Recognizing that cancer is a continuum, oncologists are increasingly expected to be knowledgeable about a diverse array of cancer-related topics including lifestyle behaviors such as diet and exercise, risk assessment, screening, other preventive interventions, in addition to current treatments for advanced malignancy. The understanding, use, and management of interventions designed to delay or reverse the carcinogenesis process have become integral components of the this role.4
DEFINING CANCER RISK–REDUCING AGENTS (CHEMOPREVENTION)
Cancer risk reduction, commonly referred to as chemoprevention, is the use of a range of interventions from drugs to isolated dietary components to whole-diet modulation to block, reverse, or prevent the development of invasive cancer.5,6 Human cancer risk reduction asserts that one can intervene at many steps in the carcinogenic process, which occurs over many years. This prolonged latency provides opportunities to intervene at many time points and at multiple events in the carcinogenic process. Successful deployment of cancer risk–reducing agent interventions requires evidence of reduced cancer-associated incidence and/or mortality.
The concept of field carcinogenesis was first described in the early 1950s as field cancerization in squamous cell carcinomas of the head and neck, and subsequently ascribed to many epithelial sites. The field carcinogenesis concept is that patients have a wide surface area of precancerous or cancerous tissue change that can be detected at the gross (oral premalignant lesions, polyps), microscopic (metaplasia, dysplasia), and/or molecular (gene loss or amplification) levels. Recent molecular studies detecting profound genetic alterations in histologically normal tissue from high-risk individuals have provided strong support for the field carcinogenesis concept. The implication of the field effect is that multifocal, genetically distinct, and clonally related premalignant lesions can progress over a broad tissue region.1 The essence of cancer risk reduction, then, is intervention within the multistep carcinogenic process and throughout a wide field.
IDENTIFYING POTENTIAL CANCER RISK–REDUCING AGENTS
Cancer risk–reducing agent identification results from the synthesis of data from population, basic, translational, and clinical sciences. Findings from all of these disciplines are combined to contribute to the identification of agents with the potential to delay or reverse the carcinogenesis process (see Fig. 33.1).
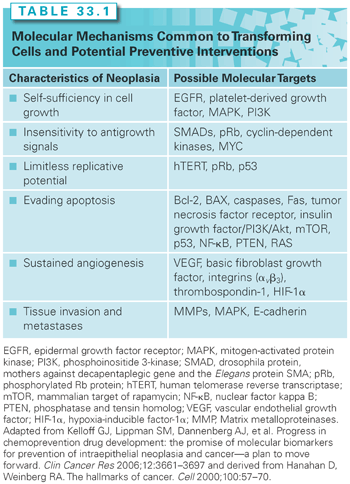
The Hanahan and Weinberg hallmarks of malignant transformation—self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis, limitless replication potential, sustained angiogenesis, and tissue invasion and metastasis7—reflect the loss of cellular signaling control. The molecular damage that results in transformation is triggered by a large array of genetic and environmental stressors such as chronic inflammation, oxidation, inherited genetic mutations, and exogenous environmental exposures. Many such signaling intermediates have common functions in multiple organ sites (see Fig. 33.1). The complexity and overlap of signal transduction pathways suggests that single molecular therapeutic/preventive targets may have limited effectiveness. Interventions aimed at preventing the occurrence of or overcoming the effects of molecular defects in multiple pathways or targets may be required to arrest or reverse carcinogenesis. Using the Hanahan and Weinberg hallmarks, examples of possible targets are shown in Table 33.1.
PRECLINICAL DEVELOPMENT OF CANCER RISK–REDUCING AGENTS
Similar to the development of therapeutic interventions, the assessment of efficacy and toxicity of single chemically synthesized entities, agents designed in silico, botanicals, nutrients/nonnutrient substances used as drugs for cancer risk–reducing agent efficacy proceeds through a translational paradigm that identifies efficacy in cell culture models, in live animal models, and in humans. Preclinical models that simulate the carcinogenesis process in target epithelia identify molecular biomarkers for modulation by interventions. These models can be used to identify potential toxicity of interventions and to assess the effect of interventions on the development and progression of preneoplasia/neoplasia.8
The U.S. National Cancer Institute’s (NCI) PREVENT Cancer Preclinical Drug Development Program is a prime example of a rational strategy to select promising agents for clinical trials through a stepwise approach of preclinical in vitro testing followed by in vivo screening.8–10 This system involves several phases: biochemical prescreening assays, in vitro efficacy models, in vivo short-term screening, animal efficacy testing, and preclinical toxicology testing.
Biochemical Prescreening Assays
Prescreening assays are a series of short-term, mechanistic assays developed to evaluate the ability of a test compound to modulate biochemical events presumed to be mechanistically linked to carcinogenesis.8 These in vitro assays are rapidly completed for potential cancer risk–reducing agents. Examples of such assays include carcinogen-DNA binding, prostaglandin synthesis inhibition, glutathione–S-transferase inhibition, and ornithine decarboxylase inhibition.
