Cancer immunotherapy
Padmanee Sharma MD, PhD  Sumit K. Subudhi, MD, PhD
Sumit K. Subudhi, MD, PhD  Karl Peggs, MD, MA, MRCP, FRCPath
Karl Peggs, MD, MA, MRCP, FRCPath  Sangeeta Goswami, MD, PhD
Sangeeta Goswami, MD, PhD  Jianjun Gao, MD, PhD
Jianjun Gao, MD, PhD  Sergio Quezada, PhD
Sergio Quezada, PhD  James P. Allison, PhD
James P. Allison, PhD
Overview
The basic principles that guide cancer immunology are immune surveillance, immune editing, and immune tolerance. Rapid increase in the knowledge of the mechanistic details of these basic principles has led to clinical success in the treatment of cancer. In this chapter, we discuss the basic principles and recent advances in the field of basic and clinical immunotherapy that has given credence to the long-held belief that the immune system can be used to treat cancer. Further, we also focus on the role of combining different types of immunotherapies and other therapeutic modalities in the treatment of cancer.
Immunosurveillance
The idea that the immune system is capable of recognizing and responding to cancer is not novel. Paul Ehrlich (1854–1915) was perhaps first among the early visionaries to speculate that the immune system played a key role in suppressing tumors and that the incidence of cancer would be much greater were it not for the ability of the immune system to identify and eliminate nascent tumor cells, a concept that would later become known as the “immune surveillance” hypothesis when revisited over 50 years later by F. Macfarlane Burnet and Lewis Thomas.1–3 However, proof of concept remained elusive. While it was established that chemically induced tumors were immunogenic in murine models, spontaneously arising tumors behaved differently and were not rejected in similar experimental systems.4 These data informed a growing consensus that naturally arising tumors were nonimmunogenic and that the antigens targeted by the immune system in chemically induced tumors were perhaps unique to this setting. Moreover, profoundly immunosuppressed athymic mice did not have an increased frequency of tumors induced by a chemical carcinogen.5 Even though a notable excess of a variety of cancers occurring in immunosuppressed organ transplant recipients was recognized as inferential that immune surveillance occurred in humans, it remained frustratingly apparent by the early 1980s that definitive support from murine models was still lacking.3 Furthermore, while certain tumors were many hundred-fold more common in immune-suppressed individuals (e.g., some skin cancers, Kaposi’s sarcoma, and lymphomas), the frequency of noncutaneous, nonvirally induced cancers was generally not increased, suggesting a potentially unique role for immune surveillance in malignancies associated with oncogenic viruses.6, 7 It was at this time that further inferential evidence in humans was beginning to emerge from the demonstration of increased rates of malignancies in patients with the newly defined acquired immunodeficiency syndrome.8, 9 However, the marked association with virally induced cancers was similarly striking.
Multiple strands of evidence would need to coalesce to rekindle enthusiasm for the therapeutic potential of immune-based strategies. Aline van Pel, Thierry Boon, and Pierre van der Bruggen demonstrated that specific immunity to spontaneous tumors could be induced by vaccinating mice with mutagenized tumor cells10 and that tumor-specific antigens could be recognized by human cytolytic T cells.11 Their studies showed that spontaneous tumors were not inherently deficient in tumor antigens but rather failed to stimulate an effective immune response. Critically, this failure could be overcome by vaccination in mouse models. The molecular definition of tumor antigens revolutionized the field of tumor immunology by legitimizing the mechanism by which the adaptive immune system discriminates between normal and neoplastic cells. The detection of tumor-specific responses in humans fueled speculation that these responses could be similarly manipulated to induce tumor eradication.12 Simultaneously, it engendered rapid development of a new field within tumor immunology searching for tumor-associated antigens. Many tumor antigens have since been cloned. They can be broadly segregated into five major categories: (1) differentiation antigens, for example, melanocyte differentiation antigens, tyrosinase, gp-100, and Melan-A/MART-1; (2) mutational antigens, for example, abnormal forms of the tumor suppressor p53; (3) over-expressed normal antigens, for example, HER-2/neu and galectin-9; (4) cancer–testis (CT) antigens, for example, MAGE, LAGE, and NY-ESO-1; and (5) viral antigens, for example, EBV and HERV.13, 14
Immunoediting
The demonstration of tumor-specific antigens in spontaneously arising tumors that could be recognized by the immune system gave further credence to the theory of immune surveillance but fell short of providing definitive proof. Antigenicity is necessary but not sufficient for immunogenicity. In 2001, Robert Schreiber in collaboration with Lloyd Old demonstrated that T lymphocytes and the immune stimulator IFN-γ cooperated to inhibit the development of spontaneous and carcinogen-induced tumors in mice genetically engineered to lack a functional immune system (RAG-2−/−),15 but that some tumor cells escape detection and eventually cause cancer. They posited that driven by the selective pressure exerted by the immune system, the tumor cell population undergoes “immunoediting” becoming serially less immunogenic than the starting population, through a Darwinian environmental selection. Tumor cells from immunodeficient mice were more immunogenic than those from immunocompetent mice. Although sustaining the concept of immune surveillance, these data raised a potentially formidable obstacle to the delivery of clinically useful immunotherapies, suggesting that by the time a cancer becomes detectable it is already beyond the capabilities of the host immune system to eradicate it. Critically from the therapeutic standpoint, however, they showed that it was possible to make these cells visible to the immune system by increasing their antigen expression. Similar findings were obtained in RAG-1−/− mice,16 and additional studies in mice specifically deficient in αβ or γδ T cells demonstrated that both cell types play critical and distinct roles in immune surveillance.17–19 Natural killer (NK) and natural killer T (NKT) cells were also implicated as mediators of protection in experiments in which they were depleted with either anti-NK1.1 monoclonal antibody or anti-asialo-GM1 before challenge with a chemical carcinogen.16 Further dissection of the critical effector mechanisms underlying host resistance to both chemically induced and spontaneous tumors has emerged from studies on IFN-γ and perforin demonstrating that deficiencies in either of these key immunologic molecules increases susceptibility to tumor growth.15, 20–26
Immunoediting consists of three processes that occur either concurrently or sequentially.27–30 First, “elimination” in which immunity functions as an extrinsic tumor suppressor (equivalent to the original concept of immunosurveillance); second, “equilibrium” in which cancer cells survive but are held in check by the immune system; and third “escape” in which variant cancer cells with either reduced immunogenicity or the capacity to attenuate or subvert immune responses grow into clinically apparent cancers. Data documenting the equilibrium process has remained relatively elusive and largely inferred from clinical observation. For example, the development of melanoma of donor origin in two recipients of renal allografts from the same donor who had been considered cured of melanoma treated 16 years previously.31 More recently, Schreiber’s group has shown clearer evidence to support the existence of equilibrium in a chemically induced primary murine cancer model.32 Following exposure to the carcinogen, approximately 20% of animals developed fatal tumors but some survived with no overt evidence of tumor growth (apparent elimination). Immune suppression in these mice, however, resulted in the unmasking of dormant tumors, which then spread, ultimately killing the host. This effect was observed either following T cell depletion or following neutralization of IFNγ or IL-12 but not following depletion of NK cells suggesting a specific role for adaptive immunity in the maintenance of equilibrium. Furthermore, examination of stable dormant lesions revealed cancerous cells with similar morphological features to those in progressive lesions but with a lower proliferative index and increased apoptosis. The lesions were infiltrated with T cells suggesting a possible ongoing interaction with the host immune system. Engraftment of these cancerous cells into immunodeficient mice following a short period of ex vivo culture resulted in tumor growth. In contrast, transfer into immunocompetent recipients failed to induce tumor growth. Finally, tumor outgrowth occasionally occurred following a period of dormancy in immunocompetent animals, and in these cases, tumor cells were able to grow following transfer to immunocompetent mice, suggesting they had become less immunogenic. These data support the idea that tumor cells are unedited in equilibrium but become edited when they spontaneously escape immune control.
