FIGURE 28.1 Examples of grade II (panel A), grade III (anaplastic: panel B), and grade IV (glioblastoma: panels C and D) astrocytoma histopathology. Arrow in panel B shows mitotic figure that is a classification criterion of grade III malignancy. Panels C and D show microvascular proliferation and necrosis with perinecrotic cellular palisading, respectively, which are diagnostic criteria of glioblastoma, grade IV. The asterisk in panel D denotes the necrotic focus.
CNS TUMOR HISTOPATHOLOGY AND MOLECULAR CORRELATES
Diffuse, Fibrillary Astrocytomas
Diffuse, fibrillary astrocytomas are the most common type of primary brain tumor in adults. These tumors are divided histopathologically into three grades of malignancy14: World Health Organization (WHO) grade II diffuse astrocytoma, WHO grade III anaplastic astrocytoma, and WHO grade IV glioblastoma (GBM) (Fig. 28.1). WHO grade II diffuse astrocytomas are the most indolent of the spectrum. Nonetheless, these tumors are infiltrative (Fig. 28.1A) and have a marked potential for malignant progression.18
TABLE 28.1
COMMON CENTRAL NERVOUS SYSTEM TUMORS AND CORRESPONDING GENE ALTERATIONSa
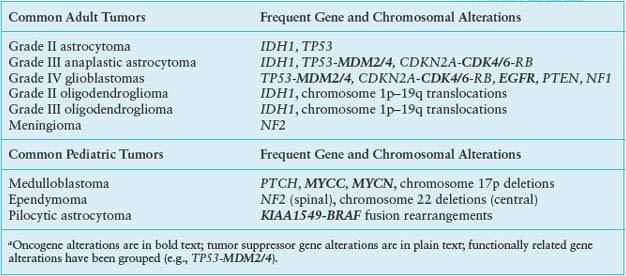
Alterations of p53, a tumor suppressor encoded by the TP53 gene on chromosome 17p, play a key role in the development of at least one-third of all three grades of adult astrocytoma.19,20 In addition, in higher-grade astrocytomas, p53 function may be deregulated by alterations of other genes, including amplification of MDM2 or MDM4 and 9p deletions that result in loss of the p14 product of the CDKN2A gene (Fig. 28.2). A recent survey of this pathway demonstrated gene alterations that compromise p53 function in 87% of glioblastoma.21
Studies revealing frequent alterations of TP53 in sporadic astrocytoma are complemented by various model system investigations that support the contribution of p53 inactivation in the early stages of astrocytoma formation. For instance, cortical astrocytes from mice without functional p53 appear immortalized when grown in vitro and acquire a transformed phenotype with sustained propagation in defined media.22 Cortical astrocytes from mice with haploid TP53 status behave more like wild type astrocytes and only show signs of immortalization and transformation after losing their sole wild type copy of TP53.22 Results associated with p53 inactivation in genetically modified mouse models also support the importance of p53 loss of function in promoting astrocytoma initiation, although such demonstrations have been reported in the context of an accompanying, second gene alteration, such as NF1 gene inactivation.23 In total, there is ample evidence provided through numerous avenues of investigation that indicate the importance of compromised p53 function to the formation of astrocytoma.
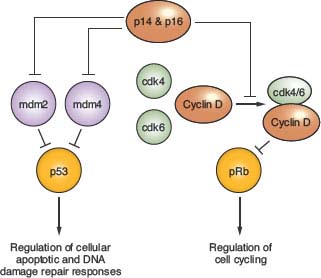
FIGURE 28.2 Regulation of p53 and pRb function. p14 and p16 function is inactivated in approximately half of glioblastoma, as well as in a significant fraction of grade III (anaplastic) astrocytoma, due to homozygous deletion of a DNA sequence at chromosomal location 9p21 that encodes each of these tumor suppressors. The genes encoding mdm 2 and 4, as well as for cdk4 and 6, are amplified in some high-grade astrocytomas, and provide alternative genetic mechanisms to the p14 + p16 gene deletions for achieving suppression of p53 and pRb function. The TP53 and RB genes that encode these tumor suppressor proteins are themselves inactivated in many high-grade astrocytomas, and in such instances the gene alterations affecting upstream regulators are not observed. Proteins indicated in green are oncogenic, whereas those indicated in red act as negative regulators of cell growth (tumor suppressors). Percent values indicate gene alteration frequencies, as defined by The Cancer Genome Atlas project.21
During the 16 months prior to this writing, it has become well established that mutations of the isocitrate dehydrogenase 1 gene (IDH1) occur in a large fraction of grade II and grade III gliomas, irrespective of relative astrocytic versus oligodendroglial histology.24 The possible consequences of this gene alteration are discussed later, but, notably, antibodies specific to the mutant form of the IDH1 protein can now be used reliably for glioma diagnosis on routine tissue sections.25 Interestingly, both TP53 and IDH1 mutations have been positively correlated with tumor MGMT methylation, which, in turn, is thought to accelerate G:C to A:T transition mutations. Others have previously noted that gliomas with MGMT methylation have a higher incidence of such mutations in TP53,26 and MGMT methylation has recently been correlated with IDH1 mutation in glioma,27 with the most common IDH1 alteration also being of the G:C to A:T transition type. Thus, it is possible that epigenetic alteration of low-grade glioma genomes, specifically methylation inhibition of MGMT expression, promotes the mutation of genes whose alteration are well established as occurring frequently in low-grade tumors.