In Vitro Efficacy Models
In vitro assays test the cancer risk–reducing activity of a screened compound in four epithelial cell systems (primary rat tracheal epithelial cells, human lung tumor [A427] cells, mouse mammary organ cultures [MMOC], and human foreskin epithelial cells). The assays measure the ability of potential cancer risk–reducing agents to reverse transformation in normal epithelial cells exposed to carcinogens. For example, after treatment with a carcinogen such as 7,12-demethylbenz(a)anthracene, MMOCs develop lesions similar to alveolar nodules that are considered precancerous in mouse mammary glands in vivo.11 Pretreatment of organ cultures before carcinogen exposure measures the effect of cancer risk–reducing agents in the initiation stage of carcinogenesis, whereas treatment after carcinogen exposure measures activity during tumor promotion. Three of these assays (using rat tracheal epithelial, A427, and MMOC cells) have shown predictive values of 76% to 83% for cancer risk–reducing agent efficacy in in vivo models.8
Preclinical In Vivo Models for Cancer Risk–Reducing Agent Efficacy Testing
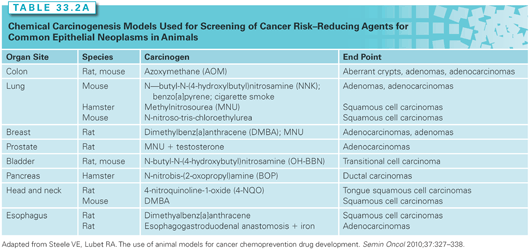
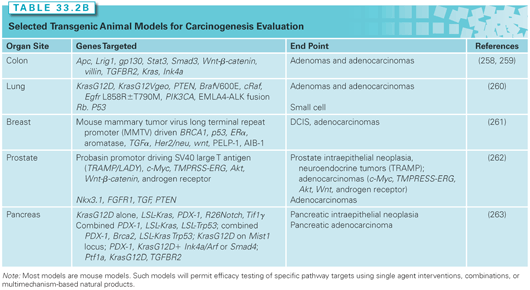
Animal models remain a crucial link in the efficacy assessment of cancer risk–reducing agents for epithelial cancer. Chemical carcinogenesis models provide the reproducible development of tumors in animals following the administration of a known chemical initiator or combination initiator/promoter and have been the primary in vivo screening tool for cancer risk–reducing agents (Table 33.2A).12,13 Carcinogenesis models employing genetically engineered mice permit the interrogation of targeted pathways and the corresponding efficacy of cancer risk–reducing agents. Although useful for mechanistic studies, knockout or genetic mutational models create accelerated neoplastic progression that does not accurately recapitulate the more complex, stepwise, human carcinogenesis process. Recombinant alleles can be driven by the addition of drug-sensitive regulatory elements, such as tetracycline or tamoxifen analogs. The drug-sensitive regulatory elements achieve temporal control over a gene promoter through the administration of the drug that binds to the regulatory element. Such a system permits the inhibition or overexpression of the organ-specific gene using Cre recombinase, a site-specific DNA recombinase that targets DNA regions flanked by loxP sequences. Tables 33.2A and 2B list representative organ-specific chemical and transgenic mouse models that may be used for cancer risk–reducing agent testing.
CLINICAL DEVELOPMENT OF CANCER RISK–REDUCING AGENTS
Special Features of Cancer Risk–Reducing Agent Development
The clinical efficacy assessment of cancer risk–reducing agents employs phased testing (phase I to III) models used for development of drugs14 but with crucial differences in study design and end points. Special features for the clinical development of cancer risk–reducing agents create the following challenges to be overcome: (1) the need for large therapeutic index (doses associated with potential toxicity of an intervention need to substantially exceed doses aimed at delaying or reversing transformation) for use in individuals who are asymptomatic yet may benefit from an extended (years) treatment course; (2) the long latency to malignant transformation (an assessment of effectiveness based on the reduction in cancer incidence requires studies lasting for years and involving thousands of participants); (3) adherence (once-daily dosing regimens using interventions that have sufficiently long half-lives may minimize the impact of a missed dose yet maintain the biologic impact on the physiologic target; minimal toxicity and strong psychological commitment to preventive goals also enhance adherence15); and (4) complex risk assessment for cancer (individuals with highly penetrant but infrequent, germ-line genetic susceptibility to breast and colon cancers16,17 are excellent candidates for cancer risk–reducing agents and are likely to accept some toxicity for reduced cancer risk). For individuals at more modestly increased risk (e.g., long-term, current smokers; persons with a family history of cancer; women with mammographically dense breasts), quantitative risk assessment algorithms may be useful in the future to identify optimal cancer risk–reducing agents. The refinement of cancer risk calculators for breast,18 colon,19 and prostate cancer20 promises to appropriately select high-risk individuals for cancer risk–reducing agents such that anticipated benefits exceed potential risks.
Biomarkers as Cancer Risk–Reducing Agent Targets and Efficacy End Points
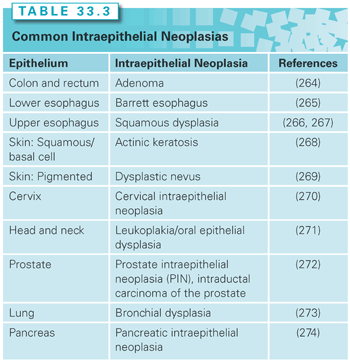
A biomarker is a characteristic that is measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to therapeutic interventions.21 A surrogate end point for cancer prevention assumes that a measured biologic feature will predict the presence or future development of a cancer outcome.22 Biomarkers enable a reduction in the size and duration of an intervention trial by replacing a rare or distal end point with a more frequent, proximate end point.23 Intraepithelial neoplasia has served and continues to serve as a biomarker for invasive malignancy (Table 33.3). Although many advocate the use of intraepithelial neoplasia-based biomarkers as regulatory surrogate end points, others caution that intraepithelial neoplasias may not serve as sufficiently robust surrogate biomarkers for cancer incidence or mortality.24
In order to be useful as end points for cancer risk–reducing agent efficacy testing as regulatory end points, any biomarker must have statistical accuracy, precision, and effectiveness of results24 that demonstrate prediction of a hard disease end point—cancer incidence or mortality. An independent validation data set must address defined standards of validation that minimize bias in the study design and the populations studied.25 The biomarker must be generalizable to the specific clinical or screening population (Table 33.4).
Phases of Cancer Risk–Reducing Agent Development
Phase I cancer risk–reducing agent trials define an optimal cancer risk–reducing agent dose. An optimal cancer risk–reducing agent dose is one that is usually nontoxic, scheduled once daily, and modulates a tissue, cellular, or serum biomarker of drug activity (e.g., the dose of aspirin that inhibits prostaglandin production in a target tissue site). The definition of a maximum tolerated dose is not an essential end point of a phase I cancer risk–reducing agent trial. Higher, yet nontoxic doses may lower cancer risk–reducing agent efficacy. For example, β-carotene at high doses has pro-oxidant activity and may enhance the carcinogenesis process, whereas at low doses, it is a potent antioxidant and differentiating agent.26
Phase II cancer risk–reducing agent trials begin to define cancer risk–reduction efficacy. These short-term (6 months to 1 year) treatment periods gather evidence of risk reduction by assessing drug effects on tissue, cellular, or blood surrogate markers of carcinogenesis. Phase IIa trials are nonrandomized, biomarker modulation trials. Phase IIb trials are randomized, placebo-controlled trials of several hundred subjects testing, for example, whether a risk-reducing agent reduces recurrence of a previously resected intraepithelial neoplastic lesion as the primary end point. Cellular dynamic (e.g., proliferation, apoptotic index), biochemical, or molecular (e.g., p53, cyclin D) end points may be used as secondary end points. Preoperative or window of opportunity trials enroll subjects for brief study periods prior to obtaining tissue by a planned resection of an invasive neoplasm. Such designs permit the exploration of biomarker modulation in the invasive neoplasm and in contiguous epithelial fields proximal and distal to the invasive neoplasm.27
Phase III cancer risk–reducing agent trials define reduction in a hard cancer end point such as cancer incidence or mortality. Such trials, using large, higher risk populations in a randomized, double-blinded intervention, are designed to identify a standard of preventive care for a given risk population. For example, trials of tamoxifen for the reduction of breast cancer incidence,28,29 finasteride for the reduction of prostate cancer incidence,30 and β-carotene for the reduction of lung cancer incidence31 serve as examples of well-conducted, definitive phase III cancer risk–reducing agent clinical trials.