Immune tolerance
In 2005, Gerald Willimsky and Thomas Blankenstein suggested that sporadic tumors do not lose immunogenicity as would be predicted by models invoking immunoediting as the major reason for tumor escape but rather induce tolerance to evade immune detection.33 The idea itself was not new and indeed had been part of the original immunoediting model. The message was simple and had major implications for the successful application of immune-based therapies. Progression of cancer may not depend solely on intrinsic adaptions of the tumor cells to evade detection but rather on changes exerted on host immunity to permit tumor growth. These mechanisms are neither mutually exclusive nor entirely separable, as upregulation of the surface expression of immunoinhibitory ligands by tumor cells can potentially abrogate immune responses just as efficiently as downregulation of immunostimulatory elements. Their studies involved the use of mouse model in which a viral cancer-promoting gene (SV40 large T) was controlled to activate rarely in random tissues. Although immune responses to the SV40 large T protein were initially detected in the mice, they subsequently developed immune tolerance, while the tumors remained capable of eliciting vigorous immunity when transferred into identical but tumor-free mice (with no clear evidence for immunoediting).
The apparent contradictions between this study and those supporting a more central importance for tumor-intrinsic editing may reflect differences in inherent tumor immunogenicity. More immunogenic tumors will, by definition, generate a more robust immune response. This likely relates partly to the ability of dendritic cells (DCs) to present tumor debris in an “immunogenic” manner, which in itself may reflect activation by secondary signals. In this setting, evolutionary pressures may be higher and immunoediting may be critical to immune evasion. For less immunogenic tumors that do not efficiently activate cellular antigen-presenting machinery, arousal of the immune system is minimal and escape variants are therefore less likely to be sculpted by immunoediting. In these cases, tolerance or immunological ignorance may play variable roles.
Early failures—lessons learnt
The apparent confirmation of the validity of the immune surveillance hypothesis led to great enthusiasm for the development of immune-based anticancer therapies. However, attempts to target human cancers by immunotherapy have been significantly less successful than initially envisaged, leading to the marginalization of immunotherapies from the mainstream of oncologic practice. In patients presenting with cancer, endogenous antitumor immunity has been insufficient to eliminate the tumor. Immunological ignorance may contribute to tumor outgrowth, but antitumor responses are detectable in many of these patients. Indeed, it is clear from the detailed studies of immunoediting in mouse models that even when immunosurveillance fails, the relationship between the immune response and cancer continues to evolve. It is, therefore, likely that failure to eradicate tumors relates to limitations of effector function of the tumor-specific T cells. For maximal antitumor responses, it is necessary for appropriately targeted and activated effectors expressing T cell receptors (TCR) of sufficient avidity to migrate into tumor sites and maintain effector function within immunologically hostile tumor microenvironments. The presence of even large numbers of T cells capable of recognizing tumors is not singularly sufficient to mediate tumor regression, as evidenced by unrestricted tumor growth in TCR transgenic mice in which all of the T cells are capable of recognizing the tumor antigen.34 Clinical studies of active immunization that have shown tumor growth can go unimpeded despite expansion of cancer cell-reactive T cells to 40% of the circulating CD8+ T cell repertoire.35 There is now ample experimental evidence that functional systemic antitumor activity may not translate into tumor rejection, either because of lack of infiltration of T cells into the tumor36 or because of local suppression of function within the tumor microenvironment. Although tumor-specific immunity is compromised in tumor-bearing mice, there is often no generalized immune deficiency,37 indicating that tumors can specifically suppress the induction of effective antitumor immunity. This concept is perhaps best highlighted by concomitant immunity, wherein a mouse injected with a tumor will reject a subsequent challenge with the same tumor at a distant site, despite continued growth at the site of initial challenge.38–40 Intriguingly, however, it has been known for some time that the initial generation of concomitant immunity is eventually subverted during primary tumor progression by the establishment of CD4+ T cell-mediated immune suppression.41, 42 Despite these findings, the majority of approaches to tumor immunotherapy have until recently remained grounded in infectious disease principles. It had been established in the 1980s that antigen-specific CD8+ cytotoxic T cells (CTLs) were able to induce fully protective immunity against the influenza virus, leading to the development of effective vaccines.43 The critical role of DCs in mediating these immune responses was recognized soon thereafter in the work of Ralph Steinman and others.44 On the basis of growing evidence that tumors express antigens that can be presented by professional antigen-presenting cells (APCs) to induce the generation of tumor-specific CTLs, tumor immunotherapists aimed to parallel the successes achieved in developing vaccines for infectious diseases. Strategies have included vaccination with peptide, DNA, or antigen-pulsed DCs. Alternate approaches have been pursued based on directly enhancing effector number or function by adoptive transfer of T cells expanded from tumor-infiltrating lymphocytes (TILs), T cells activated ex vivo with cytokines, T cells together with cytokines, and more recently, T cells engineered to express receptors specific for tumor-associated antigens [either TCR or chimeric antigen receptors (CAR)].45–49 While these approaches have resulted in some impressive responses (reviewed later), they all potentially remain limited by the locally immunosuppressive microenvironment within the tumor, as evidenced by the evolution of such strategies to incorporate lymphodepleting or nonmyeloablative conditioning to enhance responses. As tumors can be viewed as taking advantage of immunological ignorance, anergy, and suppression, effectively subjugating host responses to create isolated nodes of immune privilege within an otherwise immunologically intact host, they share many similarities with chronic pathogens such as Mycobacterium tuberculosis, Listeria monocytogenes, and Leishmania major. In this context, the challenges of delivering effective vaccines or immunotherapies are much more closely aligned with those associated with therapy of these established chronic infections than with acute infectious pathogens, in which the majority of the successes have come with prophylactic vaccination strategies. Further consideration of the mechanisms underlying the tumor escape phase of the immunoediting model may enlighten strategies to enhance the effectiveness of immunotherapies.