Progression to Anaplastic Astrocytoma
The transition from WHO grade II astrocytoma to WHO grade III anaplastic astrocytoma is accompanied by a marked increase in malignant behavior.14 Although patients with grade II astrocytomas may survive for 5 or more years, patients with anaplastic astrocytomas often die within 2 or 3 years and frequently show progression to GBM. Histologically, the major differences between grade II and grade III tumors are increased cellularity and the presence of mitotic activity (Fig. 28.1B), implying that higher proliferative activity is the hallmark of the progression to anaplastic astrocytoma.
A number of molecular abnormalities have been associated with anaplastic astrocytoma, and several studies indicate that these abnormalities converge on one critical cell-cycle regulatory complex which includes the p16, cyclin-dependent kinase 4 (cdk4), cdk6, cyclin D1, and retinoblastoma (Rb) proteins. The simplest schema suggests that p16 inhibits the cdk6/cyclin D1 and/or cdk4/cyclin D1 complexes, preventing these from phosphorylating Rb, and so ensuring that phospho-Rb (pRb) maintains its brake on the cell cycle (Fig. 28.2).
Chromosome 9p loss occurs in approximately 50% of anaplastic astrocytomas and GBMs, and these 9p alterations target the CDKN2A locus, which encodes the p16 and ARF proteins. The CDKN2A gene is inactivated either by homozygous deletion or, less commonly by point mutations or hypermethylation.28,29 Loss of chromosome 13q occurs in one-third to one-half of high-grade astrocytomas, with the RB1 gene preferentially inactivated by losses and mutations. RB1 and CDKN2A alterations in primary gliomas are inversely correlated, and rarely occur together in the same tumor. Inactivation of pRb or p16 in mouse astrocytes has been shown to lead to anaplastic astrocytomas.30 Amplification of the CDK4 gene, located on chromosome 12q13–14, provides an alternative to subverting cell-cycle control and facilitating progression to GBM in up to 15% of malignant gliomas.28 Detection of any of the gene alterations known to influence Rb protein function (CDKN2A homozygous deletion, CDK4 amplification, or RB deletion + mutation) is associated with a poor prognosis for anaplastic astrocytoma patients.31
Allelic losses on chromosome 19q have been observed in up to 40% of anaplastic astrocytomas and GBMs, indicating a progression-associated glial tumor suppressor gene that maps to 19q13.3, but the gene(s) being targeted for inactivation has is yet to be identified.
Progression to Glioblastoma
GBM is the most malignant grade of astrocytoma, with survival times of substantially less than 2 years for most patients. Histologically, these tumors are characterized by dense cellularity, high proliferation indices, microvascular proliferation (Fig. 28.1C) and focal necrosis (Fig. 28.1D). The highly proliferative nature of these lesions is most likely the result of multiple mitogenic effects. As previously mentioned, at least one such effect is deregulation of the p16-cdk4/6-cyclin D1-pRb pathway of cell-cycle control (Fig. 28.2). The vast majority, if not all, GBM have alterations of this system, whether it involve inactivation of p16 or pRb, or overexpression of cdk4.9,28,29,32
Chromosome 10 loss is a frequent finding in GBM, occurring in 60% to 95% of these tumors, and is far less commonly observed in anaplastic astrocytomas. The PTEN tumor suppressor gene at 10q23.3 is clearly one target of the chromosome 10 deletions, with PTEN mutations of the remaining allele identified in up to 30% of GBM, and a lesser percent of GBM having deletion of all or part of their remaining PTEN gene.21,33 PTEN functions as a 3’ phosphoinositol phosphatase that influences cell proliferation through modulation of the PI3-kinase signaling pathway; PTEN also has protein tyrosine phosphatase activity. Results from model system studies, involving approaches such as the introduction of wild type PTEN into glioma cells with inactivated endogenous PTEN, and which results in the suppression of cell growth,34 or the inactivation of PTEN function in genetically modified mice, which accelerates tumor formation,30,35 support the loss of PTEN function as a critical step in the development of high-grade astrocytic malignancy.
