 protein (NPC1L1), located on the brush border membrane of intestinal epithelial cells (and at sites in the liver). However, ezetimibe is not classified as an intestinal transport inhibitor, but rather maintained in a class termed intestinal absorption inhibitor. Through inhibiting intestinal cholesterol transport, ezetimibe reduces cholesterol entering the enterocyte, reduces the cholesterol packaged into chylomicrons, decreases the amount of cholesterol delivered to the liver, promotes a compensatory upregulation of hepatic LDL receptors, enhances LDL-C clearance from the circulation, and thus lowers LDL cholesterol blood levels.
This mechanism of action of ezetimibe is complementary to statins, with the combination sometimes described as representing “dual inhibition,” referring to the inhibition of cholesterol production by statins and the inhibition of intestinal cholesterol absorption by ezetimibe. In some ways, both statins and ezetimibe share the same mechanism of action in that both increase hepatic LDL receptor activity resulting in enhance clearance of LDL particles, with a greater potential to reduce cholesterol blood levels, compared to either agent alone. Increased LDL receptor activity may also increase clearance of other apolipoprotein B containing lipoproteins, such as TRL (e.g., VLDL), helping to account for why both statins and ezetimibe lower TG levels.
Ezetimibe: Effects Upon Dyslipidemia Associated with Diabetes Mellitus
Lowering LDL cholesterol remains the primary lipid treatment target for most patients with T2DM. While ezetimibe’s main clinical use is lowering LDL cholesterol, ezetimibe has other effects applicable to the dyslipidemias so often found in patients with T2DM. This may be of clinical importance because CHD risk reduction may best be achieved by modification of multiple CHD risk factors.
Non-HDL Cholesterol
Non-HDL cholesterol is the sum of cholesterol carried by all atherogenic lipoproteins such as LDL, very low-density lipoprotein (VLDL), intermediate density lipoprotein (IDL), remnant lipoprotein (RLP), lipoprotein(a) [Lp(a)], and chylomicrons. Non-HDL cholesterol is calculated as total cholesterol minus HDL cholesterol. Given that non-HDL cholesterol is more inclusive in assessing the cholesterol carried by atherogenic lipoproteins, it should not be surprising that clinical trial data suggests non-HDL cholesterol may be a better predictor of CHD risk than LDL-C levels [16]. In recognition of the clinical importance of non-HDL cholesterol, the National Cholesterol Education Program (NCEP) ATP III has recommended non-HDL-C treatment goals be set at levels 30 mg/dl above the respective LDL-C treatment goals, as a secondary target for high-risk patients with TG of more than 200 mg/dl [1]. Measurement of non-HDL-cholesterol in patients with metabolic syndrome and T2DM is particularly relevant, because many of these patients have increased levels of TRL, apolipoprotein B, and small, dense LDL particles which, while reflected in non-HDL cholesterol, may not be adequately reflected by measuring LDL cholesterol alone. In statin-treated patients, ezetimibe significantly reduces non-HDL cholesterol approximately 15–20 % in those with metabolic syndrome and reduces non-HDL cholesterol approximately 20–25 % in patients with T2DM [17, 18].
Apolipoprotein B
As with non-HDL cholesterol, apolipoprotein B may be a better predictor of CHD risk than LDL cholesterol alone [19], and apolipoprotein B levels less than 90 mg/dl may be considered an alternative secondary target for patients at high CHD risk [20]. Unlike non-HDL cholesterol, apolipoprotein B provides a direct assessment of atherogenic particle number, which is thought to be an important determinant of atherogenic burden and CHD risk. Chylomicron particles contain one molecule of ApoB-48, while lipoprotein particles such as LDL, VLDL, IDL, and other TRL contain one molecule each of ApoB-100. Some assays may measure both apolipoprotein B 48 and 100. Many other apolipoprotein assays only assess apolipoprotein B100. Either way, apolipoprotein B blood levels represent the concentration of atherogenic lipoproteins, which is a measurement of atherogenic risk beyond measuring the cholesterol carried by LDL alone (as reflected by LDL cholesterol levels). This may be especially of clinical importance among metabolic syndrome and T2DM patients who have elevated TG levels, and thus elevated TRL. Adding ezetimibe to ongoing statin therapy significantly reduces ApoB levels approximately 13 % in patients with metabolic syndrome and approximately 18 % among those with T2DM [17].