Mediators of immune escape
As previously discussed, the significant changes occurring in the escape phase may be broadly considered as those intrinsic to the tumor cells themselves and those involving the local tumor microenvironment, although the two overlap. Classic examples of the former include downregulation of costimulatory molecules (e.g., B7 molecules) by the tumor leading to activation of tumor-reactive T cells in the absence of appropriate costimulation and induction of anergy50 or tumor antigen loss or downregulation of major histocompatibility complex (MHC) molecules, which can render the tumor cells essentially invisible to the immune system.51 Further examples include mutations conferring increased resistance to apoptosis induction or cell-mediated cytotoxicity, such as overexpression of antiapoptotic molecules (e.g., FLIP and BCL-XL),52, 53 and mutations in FAS or the TRAIL-R (TNF-related apoptosis-inducing ligand receptor) death receptor 5 (DR5).54–56 Increased expression of T cell inhibitory molecules such as programmed cell death ligand 1 (PD-L1), B7-H3, B7x, HLA-G, and HLA-E by the tumor cells themselves or surrounding parenchyma (stromal or APCs) can directly inhibit effector T cell function, and in many cases, expression levels by the tumor or its microenvironment have been found to correlate inversely with tumor outcomes.57–65 Further proposed mediators of local immune suppression include soluble suppressive factors elaborated by the tumor or parenchyma such as IL-10, TGF-β, VEGF, or gangliosides.66–72 Indoleamine 2,3-dioxygenase (IDO) expression by tumor cells or IDO-competent APCs, such as some plasmacytoid dendritic cells (pDCs), can also contribute to acquired tolerance, both by direct suppression of T cells and by enhancement of local regulatory T cell-mediated suppression.73, 74 IDO catalyzes the rate-limiting step in tryptophan degradation, and the combination of local reduction in tryptophan levels (possibly via the intermediacy of causing cellular stress responses in effector T cells) and the production of immunomodulatory tryptophan metabolites appears to exert tolerogenic activity. Furthermore, IDO-expressing pDCs resident within tumor-draining lymph nodes appear to directly activate mature regulatory T cells, which can subsequently cause upregulation of PD-L1 by other DCs, which in turn inhibits effector T cell proliferation.75 It has become clear that these regulatory networks are therefore extremely complex.76 The presence of an array of other cell types capable of actively suppressing immune reactions such as CD4+CD25+ regulatory T cells (Treg), IL-10-secreting regulatory T cells, CD1d-restricted (NKT) T cells, immature and plasmacytoid DCs (iDCs and pDCs), and myeloid-derived suppressor cells (MDSCs) (noncell-autonomous suppression) within the tumor or tumor-draining lymph nodes is clearly critical to induction and/or maintenance of local immune privilege in a number of systems.74, 77 Such cells may be preferentially recruited to these sites or expanded or induced therein.
Noncell-autonomous suppression
A number of CD4+ T cell subtypes with regulatory or suppressive activity are now recognized. They fall broadly into one of two categories: those which are produced by the thymus, express CD4, CD25, GITR, OX40 and CTLA-4, and appear crucially dependent on the expression of the X-linked forkhead/winged helix transcription factor, Foxp3, for their development (so-called “naturally occurring” regulatory T cells)78–84 and those which arise from naïve CD4+ T cells as a result of “tolerogenic” encounters in the periphery. The latter “inducible” or “adaptive” regulatory T cells include interleukin (IL)-10-producing, Foxp3-negative Tr1 cells,85–87 transforming growth factor-β (TGFβ)-producing Th3 cells,88, 89 and extra-thymically generated CD4+CD25+Foxp3+ iTreg cells.90–95 In addition, CD4+CD25–Foxp3+ T cells with regulatory capabilities have been recognized.96 The acquisition of regulatory phenotype and suppressive functions by conventional nonregulatory CD4+ T cells following exposure to antigens under certain conditions is now recognized as a major contributor to the maintenance of T cell homeostasis and control of inflammation. Of particular note, as thymic involution occurs relatively early in humans compared to mice and telomere length in human Treg is considerably shorter, it is conceivable that peripheral conversion plays a far more important role in the maintenance of tolerance (and perhaps in immune subversion by tumors) in humans than in mice.97–99 Furthermore, if antigen encounter is required for conversion, it seems plausible that the regulatory pool expands at the expense of potential effector T cells, as precursors recognizing tumor antigens will be redirected into a suppressor rather than effector phenotype.96, 100, 101 Characterization of the conditions that drive such peripheral conversion is ongoing, but factors such as suboptimal antigen stimulation in combination with TGF-β appear to be important, both of which are likely to be relevant within the tumor microenvironment.93, 102 IDO produced by either tumor cells or parenchyma (e.g., pDCs) also favors conversion,75, 103, 104 and retinoic acid appears to be a key mediator in establishing intestinal tolerance.105–108 Regulatory T cells require TCR triggering to become functional, and at least some tumor-associated Treg are specific for tumor antigens,109 but once activated, they suppress in an antigen-independent manner. While adaptive Tr1 and Th3 cells exert suppression mostly through soluble factors (IL-10 and/or TGF-β), their Foxp3+ counterparts suppress via a variety of mechanisms, the relative importance of which remain to be fully elucidated in any given situation. Possible mechanisms include those involving CTLA-4, membrane-bound TGF-β, and pericellular generation of adenosine.110–116 Their expansive capacity to suppress multiple immune effector populations, including CD4+ and CD8+ T cells, B cells, and NK cells,117 makes them potentially attractive “nodal” targets for therapeutic intervention within immunosuppressive networks. To add further to this complexity, CD8+ T cells with suppressor activity have also been described.118–121 The dominant inhibitory potential of regulatory T cell populations in murine models of malignancy is well established,122 and more recently, their potential role in human malignancies has been demonstrated.123 The mechanisms driving regulatory T cell expansion and accumulation in patients with cancer are not fully understood, but both proliferation of pre-existing Treg and conversion from naïve precursors are likely to be involved.101, 124, 125 Their relative abundance predicts for tumor outcomes in murine models126, 127 and correlates inversely with outcomes in several epithelial carcinomas, including ovarian,123, 128 breast,129 and hepatocellular carcinoma.130 Intriguingly, in hematological malignancies, this association is reversed and high levels of regulatory T cells appear to confer improved prognoses (e.g., cutaneous T cell, follicular, and Hodgkin lymphoma.131–133 The level of infiltration of a tumor by regulatory T cells alone may not be the best predictor of outcome. Hodgkin lymphoma tumors contain significant populations of both IL-10-secreting Tr1 and CD4+CD25+ regulatory T cells, which induce a profoundly immunosuppressive environment.134 Combined assessment of cells expressing Foxp3+ and of cells expressing TIA-1 (cytotoxic granule-associated RNA binding protein) offers a better predictor of response than either alone,133 highlighting the potential importance of assessing the relative prevalence of multiple infiltrating cellular populations. Indeed, identification of specific immunological signatures (based on flow cytometry, PCR, or microarray analyses135) that predict outcomes or guide the institution or monitoring of therapies would be useful adjuncts to modern clinical practice. For example, it is plausible that tumors that contain few TILs, including Treg, will respond well to treatments that aim to enhance CTL numbers, function, or migration, while those that contain significant numbers of Treg would benefit from therapies aimed at reducing Treg number or function. In addition, Treg represent another regulatory mechanism that may be enhanced in response to, and hence limit the efficacy of, current immunotherapeutic interventions. For example, IL-2 has entered clinical trials for a number of human cancers such as melanoma, renal cell carcinoma, rhabdomyosarcoma, and ovarian cancer. Its initial use was based on the idea that it may directly enhance effector function of both innate and adaptive immune systems. However, IL-2 is recognized as crucial for the homeostasis and function of CD4+CD25+ regulatory T cells in vivo,136–138 and administration to patients with cancer results in increases in the numbers of peripheral Treg cells and stimulation of expression of CXC-chemokine receptor 4 (CXCR4) and CCR4 on Treg promoting their migration toward CXCL12 and CCL2 within the tumor microenvironment.139–141 As the targets of many cancer vaccination strategies are self-antigens, it is perhaps no surprise that “therapeutic” cancer vaccines can induce amplification of tumor-specific regulatory T cells.96, 142, 143 The “immunogenicity” or “tolerogenicity” of DCs thus becomes an increasingly important consideration in vaccination programs as even conventionally “mature” DCs can activate and expand autoantigen-specific Treg cells.144, 145
Multiple suppressive APC populations play a part in the generation of local immune privilege within tumors. Developing tumors may selectively recruit suppressive APCs or convert stimulatory APCs into suppressors, mirroring the situation with suppressive T cell populations. The molecular mechanisms underpinning active immune suppression by DC and myeloid populations have not been fully elucidated but include secretion of IL-10 and TGF-β, expression of FAS ligand, PDL1, and elaboration of intracellular IDO.146–150 IDO-competent DCs can induce apoptosis of activated T cells or either T cell anergy or conversion of effectors into Treg as previously outlined.75, 103, 151, 152 The local balance of stimulatory versus suppressive APCs is probably critical in determining the eventual outcome of T cell encounter with antigen in these sites. It has also become clear that the interaction between DCs and Treg is likely a two-way process.153–155 MDSCs are a heterogeneous group of cellular precursors of macrophages, granulocytes, DCs, and myeloid cells at earlier stages of differentiation, which express both the myeloid differentiation antigen Gr-1 (Ly6G and Ly6C) and CD11b in mice, and are generally defined as CD14−CD11b+ cells in humans (expressing CD33 but lacking expression of mature myeloid or lymphoid markers).156–158 Specific phenotypic markers that are reflective of suppressor function remain relatively poorly defined.159 They consist of two major subsets of Ly6G+Ly6Clow granulocytic and Ly6G−Ly6Chigh monocytic cells in mice. Numbers may correlate with clinical outcomes in human cancer.160 Several tumor-derived cytokines have been implicated in the expansion of MDSC, including VEGF, IL-1β, and GM-CSF.161–163 The mechanism of MDSC-mediated suppression is complex, involving contributions from either iNOS or arginase 1,147, 164–167 which enable MDSCs to inhibit T cell responses in various ways, including induction of apoptosis, inhibition of proliferation, or induction of a regulatory phenotype. Nitration of tyrosines in the TCR-CD8 complex appears to render CD8-expressing T cells unable to bind peptide-MHC complexes and to respond to the specific peptide, although they retain their ability to respond to nonspecific stimulation.168 Type 2 macrophages found at tumor sites have also been implicated in suppression of tumor immunity and seem to share some functional properties with immature myeloid cells.169, 170
Even in those cancer immunotherapies that succeed in inducing systemic immunity, translation into clinically significant effects has been rare. It is likely that these mechanisms will need to be addressed in order to improve outcomes. What remains wholly unclear is how effectively this aim can be met and how many of the disparate targets will need to be tackled at any one time in order to generate locally curative immunity.