Some investigators consider randomized, controlled clinical trials with an end point sufficient for regulatory review as phase III. Using such a definition, a clinical trial with an end point of reduction in adenoma recurrence is considered a phase III trial. Other investigators define phase III cancer risk–reducing agent trials as randomized, controlled clinical trials with a cancer incidence or mortality end point. This controversy causes confusion in the literature. For the purpose of clarity in this textbook, the latter definition of phase III trial is used—a prospective, randomized, controlled clinical trial with a cancer incidence or mortality end point. Randomized, controlled clinical trials with a surrogate biomarker end point such as an intraepithelial neoplasia (e.g., adenoma) are defined as phase IIb cancer risk–reducing agent trials.
Definition
Micronutrients comprise a large, diverse group of molecules typically ingested as part of the diet that play roles in normal human biology. This group of compounds has been investigated extensively as cancer risk–reducing agents in purified forms (i.e., supplements), as components of multiagent cocktails, and occasionally, as components of food extracts/other mixtures. Although retinoids are not micronutrients per se, they are related to retinol (vitamin A) and share certain properties with carotenoids, which are diet derived and, therefore, included along with micronutrients.
Retinoids, Carotenoids, and Antioxidant Nutrients
Overview and Mechanisms
Retinoids are the natural derivatives and synthetic analogs of vitamin A.32 Cancer risk–reduction intervention studies have evaluated the parent compound (retinol, typically given as retinyl acetate or palmitate), naturally occurring retinoids such as all-trans–retinoic acid (ATRA) and 13-cis-retinoic acid (13cRA), and also synthetic retinoids such as etretinate and fenretinide (4-hydroxy[phenyl]retinamide [4HPR]). These agents have been of interest for cancer risk reduction for decades. Mechanistically, retinoids have been shown to modulate cellular growth and differentiation, as well as apoptosis.32 A large body of research indicates that retinoids have activity in the promotion and progression phases of carcinogenesis, including extensive evidence of efficacy in the setting of premalignant lesions, leading to their evaluation in human Phase III trials. Nuclear retinoic acid receptors mediate many of the retinoid-signaling effects; however, retinoids interact with other signaling pathways, such as estrogen signaling in breast cancer.33
Carotenoids are a group of naturally occurring plant pigments, only some of which are found in appreciable levels in the human diet and human tissues, including beta-carotene, alpha-carotene, lycopene, lutein, and β-cryptoxanthin.34 Of these, the most widely studied carotenoids for cancer risk reduction are beta-carotene and lycopene. Beta-carotene has the highest pro–vitamin A activity of the carotenoids, but alpha-carotene and β-cryptoxanthin also possess pro–vitamin A activity. Other carotenoids, such as lycopene, do not possess vitamin A activity but are known to have potent antioxidant activity, particularly with regard to singlet oxygen quenching.35 Furthermore, eccentric cleavage products of beta-carotene,36 as well as other non-pro–vitamin A carotenoids such as lycopene (e.g., apocarotenals, apocarotenoic acids) appear to be biologically active and may also act via retinoid-signaling pathways.37
Because of the known antioxidant function of carotenoids, they are often studied for risk-reducing efficacy in combination with other antioxidant nutrients, especially vitamins E and C and selenium (sometimes as a cocktail, versus placebo). Thus, we will consider this group of nutrients first, followed by other micronutrients that are thought to act via different mechanisms and/or pathways.
Epidemiology
A large body of literature indicates that people who consume greater amounts of carotenoids from foods (primarily fruits and vegetables) and people with higher serum or plasma levels of various carotenoids have a lower risk for various cancers.38 In particular, according to the systematic review of the literature conducted by the World Cancer Research Fund/American Institute for Cancer Research,38 foods containing carotenoids are “probably” associated with lower risks of cancers of the mouth/pharynx/larynx and lung; foods containing beta-carotene are “probably” associated with lower risks of cancers of the esophagus; and foods containing lycopene are “probably” associated with lower risks of prostate cancer. Preformed retinol intake is inconsistently associated with risks of various cancers but that, in part, likely reflects confounding, because dietary sources of preformed retinol primarily include foods of animal origin (e.g., liver, eggs, milk). Vitamin C in the diet comes primarily from the consumption of fruits and vegetables; therefore, vitamin C and carotenoids often trend together in epidemiologic findings. Vitamin E, in contrast, is found in different foods, especially nuts, seeds, and vegetable oils; intake and blood concentrations are somewhat inconsistently associated with cancer risk.39 Selenium, being a trace mineral, is difficult to measure in the diet, but higher selenium status has been associated with a lower risk of certain cancers, although the results are not entirely consistent.39
Preclinical In Vivo Models
In preclinical models, retinoids induce differentiation as well as arrest proliferation33 of various cancers, making them attractive agents for cancer risk reduction.40 The International Agency for Research on Cancer reviewed the preclinical research involving beta-carotene, concluding that there was “sufficient” evidence of cancer preventive activity, particularly involving mouse skin tumor models and the hamster buccal pouch model.41 Notably, there was inconsistent evidence of efficacy in respiratory tract models. Lycopene has been evaluated in numerous cell culture systems and in a variety of models of prostate carcinogenesis, including chemically induced, orthotopic implantation, transgenic, and xenotransplantation, with mixed evidence of efficacy.42 Evidence, primarily from cell culture studies, suggests that lycopene metabolites may be at least partially responsible for anticarcinogenic activity.37
Clinical Trials: Retinoids, Carotenoids, and Antioxidant Nutrients
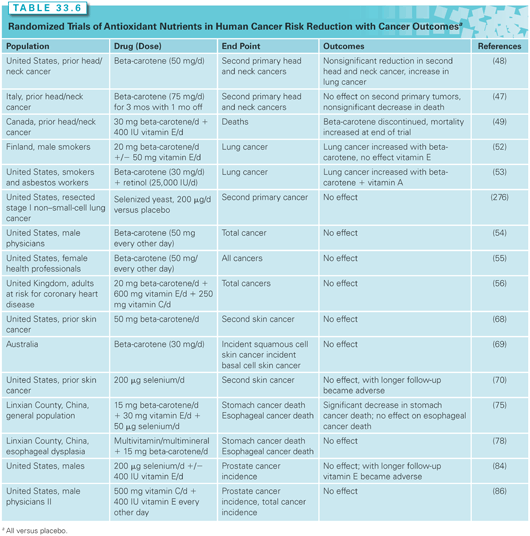
Retinoids, carotenoids, and antioxidant nutrients have been evaluated in the setting of preneoplasia/neoplasia in many different organ sites, as will be discussed. Of the many clinical trials of retinoids and carotenoids/other antioxidant nutrients, key trials with cancer incidence/recurrence as primary outcomes are tabulated (see Tables 33.5 and 33.6) and reviewed.