In contrast to the deletion of tumor suppressor genes such as PTEN, key oncogenes experience increased copy number and/or elevated expression in GBM. A signature example of this is the gene for epidermal growth factor receptor (EGFR), which encodes a transmembrane receptor tyrosine kinase, whose ligands include EGF and transforming growth factor-a. GBMs with EGFR gene amplification display overexpression of EGFR transcript and protein. EGFR amplification is consistently reported in approximately 40% of all GBM,36 while being amplified at a much lower frequency in anaplastic astrocytomas. GBMs that exhibit EGFR gene amplification have, in nearly all instances, lost genetic material on chromosome 10, and often have CDKN2A deletions.37 Approximately one-third of those GBM with EGFR gene amplification also have specific EGFR gene rearrangements,38 which produce truncated molecules that are constitutively activated in the absence of ligand and that enhance tumor cell proliferative properties.39,40 The most common of the EGFR mutants, EGFRvIII or ∆EGFR, when expressed in mouse astrocytes lacking p16, causes the formation of intracranial tumors that resemble human GBM.41
Much of our existing knowledge, reviewed in the previous sections, regarding gene alterations in GBM, was confirmed by results generated from two ambitious projects that combined detailed genome and transcriptome analyses of large series of histopathologically validated GBM. One of these identified mutation of IDH1 in 12% of GBM,32 an observation that was subsequently extended to grade II and grade III gliomas, among which IDH1 gene alterations are much more common. Our understanding of the roles of mutant IDH proteins in glio-magenesis is at an early stage, but one investigation suggests that these mutations may result in the accumulation of metabolites that activate the hypoxia-induced factor transcription factor,42 the tumorigenic effects of which are well established.43 Another suggested that the mutant and wild type IDH1 molecules may combine in a pathway that results in an oncogenic metabolite, 2-hydroxyglutarate.44
The second molecular profiling study, from The Cancer Genome Atlas (TCGA) initiative of the National Institutes of Health, confirmed and extended the importance of receptor tyrosine kinases and downstream signaling mediators in GBM by identifying frequent mutation of the NF1 tumor suppressor gene, PI3 kinase regulatory and catalytic subunits, ERBB2, and amplifications of PDGFRA (Fig. 28.3).21 Previous GBM involvement of this signaling network, as indicated through frequently occurring gene alterations, had primarily been limited to EGFR and PTEN, although each of the individual alterations had been noted in prior publications. This comprehensive study indicated that as many as 88% of GBM have one or more alterations affecting RTK-RAS-PI3K signaling.
The identification of frequent NF1 gene alterations by the TCGA consortium merits special commentary. Activating mutations of RAS, one of the most common gene alterations in human cancer, have long been known to be infrequent in brain tumors, and, therefore, activated RAS has been generally assumed to not play a significant or frequent role in CNS tumor development. Given the new information from TCGA, indicating that NF1 gene inactivation occurs in 15% to 20% of all GBMs, elevated Ras activity is now implicated in a subset of these tumors, with particularly high incidence of inactivation noted in the so-called mesenchymal GBM subtype45 (see “Subsets of Glioblastoma”).
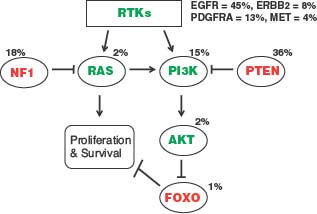
FIGURE 28.3 Alterations of RTK/RAS/PI3K signaling in glioblastoma. The third core signaling pathway, at least one element of which is altered in nearly all GBM, as confirmed by The Cancer Genome Atlas (TCGA) project.9 Receptor tyrosine kinase (RTK) alterations most frequently involve EGFR, with decreasing involvement of PDGFRA, ERBB2, and MET. Downstream signaling mediators RAS and PI3K can be activated by mutation, but are more frequently deregulated by inactivating alterations of the NF1 and PTEN tumor suppressor genes. Percent values indicate gene alteration frequencies, as defined by the TCGA project.
Subsets of Glioblastoma
Suggesting that all astrocytomas progress through identifiable genetic stages in a linear fashion is an oversimplification. Indeed, it appears as if there are biologic subsets of astrocytomas that reflect the spectrum of clinical heterogeneity observed in these tumors. For instance, approximately one-third of GBM have TP53/chromosome 17p alterations, one-third have EGFR gene amplifications, and one-third have neither change.46
GBMs with TP53 mutations that result from the malignant evolution of a lower malignancy grade astrocytic lesion are referred to as secondary (or progressive) GBM. The recently discovered IDH1 mutations, seen commonly among grade II and III gliomas, similarly show preferential association with GBM that have evolved from lower malignancy grade precursors.24 In contrast to secondary GBMs, GBMs with EGFR amplification may arise de novo or rapidly from a pre-existing tumor, without a clinically evident, lower malignancy grade precursor, and are referred to as primary GBM (Fig. 28.4).