Moving toward clinically effective immunotherapies
In 1891, William Coley (1862–1936), a surgeon at New York Cancer Hospital which later became part of the Memorial Sloan-Kettering Cancer Center, found a patient who had been seen 7 years earlier with an inoperable malignant tumor in his neck, which had disappeared following cutaneous erysipelas caused by Streptococcus pyogenes.171 Coley reasoned that somehow the infection had been the precipitant of the remarkable recovery and found more than 20 published accounts before 1890 linking infection with erysipelas and antitumor responses. On this background, he pursued his own clinical trial using live bacteria, evolving to the use of heat-killed bacteria following some infection-related deaths.172, 173 It is remarkable to consider that even a minority of these patients were cured. The first patient he infected with erysipelas was apparently cured, dying 10 years later of unknown causes, as was the first patient treated with his mixed Coley Toxins, dying 25 years later of a heart attack. Ultimately, the antitumor effects were probably attributable to the production of tumor necrosis factor (TNF) in response to bacterial endotoxins.174, 175 While TNF itself has proven too toxic for reliable clinical usage, it is perhaps fitting that members of the TNF superfamily and their receptors remain important targets for current approaches to immunotherapy.
Adoptive cell therapy and the development of personalized immunotherapies
The growing appreciation that tumor-bearing hosts often do mount antitumor responses and that the greatest concentration of accessible tumor-specific cells may reside within the tumor itself (albeit held in check by local immunosuppressive factors) has informed the development of the field of adoptive cellular therapy (ACT). Furthermore, the failure of active immunization to affect major clinical responses has clearly not always reflected a lack of systemic antitumor immunity, although it has been argued that at least part of this failure relates to stimulation mainly of low-avidity effector T cells, itself reflecting the impact of central deletional tolerance on the anti-self T cell repertoire. These cells may be detectable by conventional immune monitoring techniques but have little chance of rejecting established tumors, particularly as access to and function within the tumor microenvironment are also likely to be limiting factors. Adoptive immunotherapy allows the identification of rare cells with relatively high affinity for tumor antigen that can be selected in vitro and expanded before transfer to the host. These cells can be activated ex vivo and directly administered, in the hope of avoiding the tolerizing factors present at the tumor site. Initially based on the idea that significantly increasing the number of tumor-reactive T cells will bypass all peripheral and local regulatory mechanisms, flooding the tumor and leading to prompt tumor rejection, modern approaches now recognize the importance of further manipulation of the host to optimize the chances of therapeutic success. These approaches have been developed in parallel with, and in many cases informed, studies advancing our knowledge of the factors limiting antitumor immunity, establishing that the administration of large numbers of activated high-affinity “tumor-specific” T cells to a lymphodepleted host can overcome inhibitory factors and mediate effective cancer immunotherapy in some cases of human malignancy, particularly melanoma. Approaches to date have been largely based on the in vitro expansion of T cells obtained from tumor infiltrates and to a lesser degree from peripheral blood or lymph nodes biopsies. Transfers are often combined with administration of the T cell growth factor IL-2. It is likely that such strategies will continue to evolve, incorporating advances in immunostimulatory antibody research (see the following text).
Much of the pioneering work that laid the foundations for subsequent investigation of passive anticancer immunotherapy through ACT was performed by Nicholas Mitchison in the early 1950s,176, 177 and it is perhaps germane to reflect that the enhancing activity of host irradiation and the significance of antigen dose in determining immune response were already established in these seminal works.178–180 The further development of ACT owes much to work of the groups of Alexander Fefer, Martin Cheever, and Philip Greenberg at University of Washington, as well as those of Steven Rosenberg and Nicholas Restifo at National Cancer Institute. Fefer advanced the concept of “chemoimmunotherapy” in the late 1960s, demonstrating that an established syngeneic lymphoma (FBL-3 or Friend virus-induced lymphoma) could be eradicated after therapeutic combination of high doses of the alkylating agent cyclophosphamide and the transfer of immune cells from a mouse previously sensitized (or challenged) with the same tumor.181 Further studies helped to define some of the mechanisms underpinning this antitumor activity, demonstrating that it was either directed against tumor or virus antigens,182, 183 that T cells were the major mediators,184 and that this activity could also be isolated from nonsensitized mice, although with a much diminished potency.185 This last point is significant as it suggests that a population of tumor-reactive T cells may be present in naive animals. The work was extended to show that antitumor activity was enhanced in cells that had been resensitized and expanded with irradiated tumor in vitro,186–192 or even with cells that had been only primarily sensitized in vitro.189 An important advance during this work was the introduction of IL-2 in the in vitro system that allowed further expansion of tumor-reactive T cells.189 Finally, it was demonstrated that tumor-reactive T cells could be cultured in vitro for long periods of time and that after adoptive transfer they would proliferate and mediate specific tumor rejection that could be significantly augmented by the in vivo administration of IL-2.190–192 These studies were among the first to highlight the potential benefits of the use of IL-2 both in vitro and in vivo in mediating efficient expansion and enhancing tumor rejection.