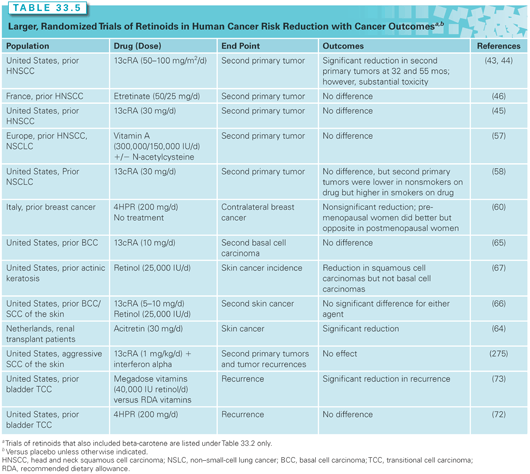
Clinical Efficacy in the Upper Airway
Many trials of cancer risk–reducing agents have been done in the setting of squamous cell carcinomas of the head and neck, in large part because of the substantial clinical problem of relatively high rates of recurrences and second primary tumors in curatively treated cancer patients. Early work demonstrated that high-dose 13cRA (50 to 100 mg/m2 per day) produced no significant differences in disease recurrence (local, regional, or distant) but significantly lowered the rate of second primary invasive neoplasms,43 with the benefits persisting for at least 5 years.44 Substantial retinoid toxicity, however, including skin dryness and peeling, cheilitis, conjunctivitis, and hypertriglyceridemia, was evident in a large proportion of patients. Subsequent trials thus used lower doses of retinoids (13cRA or a synthetic retinoid, etretinate), but failed to show efficacy in reducing second primary tumor formation (Table 33.5).45,46
Supplemental beta-carotene has also been studied as a single agent and in combination with other agents for the prevention of second primary cancers of the mouth and throat (Table 33.6). One trial of beta-carotene alone observed no harm or benefit47; another observed nonsignificantly fewer second head and neck cancers but more lung cancers48; and a third gave beta-carotene with α-tocopherol (400 IU per day).49 In the third trial, beta-carotene was discontinued early due to adverse findings from lung cancer prevention trials (see the following); however, after a median follow-up of 6.5 years, all-cause mortality was increased, which the authors attributed to the supplemental α-tocopherol. As will be discussed later, adverse effects of antioxidant nutrients are not limited to head and neck cancer patients; potential mechanisms for adverse effects are discussed further.
Clinical Efficacy in the Lung (Lower Airway)
Reversal of Metaplasia or Dysplasia. Active smokers and recent quitters have multiple preinvasive metaplastic and dysplastic lesions in the pulmonary tree. Most of these lesions resolve upon smoking cessation, but some remain and progress to invasive neoplasms. Unfortunately, micronutrient or retinoid interventions have not demonstrated preventive efficacy in most rigorous trials in patients with early lesions. For example, a US trial randomized 755 asbestos workers to receive beta-carotene (50 mg per day) and retinol (25,000 IU every other day) versus placebo; sputum atypia was not reduced after 5 years.50 As another example, eligible smokers with lung metaplasia or dysplasia were randomized to 6 months of 13cRA or placebo. The extent of metaplasia decreased similarly (in approximately 50% of subjects) in both study arms.51 Only smoking cessation was associated with a significant reduction in the metaplasia index during the 6-month intervention.
Prevention of Invasive Neoplasms. Large, phase III efficacy trials of beta-carotene plus other micronutrients for primary prevention of lung cancer have been completed, as summarized in Table 33.6. The Alpha-Tocopherol, Beta-carotene (ATBC) Trial involved 29,133 men from Finland who were heavy cigarette smokers at entry.52 In a two-by-two factorial design, participants were randomized to receive either supplemental α-tocopherol, beta-carotene, the combination, or placebo. Unexpectedly, participants receiving beta-carotene (alone or in combination with α-tocopherol) had a statistically significant 18% increase in lung cancer incidence and an 8% increase in total mortality relative to participants receiving placebo. α-Tocopherol had no effect.
The finding of an increased incidence of lung cancer in the beta-carotene–supplemented smokers was replicated in the Carotene and Retinol Efficacy Trial (CARET), a large randomized trial of supplemental beta-carotene plus retinol versus placebo in asbestos workers and smokers.53 This trial was terminated early, but, at the time of termination, overall lung cancer incidence was increased by 28% in the supplemented subjects and total mortality was also increased by 17%. In contrast, the Physicians’ Health Study (PHS) of supplemental beta-carotene versus placebo in 22,071 male US physicians reported no significant effect—positive or negative—of 12 years of supplementation of beta-carotene on total cancer, lung cancer, or cardiovascular disease (see Table 33.6).54 Two other trials involving supplemental beta-carotene alone (the Women’s Health Study55) or with other antioxidant nutrients (the Medical Research Council/British Heart Foundation Heart Protection Study56) on overall cancer incidence also failed to observe efficacy.
