CANCER GENES
Over the past 50 years, multiple discoveries have had an impact on our understanding of key genomic events that influence the development of malignant growth. After many large-scale sequencing projects of cancer genomes, we now understand that many genetic alterations in specific cancer genes are responsible for the development and progression of the disease. These alterations may occur at the level of the patient’s germline, predisposing to inherited forms of cancer that may develop in many tissues throughout the body. Genetic alterations may also be somatic, or newly acquired changes within the genes of an individual cell or group of cells over time and due to environmental stresses. Somatic alterations may come in many forms, including single-base substitutions; insertions or deletions of DNA fragments; rearrangements and rejoining of DNA from alternative locations in the genome; and copy number increases and reductions. Should these alterations effect key cancer genes, malignancy may develop.
In the early 1970s, when studying retroviruses that reverse transcribe RNA into DNA, it was found that (1) certain retroviruses, when incorporated into host cells, have the ability to transform normal cells into rapidly dividing tumors. Rous sarcoma virus (RSV), isolated by Peyton Rous, was the first retrovirus found to cause sarcoma in chickens (2). Later, hybridization studies proved that the RSV gene, termed v-src, was homologous to a highly conserved eukaryotic gene, c-src. Src became the first known viral oncogene (3). In contrast to highly transforming retroviruses, weakly transforming viruses can insert into the genome near proto-oncogenes, normal genes that when mutated give rise to an oncogene, and induce cancer. Activation of proto-oncogenes to oncogenes, through activating point mutations, gene amplification, or chromosomal translocation events, can occur independent of retroviral transformation and cause cancer.
In 1981, Shih and colleagues showed that normal NIH3T3 mouse fibroblast cells could be made cancerous by introduction of total genomic DNA from human cancers (4). Isolation of the specific DNA segment responsible for this transforming activity led to the identification of the first naturally occurring, human cancer-causing sequence change—the single-base G > T substitution that causes a glycine-to-valine substitution in codon 12 of the HRAS gene (5). These experiments demonstrated the causal relationship between oncogenic mutations and cancer. The discovery of HRAS and many other oncogenes altered our understanding of cancer and expanded our knowledge of driver mutations that can be targeted to treat disease.
Another commonly referred to class of cancer genes is tumor suppressor genes. These genes are frequently involved in cell cycle regulation, inhibition of cellular proliferation, and DNA repair. When functioning normally, they act as barriers to unregulated tumor growth. However, dysfunction of both copies of the gene are usually required to initiate tumor development as only one functioning copy is needed to regulate the cell. Alfred Knudson, in 1971, was the first to theorize about the role of tumor suppressor genes in cancer development (6). He described the two-hit hypothesis: The development of cancer was due to the loss of inherited regulatory genes that functioned to suppress tumor formation, subsequently followed by the somatic loss of the normal homologous allele. In non-hereditary forms, both alleles would be somatically affected (6,7).
The first tumor suppressor gene to be identified was the retinoblastoma (RB) gene, found to cause childhood cancers of the retina. In the hereditary form, one copy of the RB gene is usually defective, with a second mutation or deletion of the normal gene leading to early cancer development. Hereditary forms frequently affect the bilateral eyes. Sporadic retinoblastoma is much rarer and occurs when there is homozygous deletion or somatic mutation of both normal copies of the gene. Sporadic retinoblastoma usually presents later in life than the hereditary form and usually effects only one eye.
In the mid-1980s, Webster Cavanee localized the retinoblastoma gene to a small region on chromosome 13 and found that both the inherited and sporadic varieties had the same secondary abnormalities leading to homozygosity of mutations in the RB region (8). In 1986, Stephen Friend isolated human complementary DNA mapping to the RB gene (9). The following year, Wen-Hwa Lee and Yuen-Kai Fung both cloned RB using chromosome walking (10,11). Huei-Jen Su Huang and colleagues later proved a causative relationship between the defective RB gene and cancer by performing rescue experiments of the neoplastic phenotype in RB-mutated retinoblastoma cells with wild-type RB (12). Aside from retinoblastoma, many tumors have subsequently been found to have defects in the RB gene, which may play a role in the establishment of these cancers.
NEXT-GENERATION SEQUENCING
Completion of the Human Genome Project in April 2003 and subsequent publication of a human reference genome provided new opportunities for analyzing cancer. Early on, many groups conducted these studies by sequencing large segments of polymerase chain reaction (PCR) products to detect substitutions and small insertions and deletions. More recently, second-generation sequencing, or next-generation sequencing, has revolutionized the field of oncology by allowing for the entire sequencing of whole genomes in a relatively timely and economic manner (13). Commonly used next-generation platforms include the Illumina and Ion Torrent systems. In just over a week, a solitary run on an Illumina HiSeq 2000 sequencer can generate 200 gigabases of data. Over the past decade, there has been a dramatic decrease in sequencing cost with an increase in sequencing capacity that has outpaced Moore’s law of technology (14). Deep sequencing and single-cell sequencing has allowed us to identify mutations in highly admixed samples. In addition, next-generation sequencing has allowed us to query multiple genomic alterations at a time, such as somatic mutations, copy number variations, as well as some structural information common to cancer (15).