Contemporaneously, Rosenberg also demonstrated the benefits of IL-2 in cancer immunotherapy. In what would become one of the cornerstones of ACT, a series of studies were published on the use of “T cell growth factor” (TCGF or IL-2) to clone and expand T cells,45, 193, 194 including its use for the isolation and expansion of T cells infiltrating solid tumors.195 Importantly, human lymphocytes grown in TCGF were capable of killing autologous tumor cells in vitro.196 This initial observation led to a series of publications documenting the role of IL-2 in the “Lymphokine-Activated Killer” (LAK) cell phenomena197and the capacity of these cells to kill fresh tumor cells in vitro and to induce regression of established disseminated lymphoma after adoptive transfer in animal models.198 These studies were not limited to lymphoma and follow-up studies rapidly transitioned to treatment of established murine melanoma.199 One notable feature of Rosenberg’s approach to immunotherapy was its fast translation into clinical trials. In early 1984, the first phase I study on the use of LAK cells in humans was published.200 This trial demonstrated proof of concept, showing that large numbers of cells could be obtained by in vitro expansion of lymphocytes derived from peripheral blood, which could then be safely transferred back into patients with cancer. The study also documented evidence of migration of these cells into several organs and tissues including tumor. Two additional studies would be instrumental in the advancement of ACT into the clinical setting, both relating to the in vivo administration of IL-2 in murine models.201, 202 Following phase I assessment of the safety of systemic administration of IL-2 in humans,203 a much larger study combining LAK and IL-2 administration was performed204, 205 in which more than 100 patients were treated with several courses of IL-2 and very high numbers of autologous LAK cells (up to 18.4 × 1010 cells). Complete or partial responses were seen in 21% of patients.205 Despite the favorable results obtained in these early clinical trials, data from murine models would redirect research away from LAK cells and toward the transfer of highly specific tumor-reactive T cells. The new approach was based in the isolation and in vitro expansion of TILs with IL-2.195 These expanded TILs were 50–100 times more efficient than LAK cells in the treatment of various types of tumors and their potency was significantly enhanced when their transfer was combined with in vivo administration of cyclophosphamide and IL-2.45 Clinical trials mirroring the murine experience and combining cyclophosphamide, TILs, and IL-2 have yielded complete and partial responses in up to 31% of patients with metastatic melanoma.206
These early studies have helped to inform the evolution of ACT as a form of personalized immunotherapy.207–209 The transition of ACT into the clinical setting has, however, not been without difficulties. These can be considered in two groups: factors relating to difficulties in generating appropriate products for adoptive transfer and factors relating to host or tumor resistance to transferred populations.
Generation of cellular therapy products
One major limitation has been the difficulty in accessing tumor samples containing sufficient numbers of viable TILs for successful isolation and expansion, which remains a limiting step in 60–70% of cases.210 Alternative strategies for isolation of tumor-reactive lymphocytes (TRLs) would facilitate more widespread application. Such approaches include the use of T cells from peripheral blood or lymph node biopsies.211–214 Stimulation with antibodies directed against CD3 and the costimulatory receptor CD28 expressed on T cells are now widely used for the ex vivo expansion of large numbers of TRLs, while 4-1BB (see below) has recently attracted more attention both for activation and selection protocols.214–218 The identification and cloning of T cells with specificity to antigens that are either more abundant on or, less commonly, specific to tumor cells (NY-ESO-1, MART-1, tyrosinase, gp100, p53) have also contributed greatly to the development of alternatives to isolation of TILs. In vitro enrichment of such TRLs from peripheral blood by stimulation with their cognate antigen can be followed by expansion protocols as previously outlined. Much has been learnt about the optimal characteristics of the transferred cells, in terms of maximizing persistence and antitumor activity. Long-term in vitro culture has been demonstrated to be detrimental and the level of differentiation of T cells to be critical.219 IL-2 drives differentiation of T cells to intermediate and late effector stages of differentiation. Furthermore, IL-2 may not be the optimal cytokine to use to enhance activity in vivo as it also expands Treg and is associated with significant toxicity. The first issue may be addressed by the use of IL-7, IL-15, and IL-21, which seem to more specifically target effector T cells when applied either alone or in combination,220 rescuing tolerant CD8+ T cells,221 and generating less differentiated cells that appear to mediate greater antitumor immunity.222–224 IL-2 seems to be required not only for the expansion but also for the long-term persistence of the adoptively transferred T cells. More recent work in murine models suggests that cotransfer of CD4+ T cells can supply IL-2 and sustain TRLs for long periods of time therefore increasing the antitumor effect of ACT.225–227 Numerous studies have demonstrated the requirement for CD4+ T cell help in the generation and/or maintenance of CD8+ T cell memory. In addition to provision of cytokine support, they have roles in DC conditioning and in recruitment and activation of macrophages and eosinophils, which can mediate antitumor effects.224, 228, 229
More recent efforts have focused on genetic modification of T cells to engineer improved antitumor effects. Such approaches include the transfer of CAR that have antibody-based external receptor structures and cytosolic domains that encode signal transduction modules of the TCR.230 These allow redirection of T cell specificity in an MHC unrestricted manner, while delivering the equivalent intracellular signaling of TCR ligation. Furthermore, the impact of triggering can be enhanced by engineering the cytosolic domain for additional provision of counterfeit costimulatory signaling following ligation (mimicking CD28, 4-1BB, or OX40 ligation). Although incorporation of a CD28 component results in IL-2 release and limited proliferation (plus enhanced resistance to the suppressive effects of Treg231), T cell activation remains incomplete and can be further enhanced by inclusion of the 4-1BB or OX40 signaling domains within the construct.232, 233 A number of early trials have suggested that persistence of transgene-expressing cells may be limited to periods of days to weeks following transfer.49, 234–237 This may relate in part to the potential immunogenicity of the CAR. However, lessons learnt from earlier ACT studies may also be pertinent, and current approaches are focusing on the use of central rather than effector memory populations238, 239 and on the use of T cells specific for herpes viruses that are maintained at relatively high levels in humans owing to persistent stimulation via their native TCR by the “latent” viral reservoir.49 Central memory cells represent the least differentiated end of the spectrum of antigen-experienced T cells and retain the developmental options of naive T cells including the capacity for marked clonal expansion. An alternative strategy to redirect T cell specificity relies on transduction of TCR genes from tumor-reactive clones, although this has the relative disadvantage of conferring MHC-restricted targeting.240–242 In the first clinical trial using this approach, a MART-1-reactive TCR was transduced into human lymphocytes, inducing the capacity to secrete effector cytokines and display lytic activity when coincubated with MART-1+ tumor cells. Following recipient lymphodepletion, these cells were infused into patients with melanoma who subsequently received infusions of IL-2 with the suggestion that the T cells may have effected clinical responses in 2/17 patients.48 Potential factors limiting efficacy in this study include variable persistence of gene-modified cells and relatively low levels of surface expression of the introduced TCR. The latter relates to competition of exogenous TCR chains with endogenous TCR chains for assembly with CD3 components and also to the formation of mixed dimers of exogenous and endogenous TCR chains, restricting avidity.243 Indeed, gene optimization that elicits only modest increases in TCR expression may result in marked enhancement of antitumor activity.244 An alternate and perhaps complementary approach is to engineer cells in ways to enhance survival, for example, by transducing cells with chimeric GM-CSF-IL-2 receptors (designed to deliver an IL-2 signal when binding GM-CSF in an autocrine loop245), CD28,246 or the catalytic subunit of telomerase.247 The ability to further modify these lymphocytes, to make them less subject to the suppressive influences present in the tumor microenvironment such as the introduction of genes encoding dominant-negative TGF-β or inhibitory RNAs to prevent the expression of inhibitory molecules such as CTLA-4 and PD-1, could potentially further enhance the activity of the transferred cells.248 Alternatively, overexpression of selected costimulatory ligands may induce auto- and transcostimulation, resulting in potent antitumor activity.249