FIGURE 28.4 Current schemes for major subgroup classification of glioblastoma. The primary versus secondary classification is based on the presence or absence of clinical history indicating GBM development from a pre-existing glioma of lesser malignancy, with specific gene alterations found to be associated with such clinical history.81 Descriptive terms for the lower classification scheme are based on tumor molecular profiles suggesting specific stages of neurogenesis.58 This classification scheme has recently undergone modification in association with The Cancer Genome Atlas project.80 +, presence of gene alteration; −, absence of gene alteration.
Another genomic alteration that defines a significant subset of GBM is the loss of chromosome 10, and in a study using array comparative genomic hybridization (CGH) to identify tumors with chromosome 10 deletion, the analysis of corresponding expression profile data revealed that a novel gene product, the transcript for YKL-40, is significantly up-regulated in tumors with chromosome 10 loss.47 Whereas the underlying basis of this relationship is yet to be determined, regulation of YKL-40 expression is a matter of interest given the association of elevated YKL-40 with reduced survival for GBM patients.48
In contrast to the use of single biomarkers, such as chromosome 10, or EGFR copy number status, or TP53 and IDH1 mutation status for the subclassification of GBM, the comprehensive molecular screening techniques, that generate thousands of data points for each tumor specimen analyzed, and various algorithms (often referred to as hierarchical clustering) are proving useful and increasingly popular for identifying patterns associated with clinical or biological properties of interest. An example of this approach, which has served as a paradigm for GBM molecular subclassification, showed that gene expression profiling reproducibly identified three prognostically significant subsets of GBM patients.49 Descriptive terms for GBM subclassification that were introduced by this study have undergone revision as a result of the TCGA initiative, and currently include classical (proliferative), mesenchymal, proneural, and neural subgroups.21 These subgroups have strong associations with EGFR, NF1, and TP53 + IDH1 gene alterations, respectively, with the neural subgroup mostly lacking a distinct signature (Fig. 28.4).45
Taking into consideration the cumulative results of GBM molecular profiling studies to date, comprehensive molecular characterizations are clearly providing approaches for a clinically relevant classification of GBM that is more robust than achieved using conventional histopathology.3,4,50 Furthermore, results from transcriptome characterizations have provided insight regarding the molecular biology of this cancer, and have also revealed potential therapeutic targets for the treatment of GBM.
Other Astrocytomas
Pilocytic astrocytoma (PA; WHO grade I) is the most common astrocytic tumor of childhood and differs in its molecular biology, clinical behavior, and histopathology from the diffuse, fibrillary astrocytomas that affect adults. These tumors frequently occur in patients with NF1, and correspondingly, allelic loss of the NF1 gene on chromosome 17q is observed in approximately half of the PAs from NF1 patients, and nearly all NF1-associated PAs show a lack of NF1 protein expression. Observations derived from the analysis of NF1-associated PAs have shown, however, their distinct gene expression signatures relative to sporadic PAs,51 suggesting distinct molecular etiologies for sporadic PAs. The recent discovery of a gene alteration that is frequent among sporadic PAs has provided important insight regarding the molecular etiology of these tumors. Specifically, the determination of gene rearrangements and fusions between the BRAF and KIAA1549 genes, have been shown in the majority of PAs examined.52 Alternative genetic alterations involving RAF1 and BRAF have also been demonstrated in PAs.53 The demonstration of KIAA1549-BRAF fusion proteins having activated BRAF kinase activity establishes these gene alterations as being oncogenic.52 However, because PAs do not generally progress to high-grade malignancy, BRAF oncogenic activation does not apparently mark these tumors for malignant conversion.
Malignant pediatric astrocytic tumors are histologically similar to their adult counterparts (astrocytomas, anaplastic astrocytomas, and GBMs), and share some of the genetic alterations associated with the adult tumors, especially TP53 alterations.54 However, EGFR and PTEN alterations that are commonly observed among adult grade III and IV astrocytomas do not occur at high incidence among the pediatric cases.55 Consequently, the molecular basis of high-grade malignancy in many pediatric astrocytomas may well be distinct from that of corresponding adult tumors. Such a possibility is supported by results from a recent study in which a common point mutation of BRAF was identified in nearly one-fourth of grade II, III, and IV pediatric astrocytomas.56 BRAF missense mutations are uncommon among adult glioblastomas.57 Pediatric astrocytomas with BRAF alterations were shown as having frequent, accompanying CDNK2A homozygous deletion, suggesting a cooperativity between these alterations for contributing to the malignant progression of pediatric astrocytoma.