Triglycerides
Hypertriglyceridemia is generally considered a CHD risk factor. Elevated TG levels are often found in patients with metabolic syndrome and T2DM. If after LDL cholesterol goals are reached, and TG levels remain ≥200 mg/dl, then national guidelines suggest non-HDL cholesterol be assessed, with treatment to non-HDL-C treatment goals [1]. Although support via clinical trial outcome data is lacking among patients with T2DM, achieving a TG level of <150 mg/dl is considered desirable [21]. When added to statin therapy, ezetimibe significantly reduces TG levels in patients with metabolic syndrome by approximately 5–10 % and approximately 10 % in patients with T2DM [17, 18].
HDL Cholesterol
Low levels of HDL cholesterol correlates with increased CHD risk. Although support via clinical trial outcome data is lacking, raising HDL cholesterol is recommended by some organizations in patients at higher risk for CHD [22]. Some suggest that among those with T2DM, achieving HDL cholesterol >40 mg/dl in men and HDL cholesterol >50 mg/dl in women is desirable [21]. When added to statin therapy, ezetimibe significantly increases HDL cholesterol levels in patients with metabolic syndrome and T2DM by approximately 3 % [17, 18].
Remnant-Like Lipoproteins
Elevated levels TRL and their remnants are associated with an increased CHD risk [22, 23]. The cholesterol carried by remnant TRL is included in measurements of non-HDL-C levels, and the number of particles are reflected in measurements of apolipoprotein B. TRL (e.g., chylomicrons, IDL, and VLDL and their remnants) may undergo intravascular remodeling into metabolic by-products of smaller and more dense particles, which may have higher atherogenic potential than larger, more buoyant, particles. Ezetimibe reduces remnant lipoprotein cholesterol approximately 10–20 % [24].
Lipoprotein Particle Size
Both metabolic syndrome and T2DM are often associated with a disproportionate baseline number of smaller LDL particles, which is often described as increasing CHD risk (Figs. 22.3 and 22.4). However, while baseline lipoprotein particle size may have some utility in predicting CHD risk, no evidence suggests that the assessment of lipoprotein particle size is useful in determining the efficacy of lipid-altering intervention. In fact, in some circumstances, post-treatment lipoprotein particle size analyses may be misleading [25].
Mechanistically, it is suggested that smaller, more dense LDL particles may (1) have decreased affinity for tissue and liver LDL receptors, thus prolonging LDL particle presence in the blood; (2) have increased permeability through the arterial endothelium with preferential retention in the arterial wall; and (3) be more readily oxidized, further increasing their atherogenic potential [25]. However, while the lipoprotein particle size effects of a lipid-altering intervention may be scientifically intriguing, the vast majority of scientific data supports LDL cholesterol reduction, non-HDL cholesterol reduction, and atherogenic lipoprotein particle number reduction (as reflected by a reduction in apolipoprotein B) as not only what is most clinically relevant but also what represents validated lipid treatment targets. A challenge to some clinicians arises when administration of cholesterol-lowering drugs (such as statins or ezetimibe) lowers LDL cholesterol, lowers non-HDL cholesterol, and reduces apolipoprotein B levels, but also results in an increase in the proportion of remaining LDL particles that are more small and dense [25]. While lipoprotein particle size may be helpful in assessing baseline CHD risk, no evidence that such measurements are helpful to assess efficacy of lipid-altering therapy [26].
Physiologically, reduced LDL clearance due to impaired LDL receptor binding is one of the proposed reasons why smaller, more dense LDL particles are potentially more atherogenic. It might therefore be expected that when hepatic LDL receptors are upregulated through therapies such as statins and/or ezetimibe, then the larger circulating LDL particles are preferentially cleared. This leaves a disproportionate amount of smaller, more dense LDL particles. But what is most clinically relevant is that both small and large LDL particles are atherogenic and that both statins and ezetimibe reduce the number of large and small LDL particles, both reduce apolipoprotein B, both reduce LDL cholesterol, and both reduce non-HDL cholesterol, which are the lipid parameters of most clinical relevance when assessing post-treatment lipid-altering efficacy [25].