The role of lymphodepletion
Transfer of tumor-specific T cells into unmanipulated hosts is often accompanied by a failure to demonstrate engraftment or persistence of transferred cells, or only modest antitumor effects on bulky tumors,250–252 and it is likely that further attention to both intrinsic and extrinsic inhibitory factors will be critical to successful clinical application. Profound lymphodepletion of the host substantially increases the effectiveness of cell transfer therapy, likely by a number of mechanisms including elimination of cytokine sinks (lymphocytes competing with transferred cells for homeostatic cytokines such as IL-7 and IL-15), elimination of Treg and myeloid suppressor cells, and provision of an environment driving homeostatic proliferation.225, 238, 253–255 The Treg content of the adoptively transferred population may also be important.225 In addition to effects on immune cells, host irradiation can sensitize the stromal cells surrounding the tumor256 and induce upregulation of adhesion molecules on tumor vasculature.257, 258 Finally, host irradiation releases LPS from commensal gut micro-flora, which further matures DCs to activate tumor-reactive T cells,259 and escalation to ablative conditioning with hematopoietic stem cell rescue may further promote the expansion and function of adoptively transferred CD8+ T cells.260
Current position of adoptive T cell transfer
Experience with ACT is, therefore, generally encouraging, but it is important to recognize that use of TILs may favor inclusion of those with an immune system inherently more capable of mediating antitumor activity. As the level of tumor infiltration with TILs has been shown to correlate with outcome in a number of studies of human malignancy, the ability to generate a therapeutic product from TILs could be a predictive biomarker for outcome. The achievement of higher response rates in the significantly less selected groups receiving CAR- or TCR-transduced ACT will be an important step forward in this regard, but current results have demonstrated results more typical of other single-agent immunotherapeutic approaches. In the absence of large randomized studies, it is impossible to assess the relative contributions of lymphodepleting chemoradiotherapy, IL-2, and ACT to overall results. The number of randomized studies performed in the field remains limited (reviewed in211) and only one of these had a positive outcome. Intriguingly, this was in patients with hepatocellular carcinoma rather than the more commonly treated melanoma or renal cancer. Patients received peripheral blood T cells activated in vitro with anti-CD3 and IL-2 in the adjuvant setting following surgical resection of the primary tumor (n = 150). Tumor recurrence was reduced 41% in those receiving ACT with longer progression-free survival (p = 0.01), and a trend toward longer overall survival (p = 0.09).261 There was no such benefit in a more recent trial of patients with stage III melanoma randomized to receive TIL plus IL-2 or IL-2 alone.262, 263 One further major problem with the application of ACT is that it is a highly personalized treatment and does not easily fit into current modes of oncological practice. Generation of appropriate cellular products is labor-intensive and requires significant laboratory expertise. Furthermore, each patient essentially requires the generation of new reagent limiting the opportunities for easy commercialization, and suggesting that delivery may be considered more service-oriented rather than product-related (as in the case of most drugs). This aspect, combined with increasing awareness that minimizing ex vivo T cell manipulation may be advantageous in terms of clinical outcomes, is driving the current evolution of clinical strategies. The issue is also informed by topical debate concerning the nature of the best targets for immunotherapy in terms of public versus private antigens. While we have historically focused on “public” or shared tumor antigens such as MART-1 and gp100, accepting that antitumor efficacy may then be inextricably linked to tissue-specific toxicity, “private” or patient-specific antigens generated as a consequence of the evolution of the malignant phenotype and inherent genetic instability within these lesions have a number of attractive advantages,264, 265 an idea we shall return to later.266 The acceptance of tissue-specific toxicity leads to the concept of “dispensible tissues,”267 which may be acceptable in the case of vitiligo with therapies targeting melanoma or B cell deficiency following anti-CD20 therapy but will be more of an issue if the target is shared by a vital organ.235 The parallel development of “off the shelf” reagents for enhancing immunity offers a number of interesting approaches to further enhance the efficacy of ACT. The recently reported clinical successes with genetically modified T cell therapies268 and renewed interest by pharmaceutical companies to develop these strategies for eventual approval of ACT for the treatment of patients holds great promise for the future.
Promoting DC function
Detailed discussion of the newer approaches aimed at enhancing antigen presentation in efforts to improve antitumor immunity is beyond the scope of this chapter. However, a few deserve brief mention. DCs are uniquely specialized to present processed antigens to stimulate antigen-specific effector responses. Progress in our understanding of factors associated with DC-induced immunogenicity rather than tolerogenicity has informed the development of clinical therapeutics, yet it remains apparent that even conventionally “mature” DCs (with high levels of expression of costimulatory molecules) can mediate immune suppressive activity.142, 269–271 Cytokines known to enhance DC immunogenicity (e.g., IFNs, IL-15, TNF, and IL-1) are generally insufficient to induce robust adaptive immunity when given in isolation.272
Toll-like receptor (TLR) agonists are showing some early promise in clinical studies, particularly in combination with other therapeutic modalities. Unmethylated CpG oligodeoxynucleotides (ODNs) bind TLR9 on pDCs and B cells, inducing Th1 polarized immune responses and regression of established tumors in mice, as well as activity in phase I and II clinical trials.273–280 TLR9-activated APCs activate NK cells, enhance expression of Fc receptors on polymorphonuclear leukocytes, and promote CTL activation. However, it is important to remain aware that in some settings TLR ligands provoke immunosuppressive or tolerogenic responses.281, 282 The context of TLR signaling may well be important and much remains to be learnt about how the immune system discriminates between pathogenic inflammatory insults and the “beneficial” inflammation associated with commensal organisms or wound healing in order to iterate either immune activation or suppression in the light of apparently similar inflammatory signaling. The imidazoquinolone Imiquimod ligates TLR7 (and to a lesser degree TLR8) and when applied topically stimulates recruitment of pDCs and myeloid-derived DCs into tumors, both of which are implicated in subsequent antitumor activity.283 It may also directly induce upregulation of vascular E-selectin and inhibit regulatory T cell function (including both Treg and novel γδ suppressor populations),284–286 and has been demonstrated to exhibit exciting clinical activity in vulval intraepithelial neoplasias.287, 288 Activation of invariant NKT cells by the CD1d-reactive glycolipids α-galactosylceramide (a-GalCer) or its analog a-C-galactosylceramide (a-C-GalCer) provides an alternative route to DC maturation, enhancing antitumor activity in a number of murine models.289–291 Both CpG and imiquimod also induced increased levels of tumor antigen-specific T cells in melanoma patients vaccinated with recombinant protein tumor antigen NY-ESO-1.292, 293