Prevention of Second Primary Invasive Neoplasms. EUROSCAN was a multicenter trial employing a two-by-two factorial design to test retinyl palmitate and N-acetylcysteine (also a compound with known antioxidant activity) in preventing second primary invasive neoplasms in patients with early stage cancers of the head and neck or lung. None of the interventions reduced second airway primary invasive neoplasms.57 The Lung Intergroup Trial randomized patients with surgically resected lung cancer to 13cRA versus placebo and found no significant differences between the two arms in second primary tumors.58 Notably, smoking status modified the effect of the 13cRA intervention, which was harmful in current smokers yet beneficial in former smokers.
Thus, phase III trials of both carotenoids/antioxidants and retinoids indicate that these agents overall do not reduce the risk of developing invasive lung cancers, nor do they prevent the development of second primary invasive neoplasms. However, the finding that former smokers seemed to benefit from both 13cRA58 and beta-carotene53 is intriguing. Mechanistic work suggests this interaction is real rather than chance (see the following), suggesting that (1) risk-reduction in smokers is especially challenging,59 and (2) trials in former smokers may merit consideration.
Clinical Efficacy in the Breast
Moon et al.40 first showed that fenretinide was a promising cancer risk–reducing agent for the breast, having a high therapeutic index and synergistic interaction with tamoxifen in mammary carcinogenesis model studies. This laboratory work led to a large-scale randomized trial of fenretinide (versus no treatment) for 5 years to prevent contralateral breast cancer in women aged 30 to 70 years with a history of resected early breast cancer and no prior adjuvant therapy.60 The intervention produced no significant overall effect, although fenretinide reduced contralateral and ipsilateral breast cancer rates in premenopausal women, with an opposite (adverse) trend observed in postmenopausal women. The reduced incidence of second breast cancer in premenopausal patients persisted with longer follow-up.61
Retinoid X receptor (RXR)–selective retinoids are also being evaluated in preclinical and clinical studies. Ongoing work suggests that combination treatment may represent a promising new strategy to suppress both estrogen receptor–negative and estrogen receptor–positive breast tumors, and the combination of retinoids with antiestrogens may be particularly effective.62
Clinical Efficacy in the Skin
Retinoids have been widely studied for cancer risk–reducing efficacy in skin. Early work was done in patients who have substantial skin cancer risk either due to xeroderma pigmentosum or medication-induced immunosuppression for transplants. For example, 13cRA reduced skin cancer by 63% in patients with xeroderma pigmentosum; however, severe, acute mucocutaneous toxicity with the 13cRA occurred.63 Also, the preventive effect of the retinoid was lost after stopping retinoid therapy. In renal transplant patients, acitretin (30 mg per day) reduced the numbers of premalignant lesions, the number of patients with skin cancer, and the cumulative number of skin cancers.64
In lower risk populations, low-dose 13cRA (10 mg per day)65 and retinol or 13cRA alone did not reduce the recurrence of basal or squamous cell skin cancers,66 although retinol alone reduced squamous but not basal cell carcinomas in patients with prior actinic keratoses.67
Beta-carotene (50 mg per day), in a randomized trial did not reduce the recurrence of nonmelanoma skin cancers.68 Consistent with findings in the lung, the risk was increased by 44% in current smokers randomized to beta-carotene but not in never smokers randomized to beta-carotene as compared with placebo. Supplemental beta-carotene (30 mg per day) also did not prevent basal cell carcinoma or squamous cell carcinoma of the skin in an Australian trial.69
Clark et al.70 randomized patients with a history of nonmelanoma skin cancer to 200 μg per day selenium or placebo.70 Selenium did not reduce the incidence of second skin cancers; a further report of this trial with longer follow-up71 indicated that there was instead a significant increase in total nonmelanoma skin cancer (hazard ratio [HR] = 1.17; 95% confidence interval [CI], 1.02 to 1.34) and squamous cell skin cancer (HR = 1.25; 95% CI, 1.03 to 1.51).
Clinical Efficacy in the Bladder
A trial of fenretinide (200 mg per day orally for 12 months) versus placebo was conducted for preventing tumor recurrence in patients with nonmuscle-invasive bladder transitional cell carcinoma after transurethral resection with or without adjuvant intravesical bacillus Calmette-Guérin; recurrence rates were similar in both groups.72 Another trial randomized 65 patients with biopsy-confirmed transitional cell carcinoma of the bladder to a multivitamin (recommended dietary allowance [RDA] levels) alone or supplemented with 40,000 IU retinol, 100 mg pyridoxine, 2,000 mg ascorbic acid, 400 U of α-tocopherol, and 90 mg zinc.73 The 5-year estimate of tumor recurrence was 91% in the RDA arm versus 41% in the higher-dose nutrient arm (p = 0.0014).
Clinical Efficacy in the Cervix
Randomized trials include four with beta-carotene (alone or with other antioxidant nutrients), and five with retinoids. Only one of these trials, involving ATRA,74 found a significant treatment effect. This trial administered a 0.372% ATRA solution by collagen sponge in a cervical cap delivery system. There was a higher complete response rate in the ATRA group (43%) than the placebo group (27%; p = 0.041) among the 141 patients with moderate dysplasia; no significant differences in dysplasia regression rates between the two study arms were detected in patients with severe dysplasia. The investigators experienced substantial losses to follow-up in this patient population.
Clinical Efficacy in the Esophagus and Stomach
Certain regions of China (Huixian and Linxian) have strikingly high incidence rates of esophageal and gastric cancers. Two trials were done in Linxian County; one was a general population trial that tested the efficacy of four different nutrient combinations at inhibiting the development of esophageal and gastric cancers.75 Those who were given the combination of beta-carotene, vitamin E, and selenium had a 13% reduction in total cancer deaths, a 4% reduction in esophageal cancer deaths, and a 21% reduction in gastric cancer deaths (see Table 33.6). None of the other nutrient combinations reduced gastric or esophageal cancer deaths significantly in this trial. The treatment benefit has been shown to persist for 10 years postintervention, with greater efficacy seen in participants under age 55 years.76 This finding stands in contrast to most other antioxidant nutrient supplement intervention trials, suggesting that the applicability of these results for populations with adequate nutritional status and for other tumor sites may be limited.77
The other Linxian trial evaluated a multivitamin/multimineral preparation plus beta-carotene (15 mg per day) in residents with esophageal dysplasia.78 There was no clear evidence of efficacy, although confidence intervals were wide.