High-Sensitivity C-Reactive Protein
Atherosclerosis is promoted by inflammation. C-reactive protein is an acute phase reactant and biomarker of inflammation whose increase is associated with increased CHD risk. The reduced progression of CHD associated with intensive statin treatment somewhat correlates with reductions in hs-CRP levels [27]. While ezetimibe monotherapy may reduce hs-CRP compared to placebo, these modest differences are generally not statistically significant [28]. However, when ezetimibe is added to ongoing statin therapy, this is when hs-CRP is found to be most consistently and significantly reduced [28, 29].
Ezetimibe: Clinical Trials in Patients with Diabetes Mellitus
In a post hoc assessment of metabolic syndrome or T2DM patients treated with ongoing statin therapy, adding ezetimibe significantly lowered LDL cholesterol, non-HDL cholesterol, total cholesterol, apolipoprotein B, and TG levels, irrespective of the presence of metabolic syndrome or T2DM [17]. In a pooled post hoc analysis of 27 clinical trials (n = 6,541 with T2DM; n = 15,253 without T2DM), ezetimibe combined with statin was more effective than statin monotherapy in improving LDL cholesterol, total cholesterol, HDL cholesterol, TGs, non-HDL-cholesterol, apolipoprotein B, and hs-CRP in the overall population, as well as both subgroups with and without T2DM. The safety profile was also similar between groups. This analysis also suggested that ezetimibe combined with statin may lower LDL cholesterol, total cholesterol, and non-HDL cholesterol more among those with T2DM, compared to those without T2DM [30]. In a study of 1,229 hypercholesterolemic T2DM patients comparing ezetimibe 10 mg/simvastatin 20 mg per day versus atorvastatin 10 or 20 mg per day or ezetimibe 10 mg/simvastatin 40 mg per day versus atorvastatin 40 mg per day, ezetimibe/simvastatin generally provided additional improvements over atorvastatin with regard to LDL cholesterol, total cholesterol, HDL cholesterol, non-HDL cholesterol, TG, and hs-CRP, although these findings were not statistically significant at all dose comparisons. Ezetimibe/simvastatin was also superior to atorvastatin in allowing T2DM patients to attain LDL cholesterol levels less than 70 mg/dl (P < 0.001 for all dose comparisons) [31].
Ezetimibe can be prescribed as monotherapy or combined in a single pill with either simvastatin or atorvastatin. Compared to simvastatin alone, a subgroup analysis of three similarly designed, randomized, double-blind, placebo-controlled studies in patients with primary hypercholesterolemia revealed that ezetimibe plus simvastatin significantly reduced LDL cholesterol, non-HDL cholesterol, apolipoprotein B, TG, and C-reactive protein. These effects were similar among those with and without metabolic syndrome [32]. When compared to doubling of the atorvastatin dose in hypercholesterolemic patients at high CHD risk, T2DM, and metabolic syndrome, a post hoc analysis of a double-blind, parallel group trial of hypercholesterolemia at high CHD risk demonstrated that atorvastatin plus ezetimibe resulted in greater reductions in LDL cholesterol, TG, apolipoprotein B, non-HDL cholesterol, total cholesterol, and lipid ratios in the T2DM, metabolic syndrome, and neither groups [33]. When ezetimibe plus simvastatin was compared to atorvastatin or rosuvastatin in patients with metabolic syndrome or T2DM, subgroup analyses supported ezetimibe plus simvastatin as providing greater improvements than atorvastatin or rosuvastatin in LDL cholesterol, total cholesterol, HDL cholesterol (versus atorvastatin only), non-HDL cholesterol, LDL-C:HDL-C ratio, TC:HDL-C ratio, and apolipoprotein B in all subgroups. A greater percentage of patients receiving ezetimibe plus simvastatin attained their individual National Cholesterol Education Program Adult Treatment Panel III LDL cholesterol goals of <100 mg/dl or LDL-C <70 mg/dl [1, 34], as well as non-HDL cholesterol treatment goal, again, regardless of subgroup [35].
Related posts:
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
 Insulin Resistance and Atherosclerosis
Insulin Resistance and Atherosclerosis
 Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
 Lipoprotein (LDL and HDL) Oxidation in Diabetes Mellitus
Lipoprotein (LDL and HDL) Oxidation in Diabetes Mellitus
 Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
 Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