Another attractive target for enhancing APC performance is CD40. This molecule belongs to the tumor necrosis factor receptor (TNFR) superfamily (see below), and ligation is crucial for development of competent cellular immune responses, at least in part through the induction of IL-12 secretion by DCs. It is also critical for affinity maturation and heavy chain class switching during humoral responses.228, 294 It is constitutively expressed by B cells and DCs and is also expressed by macrophages, T cells, and nonhematopoietic cells such as vascular endothelium.295 Its ligand CD40L is principally expressed by activated CD4+ T helper cells, although it is also expressed by plasmacytoid DCs, activated NK cells, and platelets. CD40 ligation is critical for licensing of DCs, providing the temporal bridge between CD4+ T cell help and effective generation of CD8+ CTL responses by “conditioning” DCs (enhancing activation and costimulatory capacity). This activity may be reliant on CD27:CD70 interactions, as blocking CD70 during anti-CD40 treatment abrogates protection in a murine lymphoma model.296 CD40 stimulation can potentially effect antitumor responses in a number of ways.297, 298 Firstly, the expression of CD40 by a number of lymphoma and carcinoma tumor cells suggests that monoclonal antibodies to CD40 could elicit complement-dependent cellular cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC). Secondly, CD40 signaling has been reported to directly induce apoptosis in some tumor cells, notably in high grade B cell non-Hodgkin lymphomas and epithelial carcinomas.299–301 Finally, by enhancing antigen-presenting capacity CD40 ligation may directly, particularly in the case of B cell lymphomas, or indirectly enhance presentation of tumor antigens to the immune system, at least partially overcoming the requirement for CD4+ T cell help in the generation of effective and durable antitumor CTL responses.302–304 CD40 ligation by either CD40L or stimulatory anti-CD40 monoclonal antibodies in vitro or in vivo enhances antitumor vaccine efficiency in a number of murine models of malignancy.297, 305 By extension, forced expression of CD40L by either DC vaccines or B cell tumors such as chronic lymphocytic leukemia using recombinant adenoviral vectors can enhance antitumor T cell responses and induce some level of clinical response.306, 307 Of note, however, these responses may be transient and associated with rapid engagement of host immune inhibitory circuits (e.g., Treg), suggesting that combinatorial approaches might be more effective.143 Clinical trials with recombinant CD40L trimer or anti-CD40 antibodies have suggested that in vivo triggering of CD40 on tumors and/or APCs can induce responses in a minority of patients, particularly in B cell lymphomas or melanoma, although toxicities including systemic inflammatory syndromes and venous thromboses have been documented.308–310 A novel approach utilized electroporation to introduce mRNA encoding CD40 ligand, constitutively active TLR4, CD70, and multiple melanoma tumor antigens into autologous DCs (TriMix-DC). Tumor regressions were observed in 6 of 17 patients who had received interferon-α-2b in combination with TriMix-DC.311 Further, combination of anti-CD40 antibody and chemotherapy agent, gemcitabine, showed tumor regression in both human and mice.312 Antitumor responses were attributed to activated macrophages that infiltrated tumor stroma. These agents may prove to be valuable components of future therapeutic combinatorial strategies.313
Directly promoting T cell function
Immune activation is critically regulated by two major families of coreceptors expressed by T cells: the immunoglobulin-like (Ig) superfamily and the TNFR superfamily.314, 315 Costimulatory members of the former include CD28 and inducible T-cell costimulator (ICOS), while OX40, CD27, 4-1BB, CD30, GITR (glucocorticoid-induced TNFR family related gene), and HVEM (herpes-virus entry mediator) are members of the latter. The most well-established inhibitory members of the immunoglobulin “costimulatory” family include cytotoxic T-lymphocyte-associated-antigen 4 (CTLA-4) and programmed cell death 1 (PD1). B- and T-lymphocyte attenuator (BTLA) is the most recently described member of the family and also appears to mediate inhibitory effects on T cell activation.316 The identities of the receptors for the newer members of the B7 ligand family (B7-H3 and B7x/B7-H4) remain elusive, but these receptors may also mediate significant inhibitory activity, perhaps more so in the periphery given the tissue distribution of these more recently identified ligands. Stimulatory or blocking monoclonal antibodies (Figure 1) are being extensively investigated for their abilities to enhance T cell numbers, function, and maintenance of immunological memory.317, 318
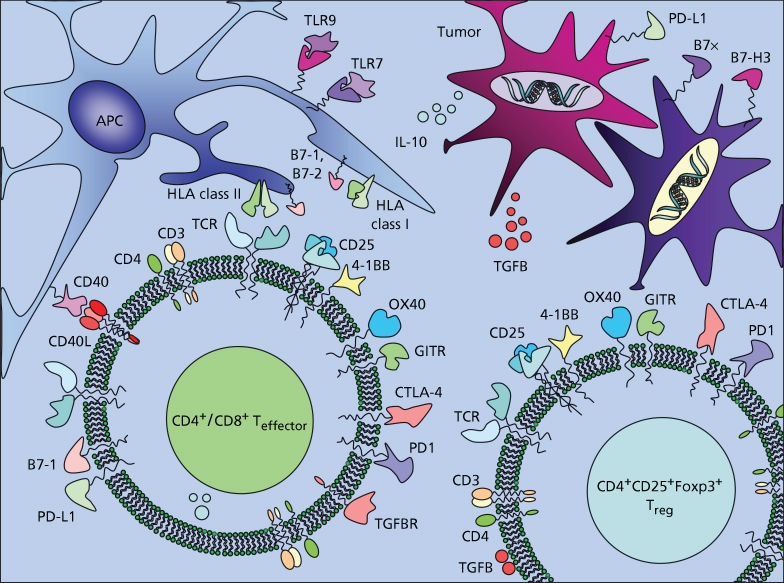
Figure 1 Potential targets for immunostimulatory therapies. The majority of T cell coreceptors that are upregulated upon activation of effector cells are also expressed by regulatory T cell populations and both cell types may be important targets for therapeutic interventions.
Stimulatory antibodies to 4-1BB (CD137), OX40 (CD134), and GITR—accentuating the positive?
A number of the members of the TNFR family are appealing candidates for the development of targeted therapeutics. To date, there has been relatively little data regarding agonistic anti-CD27 antibodies in murine tumor models, although it does appear to be a potentially interesting target for boosting antitumor immunity.296 Greater attention has been focused on 4-1BB and OX40. 4-1BB is expressed on activated T cells (including Treg and NKT cells), activated NK and DCs, eosinophils, mast cells, and endothelial cells in some metastatic tumors.315, 319–321 Its ligand 4-1BBL is expressed on activated DCs, B cells, and macrophages. Ligation on T cells results in upregulation of antiapoptotic genes and protection from activation-induced cell death (AICD),322 enhancing establishment of durable memory CTLs.323 The upregulation of 4-1BB on antigen-experienced T cells suggests that anti-4-1BB may differentially target these primed T cells, preferentially influencing those T cells with highest avidity receptors and partially explaining why 4-1BB costimulation may be superior to CD28 costimulation for the generation of antigen-specific cells for adoptive therapies.217 While it is assumed that costimulation of CD8+ T cells is the principal mechanism of action of anti-4-1BB, various other immunomodulatory activities may contribute. In this respect, a common theme developing in our understanding of the function of immunostimulatory antibodies is their possible multiplicity of function, reflecting the cellular distribution of the receptors. Thus, (1) activation of APCs,324 (2) reduction in Treg suppressive capacity325 or enhancement of effector resistance to suppression,326 and (3) costimulation of CD4+ and CD8+ T cells are all supported by experimental data. Furthermore, activated NK cells (possibly equivalent to interferon-producing killer DCs), IKDC,327–329 and NKT cells may be relevant targets for antitumor activity.320, 321, 330, 331 Reverse signaling into cells expressing the ligands for a number of Ig or TNFR superfamily members (see also GITR:GITRL, CTLA-4:B7, and PD-1:PD-L1 below) is another recurring theme of recent investigations into immune modulating functions of these molecules. In the case of 4-1BBL, this may result in enhanced production of inflammatory mediators or enhanced cell adhesion, facilitating egress of immune effectors into sites of inflammation.332, 333 There are conflicting data as to whether 4-1BB ligation on Treg results in enhanced or reduced suppressive capacity,325, 334, 335 and synergy of antitumor activity with approaches that are thought to target Treg number or function has been taken as evidence that any 