Clinical Efficacy in the Colon/Rectum
Of the randomized trials aimed at the prevention of recurrent colorectal adenomas with micronutrients that have been completed, some used beta-carotene alone79 or with other nonmicronutrient interventions.80 Others evaluated beta-carotene with and without supplemental vitamins C and E.81 None of the trials observed benefit with supplementation. A subsequent report from one trial noted that alcohol intake and cigarette smoking modified the efficacy of beta-carotene.82 Among nonsmokers and nondrinkers, beta-carotene was associated with a significant decrease in the risk of one or more recurrent adenomas (relative risk [RR] = 0.56). Among persons who smoked and also drank more than one alcoholic drink per day, beta-carotene significantly increased the risk of recurrent adenoma (RR = 2.07).
Clinical Efficacy in the Prostate
Because oxidative stress may play a role in the etiology of prostate cancer, several antioxidant nutrients, including vitamin E, selenium, and lycopene, have been of interest for preventing prostate cancer. The largest trial to date of these nutrients is the Selenium and Vitamin E Cancer Prevention Trial (SELECT), which tested selenium and vitamin E in a two-by-two factorial design for the primary prevention of prostate cancer. Despite preliminary indications of prostate cancer risk–reducing efficacy for selenium (from a trial of selenium for skin cancer70) and vitamin E (from a trial of vitamin E to prevent lung cancer83), there was no evidence of efficacy.84 With extended follow-up, the nonsignificant adverse effect of vitamin E became significantly adverse.85 Negative/neutral findings also were reported for vitamins E and C and prostate and total cancer in the PHS II randomized controlled trial.86
The carotenoid lycopene has generated much interest with regard to prostate cancer risk, and several intervention trials have been conducted based on lycopene supplements. These studies have been small, short term, based on intermediate end points, and often lack adequate control groups. The use of a tomato sauce–based intervention is arguably a better approach to evaluate, based on animal data indicating that tomato powder (which includes lycopene along with other phytochemicals), but not lycopene alone, was effective at inhibiting prostate carcinogenesis.87
Mechanisms for Ineffective Retinoid and Carotenoid Cancer Risk–Reducing Activity
There are now a number of trials demonstrating that supplemental beta-carotene/retinoids given to current smokers can produce increases rather than reductions in cancer incidence. In tobacco users, beta-carotene and other carotenoids may produce oxidative carotenoid breakdown products that alter retinoid metabolism and signaling pathways, along with pro-oxidation.88 For retinoids such as 13cRA, smoking may induce genetic and epigenetic changes in the lung that affect retinoid activity; for example, tobacco smoking can affect RAR-β expression.58 The adverse effects of supplemental nutrients are not limited to smokers; α-tocopherol increased rather than reduced prostate cancer in SELECT (which had relatively few smokers) and selenium increased prostate cancer among men without a baseline selenium deficiency.89 This may be a consequence of the relatively high doses used in SELECT,77 but certainly calls into question the notion that reducing oxidative stress is a pivotal cancer risk–reduction strategy, even in nonsmokers.
It has become clear that reactive oxygen species (ROS), such as hydrogen peroxide, can act as important physiologic regulators of intracellular signaling pathways.90,91 Data in mouse models have shown that vitamin E accelerates lung tumor growth by disrupting the ROS–p53 axis, potentially by removing oxidative damage to DNA, which can serve as a potent stimulus for p53 activation.92 Although some of the large cancer risk–reducing trials may have failed in their primary objective, they may indirectly contribute to a clearer understanding of cancer biology, leading to the recognition that the role of oxidative stress and ROS in human disease is much more nuanced than originally hypothesized.93
As for the retinoids, these agents are generally too toxic to be used as single agents for risk-reducing efficacy; however, a major area of ongoing research is examining retinoids (low doses) given in combination with other agents, especially those that regulate the epigenome, such histone deacetylase (HDAC) inhibitors.33
Folic Acid and Other B Vitamins
Overview and Mechanisms. Folate is a water-soluble B vitamin found in foods, whereas folic acid is the synthetic form found in supplements and fortified foods. Adequate folate is critical for DNA methylation, repair, and synthesis.94,95 The methylation status of genes can play a key role in gene silencing and gene expression, lending plausibility to the idea that folate could be a key nutrient in regulating cell growth and proliferation.
Epidemiology. Epidemiologic studies have linked low folate intake with higher risk of several cancers, most notably colorectal cancer.96 Long-term use of multivitamin supplements, which are a major source of folate and other B vitamins, has been associated with a reduction in the risk of colon cancer in some studies, including recent (postfortification) findings.97–99 Supporting an anticancer role of folate is that genotypes for methylene tetrahydrofolate reductase, an enzyme known to be involved in folate metabolism, predict the risk of colon cancer dependent on folate intake or status.100 Vitamin B6 has been less studied in relation to cancer than folate, but some epidemiologic studies suggest that vitamin B6 may be important for colorectal cancer.101,102 A higher risk of cancer related to deficiencies of these vitamins has been suggested for alcohol drinkers.103
Clinical Trials: Folic Acid and B Vitamins. Risk-reducing efficacy for supplemental folic acid has been primarily evaluated in the setting of prevention of recurrent colorectal adenomas (i.e., in patients with prior adenomas). Of six randomized trials of folic acid, two small trials reported suggestions of benefit of folic acid supplementation.104,105 However, benefits were not observed in two much larger trials, the Aspirin/Folate Polyp Prevention Study (AFPPS) (dose: 1 mg of folic acid daily)106 and the United Kingdom Colorectal Adenoma Prevention (ukCAP) trial (dose: 500 μg of folic acid daily).107 AFPPS found indications of an increased risk for advanced lesions and multiple adenomas with prolonged treatment and follow-up. A third large trial, the Nurses Health Study/Health Professionals Follow-up Study (NHS/HPFS) folic acid polyp prevention trial, showed no overall risk reduction.108 The most recent trial, done in a Chinese population >50 years of age,109 reported that 1 mg folic acid per day reduced sporadic colorectal adenomas when compared to no intervention (not a placebo-controlled study). One possible explanation for the discrepancy of the Chinese trial versus North American and European trials is the baseline plasma folate status. In the Chinese trial, the mean baseline folate concentration of 5 ng/mL109 was half of the reported 10 ng/mL in a United States trial,106 where folate fortification of the food supply occurs.