Oligodendrogliomas and Oligoastrocytomas
Oligodendrogliomas and oligoastrocytomas (mixed gliomas) are diffuse, usually cerebral tumors that are clinically and biologically related to the diffuse, fibrillary astrocytomas.14 These tumors are less common than diffuse astrocytomas and have generally better prognoses. Patients with WHO grade II oligodendrogliomas, for instance, may have mean survival times of 10 years. In addition, oligodendroglial tumors appear to be differentially chemosensitive, when compared with the diffuse astrocytomas.
Microdissection of the oligodendroglial and astrocytic portions of oligoastrocytomas has shown identical genetic alterations in morphologically distinct portions of individual tumors, suggesting a common underlying molecular biology despite an apparent heterogeneous cellular composition. Allelic losses in oligodendrogliomas and oligoastrocytomas occur preferentially on chromosomes 1p and 19q, affecting 40% to 80% of these tumor types.58 Because of the frequent loss of these loci in low-grade as well as anaplastic tumors, inactivation of the inferred 1p and 19q tumor suppressor genes has been generally regarded as important to the early stages of oligodendroglial tumorigenesis. The discovery of the majority of these losses being caused by unbalanced translocations between chromosomal arms 1p and 19q implies a scenario in which two tumor suppressor genes could be inactivated through a single event,59 although target loci have yet to be demonstrated. Alternatively, it has been suggested that the 1p–19q translocations reflect a process of chromatin remodeling that exposes regions of homology that are prone to recombination.59 In this scenario, the loss of DNA from 1p and 19q would be associated with global chromatin methylation changes, and would suggest an underlying molecular biology of the translocations that is not specifically directed at the deletion of 1p and 19q sequences.
In addition to 1p and 19q deletions, oligoastrocytomas may suffer allelic losses of chromosome 17p, although these losses are not consistently associated with TP53 mutations. Oncogene amplifications are rare in tumors with oligodendroglial composition, and in total, as well as in contrast to malignant astrocytomas, there is relatively little known regarding the genetic etiology of these tumors, with the exception being the frequent occurrence of IDH1 mutations, which have been demonstrated as occurring in all grade II and III gliomas, including oligodendrogliomas.24 Potential associations of such mutations with epigenetic alterations were discussed previously, and it may well be that further insight regarding the molecular etiology of low and intermediate malignancy gliomas will be forthcoming with the maturation of methods for characterizing tumor epigenomes.60
Anaplastic oligodendrogliomas have proven to be the first brain tumor for which molecular genetic analysis has had practical clinical ramifications: anaplastic oligodendrogliomas with translocation-associated allelic losses of chromosomes 1p and 19q follow different clinical courses from those tumors that do not have this genetic alteration.59 Tumors with 1p and 19q loss are usually sensitive to procarbazine, CCNU and vincristine chemotherapy, with nearly 50% of the cases demonstrating complete neuroradiologic responses; correspondingly, patients whose tumors have 1p and 19q loss have median survivals of approximately 10 years.61 These tumors also appear to have better responses to radiation therapy as well as to temozolomide.62 On the other hand, anaplastic oligodendrogliomas that lack 1p and 19q loss are only vincristine-sensitive about 25% of the time, and only rarely have complete neuroradiologic responses. As a result, patients whose anaplastic oligodendrogliomas lack 1p and 19q loss have median survivals of approximately 2 years.61 Recent studies have demonstrated intermediate recurrence-free survival times in those anaplastic oligodendrogliomas that have polysomy for chromosomes 1 and 19 in addition to the typical 1p and 19q losses.63 At the present time, molecular genetic testing is recommended for: patients diagnosed with anaplastic oligodendrogliomas, small cell malignant tumors in which the differential diagnosis is anaplastic oligodendroglioma versus small cell glioblastoma, and for patients with grade II oligodendrogliomas for whom therapeutic decisions might be influenced by additional knowledge of probable tumor behavior.58 Thus, molecular genetic analysis of 1p/19q allelic status has become a clinically useful test in neuro-oncology.64,65
Ependymomas and choroid plexus tumors
Ependymomas are a clinically diverse group of gliomas that vary from aggressive intraventricular tumors of children to benign spinal cord tumors in adults. Chromosome 22q loss is common in ependymomas, and in spinal ependymomas these losses are associated with mutations of the NF2 gene that resides on chromosome 22.66 For cerebral ependymomas, the paucity of NF2 mutations66 suggests that another, as yet unidentified, chromosome 22q gene is critical to the development of the intracranial form of this cancer.