4-1BB-mediated inhibitory effects on Treg function may be relatively modest.336–338 Forced expression of 4-1BBL in murine tumors enhances immunogenicity, reducing engraftment rates in immune competent recipients, although growth of untransfected cells is only modestly affected in relatively poorly immunogenic tumors.320 Agonistic anti-4-1BB monoclonal antibodies enhance antitumor CTL responses, enabling rejection of established syngeneic tumor cell lines.339–341 Activity appears critically dependent on CD8+ T cells and (in most studies) NK cells, with the role of CD4+ T cells varying in different tumor models.320, 340–343 Just as with other immunostimulatory antibodies, combination with vaccination strategies enhances activity in poorly immunogenic tumor models.344, 345 Coadministration with transgenic tumor-specific CTL enhances antitumor activity, apparently via a reduction in AICD rather than enhanced proliferation.346 These data demonstrate the activity of anti-4-1BB antibodies in the absence of CD4+ T cell help, albeit in immunodeficient hosts (RAG2−/−) lacking normal regulatory mechanisms. Early experience with a humanized clinical grade antibody (BMS-663513/Urelumab) has targeted mainly patients with melanoma and renal cell carcinoma.347 The antibody initially appeared to be well tolerated with some evidence of activity (6% partial responses in melanoma patients), once again demonstrating the probable need to evaluate combinatorial approaches (and also define the optimal dosing strategy).336, 348, 349
Two particularly intriguing and seemingly paradoxical features of anti-4-1BB monoclonal antibodies are their ability to ameliorate autoimmunity350–352 and to suppress humoral immunity in mice.353 Although the precise mechanism(s) remain obscure, possibilities include effects on Treg function, interference with CD4+ T cell activation (possibly via the intermediacy of expansion of IFNγ-secreting CD11c+CD8+ T cells354), or IFNγ-dependent induction of IDO.351, 352 This highlights the potential importance of the timing of immunomodulatory interventions, as CD4 downregulation may be induced by AICD, which can also occur in CD8+ T cells if exposure to anti-4-1BB occurs immediately after antigen encounter.355, 356 While this suppressive activity has been proposed to be advantageous in terms of limiting possible antibody-mediated toxicities, it is recognized that it might also be deleterious to the development of optimal anticancer immunity. Preclinical studies with anti-CD137 in murine models showed promising activity in combination with antibodies directly targeting the tumor. Most recently, it was shown that anti-CD137 increases efficacy of cetuximab (anti-EGFR mAb) in murine xenograft models,357 making it another suitable target for combination therapy. Clinical studies with PF-05082566 (Pfizer, anti-CD137) in combination with rituximab in B cell lymphoma and MK-3475 (anti-PD-1) in solid tumor is currently ongoing.
OX40 is expressed transiently on activated CD4+ and CD8+ T cells, functioning as a late costimulatory receptor.315, 358 It is also expressed by NKT cells, where triggering may be required for optimal activation by a-GalCer,359 and Treg. Its ligand OX40L is expressed in a similar distribution to 4-1BBL, on activated DCs, B cells, and macrophages, as well as activated T cells and endothelial cells.315 OX40 ligation regulates CD4+ and CD8+ T cell survival and memory generation, preventing T cell tolerance.360–363 It also impairs the suppressor functions of Treg.364, 365 Furthermore, OX40 triggering appears to be antagonistic for Foxp3 induction in antigen-responding naïve CD4+ T cells, effectively suppressing the generation of iTreg,366, 367 and blocks the generation of IL-10-producing Tr1 cells,368 suggesting that OX40 may antagonize the generation of a number of different inducible regulatory T cell populations. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses, overlapping in their intracellular signaling pathways,369 yet neither 4-1BB nor GITR signaling seem to block the generation of Tr1,368 and there are currently no reports illustrating whether they influence Foxp3+ iTreg induction. As with 4-1BBL, forced expression of OX40L by tumor cells increases immunogenicity, with tumor rejection dependent on both CD4+ and CD8+ T cells.370 Furthermore, intratumoral injection of DCs modified to have enhanced expression of OX40L can effect tumor rejection in murine models that is dependent on CD8+ CTL responses, themselves dependent on CD4+ T cells and NKT cells.359 Agonistic anti-OX40 antibodies also increase antitumor activity in a number of transplantable tumor models.371 Concomitant activity on both effector and regulatory compartments may be a prerequisite of effective rejection of established tumors.81 In preclinical models, OX40 ligation enhances several other immunostimulatory approaches.372–376 A phase I clinical trial using a mouse monoclonal antibody that agonizes human OX40 signaling in patients with advanced cancer showed that patients treated with one course of the anti-OX40 mAb (9B12) had an acceptable toxicity profile and regression of at least one metastatic lesion in 12 of 30 patients.377
In common with both 4-1BB and OX40, GITR is transiently expressed on activated T cells.378, 379 It is also constitutively expressed at high levels on Treg with further induction following activation.380, 381 Its ligand GITRL is expressed at low levels on B cells, macrophages, and some DCs, transiently increasing following activation. GITR ligation stimulates both proliferation and function of CD4+ and CD8+ T cells.382 Its activity on Treg has remained more contentious.383, 384 Anti-GITR antibodies reduce suppression in cocultures of CD4+CD25− effectors and CD4+CD25+ Treg, but whether this relates to reduced Treg suppressor function, increased resistance of effectors to the preserved suppressor function of Treg, or a combination of both, has yet to be definitively demonstrated. Experiments using mixtures of GITR+/+ and GITR−/− effector and regulatory cells suggest that ligation of GITR on the effector population rather than the regulatory population is critical for abrogating suppression,384 suggesting that enhanced effector resistance to suppression may be key in in vitro assays. Injection of adenovirus expressing recombinant GITRL into B16 melanoma promotes T cell infiltration and reduced tumor volumes,385 while agonistic anti-GITR antibodies have been shown to enhance both rejection of established methylcholanthrene-induced fibrosarcomas and to enhance systemic antitumor responses and concomitant immunity when given following B16 melanoma challenge.386, 387 Furthermore, the same antibody also enhances the impact of DNA vaccination in terms of generation of systemic immunity and enhancing resistance to challenge with murine melanoma.388
Stimulation through checkpoint blockade of CTLA-4 (CD152), PD-1 (CD279), PD-L1 (CD274)—eliminating the negative
In contrast to the TNFR superfamily, the existence of coinhibitory receptors mediating direct downregulation of lymphocyte activation and/or effector function has been a recognized feature of the Ig superfamily for some time. Indeed, the coinhibitory receptor–ligand members outnumber the costimulatory members within this superfamily, engendering the idea of regulatory or inhibitory checkpoint blockade as a therapeutic anticancer strategy by James Allison two decades ago.389 Blockade of inhibitory immune checkpoints for therapeutic benefit offers considerable promise, particularly as combination with other treatment modalities that promote cross-priming of antitumor immunity may yield additive or synergistic activity. The first strategy shown in both murine models and patients to elicit regression of tumors was antibody blockade of CTLA-4.389, 390
CTLA-4 is expressed by activated CD4+ and CD8+ T cells, although its surface expression is tightly regulated with a short half-life. While expression is difficult to detect on resting T cells, it influences some of the earliest events in T cell activation,391, 392 being rapidly mobilized from intracellular vesicles in the proximity to the MTOC to the immune synapse after TCR engagement.393 In the unphosphorylated state, an intracellular localization motif mediates rapid binding to AP-2, endocytosis, and lysosomal targeting.394 It is constitutively expressed by natural and inducible Foxp3+ Treg
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