Calcium and Vitamin D
Overview and Mechanisms. There are two major forms of vitamin D: ergocalciferol (D2) and cholecalciferol (D3). Vitamin D2 is absorbed through dietary sources such as fortified milk products, and D3 is synthesized via ultraviolet (UV) B light isomerization of 7-dehydrocholestrol in the epidermis.110 Vitamin D3 is converted to calcitriol (1, 25-[OH]2 D3) in a two-step process requiring both hepatic and renal hydroxylation. Calcitriol binds to the vitamin D receptor, which translocates to the nucleus and binds to multiple gene promoter sites. Through this mechanism, vitamin D regulates cytoplasmic signaling pathways that impact cellular differentiation and growth through proteins such as Ras and mitogen-activated protein kinase (MAPK), protein lipase A, prostaglandins, cyclic adenosine monophosphate (AMP), protein kinase A, and phosphatidyl inositol 3 kinase.110 1,25(OH)2D3 regulates cellular proliferation and apoptosis. For example, 1,25(OH)2D3 can induce cleavage of caspase 3, poly (ADP-ribose) polymerase (PARP), and MAPK, leading to apoptosis. 1,25(OH)2D3 inhibits the expression and phosphorylation of Akt, a key regulator of cellular proliferation. The differentiation properties of 1,25(OH)2D3 are mediated through transcriptional activation of the CDK inhibitor p21. The effects of vitamin D on multiple signal transduction pathways operational in cancer cells are reviewed by Deeb et al.111
Epidemiology. Observational epidemiologic studies have shown a relatively consistent inverse association between low calcium intake, including that from supplements, and increased colorectal and colon cancer risk.112,113 Vitamin D exposure is typically assessed by measuring 25(OH)vitamin D in plasma because exposure is derived not only from diet and supplements, but also from cutaneous synthesis following dermal exposure to UV radiation. A large number of observational studies have evaluated the association between vitamin D status and cancer risk, as systematically reviewed by the Agency for Healthcare Research and Quality (AHRQ).114 The evidence is inconsistent for most cancer sites, with the exception of studies showing that individuals with lower blood vitamin D levels have a higher risk of colorectal cancer or adenoma. Although some observational studies have reported that higher serum vitamin D is associated with lower breast cancer risk, the association is inconsistent.114,115 Also, there are some studies suggesting high serum vitamin D is associated with increases in certain cancers, particularly pancreatic cancer.116
Clinical Trials: Calcium and Vitamin D
Clinical Efficacy in the Colon. Baron et al.117 randomized subjects with a recent history of colorectal adenomas to either calcium carbonate (1,200 mg per day of elemental calcium) or placebo. Results showed significant benefit for the calcium arm (adjusted RR = 0.81; 95% CI, 0.67 to 0.99; p = 0.04). In a smaller, similar study of calcium gluconolactate and carbonate (2 g elemental calcium daily), the adjusted odds ratio (OR) for adenoma recurrence was 0.66 (95% CI, 0.38 to 1.17; p = 0.16) for calcium treatment,118 and while not statistically significant, it was similar to the data of Baron et al.117
In the largest trial of calcium and vitamin D with primary cancer end points (e.g., colon, breast), the US Women’s Health Initiative (WHI) evaluated the combination of 400 IU of vitamin D per day plus 1,000 mg of calcium per day in 36,282 postmenopausal women. For colon cancer, there was no benefit observed,119 although the mean baseline intake of calcium was already very high (more than 1,151 mg per day). With regard to vitamin D as a single agent, there was also no suggestion of benefit for colon cancer incidence in a 5-year British trial of vitamin D (100,000 IU every 4 months) that reported colon cancer incidence,120 although this was not a primary end point.
Clinical Efficacy in the Breast. Vitamin D has received considerable attention for a possible role in the prevention of breast cancer,121 although no trials have yet investigated vitamin D as a single agent for breast cancer risk reduction. The large WHI trial gave a combination of calcium and vitamin D, as noted previously, and there was no significant effect of this combination on breast cancer risk (HR, 0.96; 95% CI, 0.85 to 1.09).122 Lappe et al.123 conducted a trial that examined the relation between calcium plus vitamin D (1,100 IU per day) supplementation (versus calcium alone or placebo) in 1,179 healthy postmenopausal women in Nebraska.123 Although fracture was the primary outcome of the trial, total cancer incidence was reportedly lower in the calcium plus vitamin D group, although the number of end points was very small (n = 50 total cancers observed during the follow-up). An ongoing randomized trial of vitamin D and omega-3 fatty acids (the VITamin D and OmegA-3 TriaL [VITAL]) among 20,000 participants is expected to provide more definitive data on a possible role of vitamin D in the prevention of breast and other cancers.124
Summary and Conclusion: Micronutrients
Certain agents, including the retinoids, beta-carotene, folic acid, calcium plus vitamin D, vitamin E, and selenium, have received substantial attention for a possible role in reducing the risk of cancer in humans. As reviewed herein, some of the trials have observed statistically significant reductions in the risk of the primary end point (e.g., retinoids in skin carcinogenesis models, calcium in colorectal adenomas, antioxidant nutrients in Linxian, China, for gastric cancer prevention), whereas others have observed statistically significant increases in the risk of the primary end points (beta-carotene and retinoid lung cancer prevention trials in smokers, vitamin E and prostate cancer, selenium and nonmelanoma skin cancer). Considering the completed trials, there is clear evidence against the general use of nutrient supplements for cancer prevention, which is the conclusion also reached by the World Cancer Research Fund/American Institute for Cancer Research.38 Note that there is no evidence that food sources of these nutrients increase risk.