Choroid plexus tumors are also a varied group of tumors that preferentially occur in the ventricular system, ranging from aggressive supratentorial intraventricular tumors of children to benign cerebellopontine angle tumors of adults. Choroid plexus tumors have been reported occasionally in patients with Li-Fraumeni syndrome and rhabdoid predisposition syndrome, raising the possibility of the involvement of the TP53 gene on chromosome 17p or the hSNF5/INI1 gene on chromosome 22q. However, there is no evidence of hSNF5/INI1 point mutations in patients with choroid plexus papilloma, and the mutational status of TP53 have not been extensively studied in these tumors.
CGH studies of ependymoma have confirmed frequent deletions of chromosome 22, and have additionally revealed frequent losses of 6q.67,68 Recently, array CGH was used in identifying 9q33–34 gains as being associated with ependymoma recurrence and malignant progression.69 Transcriptome analysis has been applied to multiple series of ependymomas, with the results from one such study indicating potential target genes for the 6q and 22q deletions,70 the latter of which being especially important to tumors having no apparent involvement of NF2. Another report has shown increased ependymal tumor expression of genes having defined roles in cell proliferation, and suggests that the expression level of these genes may reliably distinguish between ependymomas of grade II and grade III malignancy.66−71 As has been the case for other types of CNS cancer, gene expression profiling has been used in attempts to identify clinically distinct subsets of ependymoma, with results identifying transcriptional profiles that could distinguish long vs. short-term survivors.72
Medulloblastomas
Medulloblastomas are highly malignant, primitive tumors that arise in the posterior fossa, primarily in children. One-third to one-half of all medulloblastomas have an isochromosome 17q on cytogenetic analysis, and corresponding allelic loss of chromosome 17p has been noted on molecular genetic analysis. TP53 mutations, however, are less common than 17p deletions in medulloblastoma, with the majority of 17p losses occurring preferentially at regions telomeric to the TP53 locus, implying the presence of a second, more distal chromosome 17p tumor suppressor gene. For the fraction of medulloblastomas with TP53 mutation, clinical outcomes appear to be unfavorable,73 thereby supporting determination of medulloblastoma p53 status as an important diagnostic test. In addition to allelic losses on 17p, deletions of chromosome 6q, 8p, 10q, 11, and 16q have also been noted in these tumors. Oncogene amplifications are not common in medulloblastomas, with only MYCC and MYCN shown to be amplified in significant numbers of cases.74 Copy number gains of the OTX2 gene, which encodes a transcriptional regulator of MYCC,75 occur in a small proportion of medulloblastomas.75,76 Gene amplifications and copy number increases may be restricted to the more aggressive large cell and anaplastic subclasses of medulloblastoma.
The discovery of genes underlying two hereditary tumor syndromes has directed attention to signaling pathways involved in medulloblastoma tumorigenesis.77 Gorlin syndrome (also termed nevoid basal cell carcinoma syndrome), a condition characterized by multiple basal cell carcinomas, bone cysts, dysmorphic features, and medulloblastomas arises from defects in PTCH (a homolog of the Drosophila patched gene) on the long arm of chromosome 9. Medulloblastomas, particularly the nodular desmoplastic variants that are characteristic of Gorlin syndrome, can show allelic loss of chromosome 9q and PTCH mutations, and mice that have only one functional copy of the murine Ptch gene are predisposed to the development of tumors that are histologically identical to medulloblastoma. The protein encoded by PTCH functions in the pathway regulated by the Sonic hedgehog protein (SHH). Other molecules in this pathway include smoothened (SMO), and rare SMO mutations have been documented in sporadic medulloblastomas. Intriguingly, both germ line and somatic mutations, along with allelic loss, have been identified in SUFU, another member of the SHH pathway that maps to 10q.78 Alterations in various members of the SHH pathway likely account for the majority of desmoplastic medulloblastomas.
Turcot syndrome, a condition characterized by colonic tumors and brain tumors, is also associated with medulloblastoma; patients with the adenomatous polyposis phenotype may develop medulloblastomas and these patients often have mutations of the APC gene on chromosome 5q.77 The Apc protein operates in the Wnt signaling pathway that includes b-catenin and axin-1, and rare mutations of these genes have been found in sporadic medulloblastomas.