MUTATIONAL BURDEN OF CANCER
Some cancers may be driven by a single mutation alone; however, other tumors may contain alterations in multiple driver genes, leading to overproliferation. On the low end of the range, leukemias and liquid malignancies harbor far fewer coding single mutations, about 9.6 per tumor that would alter protein-coding sequence, on average (16,17). Some more common solid tumors, such as colon, breast, brain, or pancreas, contain 33 to 66 genes, on average, with coding somatic mutations (16). The large majority, about 95% of mutations, are single-base substitutions (90.7% missense, 7.6% nonsense, and 1.7% alterations of splice sites or untranslated regions), the minority being insertions or deletions of one or more bases (16).
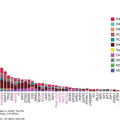
Toxic environmental factors may cause some tumors to have markedly more mutations than others with distinct mutational signatures. In addition, defects in DNA repair proteins such as dysfunction in the Fanconi pathway, the DNA mismatch repair pathway (Lynch syndrome), or the proofreading domains of DNA polymerases POLE or POLD1 may increase mutational burden (18,19). Recently, it was also found that the median frequency of mutations varies within cancer types as well. In 2013, Lawrence et al published a study examining 27 different cancer types. This study showed a wide variance in the frequency of nonsynonymous mutations that ranged more than 1,000-fold across cancers within a given subtype (Fig. 47-1) (20). As previously described, this study showed a high frequency of mutations in melanoma and lung cancer, thought to be caused by UV radiation and tobacco carcinogen exposure, respectively, with over 100 mutations/Mb. The frequency of mutations in melanoma and lung cancers was also quite variable, however, ranging from 0.1 to 100 mutations/Mb. Although less extreme than the latter cancers, acute myelogenous leukemia’s (AML’s) frequency of mutations also ranged widely from 0.01 to 10 mutations/Mb. This was despite the overall low number of mutations (0.37/Mb) (20). Alexandrov and colleagues analyzed the sequencing data from 7,042 cancers and demonstrated that these tumors exhibited greater than 20 discrete mutational signatures. Some signatures could be found across cancer types (ie, APOBEC cytidine deaminase signature), and other signatures were found in a single cancer class. Kataegis, or regional hypermutation, was also found in many cancers (21).
Figure 47-1
Whole-exome somatic mutation frequencies in 3,083 tumor-normal pairs. Each dot represents a tumor-normal pair. The y axis is the total somatic mutation frequency. Tumor types are ordered on the x axis (lowest to highest) by their median somatic mutation frequency. (Reproduced with permission from Lawrence MS, Stojanov P, Polak P, et al: Mutational heterogeneity in cancer and the search for new cancer-associated genes, Nature. 2013 Jul 11;499(7457):214-218.)

Despite the number of mutations present, not all mutations contribute to the production and growth of tumor cells. Identifying the background mutation rates for tumors as described previously was integral to our understanding of those genes that play a key role in the development of the tumor and confer a selective growth advantage. These alterations in genes are termed driver mutations. Those genes that are present within the tumor but do not contribute to tumor formation are deemed passenger mutations (22). Driver mutations may not all contribute to tumor growth in the same manner, and some driver mutations may be integral to certain steps of tumor development (proliferation vs invasion). Common driver mutations that occur in cancer genes include PTEN, EGFR, TP53, IDH1, RB1, KRAS, and BRAF (23). In addition, some mutations originally deemed passenger mutations may become drivers once treatment leads to eradication of sensitive clones and provides a niche for already present, resistant, clones to develop (24). Work is currently under way to target these genes for treatment options.
CHROMOSOMAL GAINS, LOSSES, AND TRANSLOCATIONS
Chromosomal gains, losses, and translocations are some commonly observed hallmarks of cancer alterations. Somatic copy number alterations may span the entire chromosome or one arm of a chromosome, although they may be limited to particular regions of the genome (25). It has been reported that an average cancer cell may have gains or losses involving a quarter of its chromosomes, with smaller, local events affecting about 10% of the genome (26). Many of these focal events occur in ‘‘peak’’ regions, affecting a median of 6 to 7 genes (although up to 150-200 genes in some cases). Due to the broader effect of amplification and deletion events, it is difficult to interpret which genetic alterations contribute to carcinogenesis (15,27).
Chromosomal translocations are associated with both liquid and solid malignancies, particularly leukemias, lymphomas, and sarcomas. In some tumors, translocations can result in the fusion of genes normally found at a distance from each other or bring genes closer to enhancer or promoter elements, resulting in alteration in their normal expression patterns. The number of translocations may differ between cancers depending on their degree of genomic instability (28). As discussed further in the chapter, translocations can become targets for cancer treatment.
TUMOR HETEROGENEITY
Not only have tumors been found to have a varied number of mutations, but also tumors display intertumoral heterogeneity, with diverse mutations within a given tumor type. In melanoma, for example, BRAF mutations are present in about 50% to 60% of patients, allowing for the majority, but not all, patients to benefit from BRAF inhibitors (29,30
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