Having noted that, there are other key themes emerging from this growing body of research. One such theme is that nutrient supplementation may be of benefit to some but not all. One such population that may benefit includes persons who are low in the nutrient of interest at baseline.77 This was initially suggested in the Linxian Country trial (done in a micronutrient-deficient population), with growing support from subgroup analyses of several completed trials.77 However, the hypothesis that nutrient supplementation can reduce cancer risk in subgroups selected based on inadequate nutritional status has, to date, not been formally evaluated in intervention trials.
Another consistent theme is that lifestyle factors (e.g., smoking) and genetics (polymorphisms) may determine who is most likely to benefit from supplementation. Trial data will likely be increasingly mined to identify genetic profiles associated with both better outcomes (risk prediction) and response to intervention.125,126 Ultimately, a more personalized approach to cancer risk reduction may emerge, consistent with the movement toward a more personalized approach for cancer treatment.
Finally, nearly all of these trials initiate intervention with older adults (who are more likely to develop cancer end points during the follow-up); but, animal models suggest that the timing of exposure may likely be quite relevant. For example, folic acid may protect against initiation, but may also promote the proliferation of existing neoplasms.127 Thus, the dose, form (food versus supplement), timing, and nutritional and lifestyle characteristics may all be relevant in affecting the efficacy of risk-reducing interventions involving nutrients and related substances. Further research, drawing upon newer tools now available through the field of nutritional genomics, will be needed to gain greater clarity on the heterogeneous biologic effects observed in nutrient-based risk reduction.
Mechanism
Nonsteroidal anti-inflammatory drugs (NSAIDs) represent a class of drugs that reduce cellular inflammation through multiple mechanisms, the most prominent of them being the modulation of eicosanoid metabolism.128 Eicosanoids are metabolites of dietary fatty acids, primarily linoleic acid. Linoleic acid is metabolized to arachidonic acid, which is stored in the lipid membrane and, once mobilized from the membrane, further metabolized by prostaglandin-H synthases (PGHS) 1 and 2 to PGD2, PGE2, PGF2α, PGI2, or thromboxane A2 (TxA2) by specific synthases. Leukotriene pathways involve the conversion of arachidonic acid to leukotriene A4 by 5-lipoxygenase and subsequent hydrolysis of leukotriene A4 to other downstream leukotrienes. Newly formed prostaglandins function primarily through binding to prostaglandin receptors (EP receptors), releasing coupled G-proteins to elicit responses in the same or neighboring cells.129
Prostaglandins (PG) play crucial roles in controlling cellular proliferation, apoptosis, cellular invasiveness, and angiogenesis and in modulating immunosuppression.129 Because PGE2 is the most abundant PG in tumors, reducing local concentrations of PGE2 may be a pivotal cancer preventive strategy.129
PGHS-independent mechanisms of NSAID action may, at least in part, explain NSAID preventive efficacy.130 A diverse group of NSAIDs inhibit apoptosis via multiple mechanisms. Among the more prominent of these mechanisms is the inhibition of cyclic guanosine monophosphate (cGMP) phosphodiesterase activity, attenuation of beta-catenin mRNA through suppressing transcription of the CTNNB1 gene, and activation of c-Jun N-terminal kinase 1.130 NSAIDs activate peroxisome proliferator–activated receptor (PPAR)γ, leading to increased E-cadherin expression and reduced colony formation in vitro, while reducing PPARδ, leading to reduced resistance to apoptosis.130 Selective cyclooxygenase 2 (COX-2) inhibitors inhibit Akt signaling and induce apoptosis of human colorectal and prostate cancer cells in vitro in a COX-2–independent manner via the inhibition of phosphoinositide-dependent kinase-1 (PDK-1). NSAIDs inhibit nuclear factor kappa B (NF-κB) at pharmacologic concentrations and key cellular proliferation signaling intermediates such as activator protein 1 (AP-1) and other intermediates of the MAPK pathway.130 The impact of NSAIDs on carcinogenic events driven by these upstream pathways in humans as opposed to preliminary in vitro or in vivo models remains unclear.
Epidemiology
Pooled analyses of 34 controlled trials of aspirin 75 mg to 100 mg daily (69,224 participants), conducted primarily for cardiovascular disease reduction, observed reduced cancer deaths (OR, 0.63; 95% CI, 0.49 to 0.82). Most of the benefit occurred after 5 years follow-up.131 In a pooled analysis of 150 case control and 45 cohort studies, in addition to a reduced risk of death from colorectal cancer (OR, 0.58; 95% CI, 0.44 to 0.78), chronic and frequent (once daily or more) use of aspirin also reduced the risk of death from esophageal (OR, 0.58; 95% CI, 0.44 to 0.76), gastric (OR, 0.61; 95% CI, 0.40 to 0.93), and breast (OR, 0.81; 95% CI, 0.72 to 0.93) cancers.132 An analysis of 662,624 men and women enrolled in the American Cancer Society’s Cancer Prevention Study II found that aspirin taken at least 16 times per month over a 6-year period conferred a 40% reduced risk of colorectal cancer mortality.133 Both the 46,363 male patients of the Health Professional Study134 and the 82,911 patients of the Nurse’s Health Study135 suggest that prolonged use (>10 years) of 325 mg of aspirin twice weekly or more reduces colorectal cancer risk (RR, 0.77; 95% CI, 0.67 to 0.88, from the Nurse’s Health Study). Daily NSAID intake is associated with a 40% reduction (OR, 0.56; 95% CI, 0.43 to 0.73) in the risk of esophageal adenocarcinoma.136
Evidence in Preclinical In Vivo Carcinogenesis Models
NSAIDs, including aspirin, indomethacin, piroxicam, sulindac, ibuprofen, and ketoprofen, suppress colonic tumorigenesis induced chemically (1,2-dimethylhydrazine or its metabolites) or transgenically (Min+).137,138 The selective COX-2 inhibitors were the most efficacious colon tumorigenesis inhibitors in both chemical and transgenic rodent models.139,140 In preclinical models, NSAIDs affect the onset and progression of cancers in the stomach, skin, breast, lung, prostate, and urinary bladder, although the evidence is more limited than for colon cancers.141
Clinical Trials
Key clinical trials of NSAIDs for the prevention of colorectal cancer are summarized in Table 33.7
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