In spite of the contribution of human genetics to our understanding of medulloblastoma development, PTCH and APC gene alterations, as well as alterations of other genes encoding components of their signaling pathways, are associated with less than 15% of sporadic medulloblastomas, suggesting that the genetic etiology of a significant fraction of these tumors has yet to be determined. The question of whether molecular profiling will provide needed information regarding the genetic etiology of these tumors, as well as provide guidance for the management of medulloblastoma patients, has received substantial attention. A recent study evaluated candidate prognostic markers, selected from medulloblastoma expression data, that were immunohistochemically analyzed for their prognostic value using medulloblastoma tissue arrays. Combined expression of three genes, MYCC, LDHB, and CCNB1, was able to predict survival in medulloblastoma patients.79
Collectively, the results from studies of medulloblastoma genome and transcriptome characterizations suggest that these comprehensive approaches are generating information that will be useful in tumor diagnosis and treatment, and for increasing our understanding of the molecular biology of this cancer.
Meningiomas
Meningiomas are common intracranial tumors that arise in the meninges and compress the underlying brain. Meningiomas are usually benign, but some “atypical” meningiomas may recur locally, and some are frankly malignant.14 Homozygous inactivation of the chromosome 22 localized NF2 gene is the rule in the meningiomas of NF2 patients, and NF2 inactivation is observed in the majority of sporadic fibroblastic and transitional meningioma subtypes.80 Inactivating NF2 gene alterations primarily involve immediate truncations, splicing abnormalities or altered reading frames, and result in a lack of, or grossly truncated, proteins.
Approximately 40% of meningiomas have neither NF2 gene mutations nor allelic loss of chromosome 22q. For these tumors, it is likely that alternative tumor suppressor genes are involved, with potential genomic locations of such genes, including chromosomes 1p, 3p, 5p, 5q, 11, 13, and 17p, as suggested by the results of numerous genetic investigations.
Atypical and malignant meningiomas are not as common as benign meningiomas. Atypical meningiomas often show allelic losses for chromosomal arms 1p, 6q, 9q, 10q, 14q, 17p, and 18q, suggesting the presence of progression-associated genes. Chromosome 10 loss, in particular, has been associated with meningiomas displaying clinically malignant behavior beyond that associated with normal brain invasion alone,81 which can occur with tumors that are otherwise benign. Chromosome 10 loss as a predictor of meningioma malignancy provides another example of how molecular genetic investigations have clarified grading issues in neuro-oncology. Another study showed a potential correlation between gain of 1q and shorter progression-free survival for patients with atypical meningioma.10
Hierarchical clustering of expression profiles has also been used to distinguish high-grade from benign meningiomas.82,83 Consistent with cytogenetic-based interpretations, the results of such studies have shown that genes on chromosome 1p and 14q are commonly down-regulated in anaplastic meningiomas, and the 14q11.2-localized NDRG2 gene has been of particular interest because of its decreased expression being associated with meningioma progression.84 Increased expression of the reverse transcription subunit hTERT (reverse telomerase transcriptase), whose enzymatic activity is critical to chromosome telomere maintenance, may also serve as a potential predictor of meningioma malignancy by its association with tumor recurrence and reduced progression-free survival.
CURRENT BASIS OF CNS TUMOR TREATMENT AND RESPONSE TO THERAPY
Most therapeutic approaches fail to eradicate entire CNS tumors, and tumor cells that evade primary therapies are responsible for “recurrent” cancer. One concept that is relevant to acquired therapeutic resistance involves tumor initiating cells (also referred to as tumor stem cells), which exist as a subpopulation of cells within individual tumors and that are capable of sustained self-renewal, thereby promoting unlimited tumor growth.85 Therapeutic targeting of these tumor stem cells may be critically important for the development of curative treatment approaches.86
An additional perspective that is proving highly influential for the treatment of cancer involves the individualization of therapy based on specific tumor genome alterations: two examples of this approach that involve the treatment of glioblastoma are described here.
The first example involves a landmark study that demonstrated an association between glioblastoma MGMT gene methylation status and tumor response to treatment with the DNA alkylating agent temozolomide.87 The molecular basis of this relationship may be attributable, in part, to methylguanine methyltransferase (MGMT) gene promoter methylation, observed in approximately 40% of GBM, which markedly down-regulates the synthesis of MGMT transcript and protein. Patients whose glioblastomas had methylated MGMT who received combined radiation and temozolomide therapy survived significantly longer than patients with MGMT-methylated tumors receiving radiation only, or patients whose tumors lacked MGMT methylation and received mono or combination therapy. However, it is also clear that a significant number of glioblastomas treated with temozolomide develop mismatch repair gene defects, most commonly MSH6 mutations or down-regulation, that lead to a hypermutation phenotype and more rapid progression of disease while being treated with temozolomide.21,88,89
Other studies have addressed the relationship between a molecular subset of GBM and therapeutic response involved therapy with EGFR kinase inhibitors, implicating EGFR and PTEN status in predicting response to these inhibitors.90,91 This interpretation is tenuous, however, as subsequent clinical trials have not demonstrated significant therapeutic effects of these inhibitors, potentially because multiple receptor tyrosine kinases are activated in individual tumors, and the inhibition of one receptor tyrosine kinase, such as EGFR, may contribute to the activation of others.92,93
Nonetheless, such studies suggest that current approaches should combine individual tumor biomarker status with corresponding therapies directed against specific biomarkers, or against signaling mediators of the biomarkers. The wealth of GBM biomarker information made available through the TCGA initiative21 will undoubtedly contribute to the further popularization of this conceptual approach to CNS tumor treatment.
Selected References
The full list of references for this chapter appears in the online version.
1. Louis DN, Pomeroy SL, Cairncross JG. Focus on CNS neoplasia. Cancer Cell 2002;1: 125.
4. Nutt CL, Mani DR, Betensky RA, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 2003;63:1602.
9. Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002;415:436.
13. Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol 2009;19:132.
14. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Lyon: IARC Press, 2007.
17. Wrensch M, Jenkins RB, Chang JS, et al. Variants in the CDKN2B and TEL1 regions are associated with high-grade glioma susceptibility. Nat Genet 2009;41:905.
19. Louis DN, von Deimling A, Chung RY, et al. Comparative study of p53 gene and protein alterations in human astrocytic tumors. J Neuropathol Exp Neurol 1993;52:31.
21. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455:1061.
22. Bogler O, Huang H-JS, Cavenee WK. Loss of wild-type p53 bestows a growth advantage on primary cortical astrocytes and facilitates their in vitro transformation. Cancer Res 1995;55:2746.
23. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 2000;26:109.
24. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765.
27. Weller M, Felsberg J, Hartmann C, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 2009;27:5743.
28. Ichimura K, Schmidt EE, Goike HM, and Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996;13:1065.
29. Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 1996;56:150.
30. Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell 2002;1:157.
32. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321:1807.
34. Furnari FB, Lin H, Huang HS, and Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 1997;94:12479.
35. Kwon CH, Zhao D, Chen J, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res 2008;68:3286.
36. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res 1991;51:2164.
37. Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol 1997;7:871.
38. Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A 1992;89:4309.
40. Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A 1994;91:7727.
41. Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 1998;12:3675.
42. Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009;324:261.
44. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009462:739.
45. Verhaak RG, Hoadley KA, Purdom E, et al; Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 201017:98.
46. von Deimling A, von Ammon K, Schoenfeld D, Wiestler OD, Seizinger BR, Louis DN. Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain Pathol 1993;3:19.
49. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006;9:157.
50. Maher EA, Brennan C, Wen PY, et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res 2006;66:11502.
52. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673.
54. Pollack IF, Finkelstein SD, Burnham J, et al; Children’s Cancer Group. Age and TP53 mutation frerequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res 200161:7404.
56. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res 70:512–9, 2010.
58. Reifenberger G, Louis DN. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol 2003;62:111.
59. Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006;66:9852.
61. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific chromosomal losses predict chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998;90:1473.
64. Ino Y, Betensky RA, Zlatescu MC, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res 2001;7:839.
65. Yip S, Iafrate AJ, Louis DN. Molecular diagnostic testing in malignant gliomas: a practical update on predictive markers. J Neuropathol Exp Neurol 2008;67:1.
66. Ebert C, von Haken M, Meyer-Puttlitz B, et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol 1999;155:627.
71. Korshunov A, Neben K, Wrobel G, et al. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am J Pathol 2003;163:1721.
73. Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 2010;28:1345.
77. Raffel C. Medulloblastoma: molecular genetics and animal models. Neoplasia 2004;6:3102004.
79. de Haas T, Hasselt N, Troost D, et al. Molecular risk stratification of medulloblastoma patients based on immunohistochemical analysis of MYC, LDHB, and CCNB1 expression. Clin Cancer Res 2008;14:4154.
80. Wellenreuther R, Kraus JA, Lenartz D, et al. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. Am J Pathol 1995;146:827.
82. Watson MA, Gutmann DH, Peterson K, et al. Molecular characterization of human meningiomas by gene expression profiling using high-density oligonucleotide microarrays. Am J Pathol 2002;161:665.
85. Singh SK, Hawkins C, Clarke ID, S et al. Identification of human brain tumour initiating cells. Nature 2004;432:396.
86. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756.
87. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997.
88. Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 2007;13:2038.
90. Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005;353:2012.
93. Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007;318:287.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


