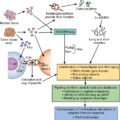
Summary of key facts
- •
The innate immune system is the host’s first line of immune defense against challenges of the environment.
- •
The innate immune system response is a nonrestricted, rapid response.
- •
Innate immunity is required for the establishment of adaptive immunity, or immune memory.
- •
Cellular components comprising the innate system include monocytes, macrophages, dendritic cells, and granulocytic cell types such as neutrophils, mast cells, eosinophils, and basophils.
- •
Myeloid cells are phenotypically plastic, modulated by multiple systemic signals associated with inflammation.
- •
In a cancer setting, myeloid cells can either promote tumor growth or have antitumor functions.
- •
Chronic inflammation associated with cancer results in emergency myelopoiesis and emergence of myeloid precursors from the bone marrow and other extramedullary sites.
- •
Tumors, tumor-produced cytokines and chemokines, tumor-associated leukocytes, restricted nutrient supply, and oxidation of lipids alter the phenotype and function of myeloid cells entering the tumor microenvironment.
- •
Therapies aimed at genetically or epigenetically reprogramming myeloid cells in vivo or in vitro have shown promise as novel immune therapies.
Introduction
Innate immune cells are extraordinarily sensitive to the environment of the tissues in which they reside, phenotypically and functionally adjusting according to the chemokine and cytokine milieu, changes in tissue metabolism, and changes in the tissue architecture. Because of their functional adaptability, innate immune cells are central for the protection and maintenance of tissue homeostasis in response to inflammation, infectious disease, and cancer. This chapter will highlight critical developmental, transcriptional, and functional aspects of the innate immune system and how each of these pathways can be coopted by or contribute to cancer initiation, tumor growth and metastasis, antitumor immunity, and therapy response. Finally, this chapter will highlight therapeutic targets within the innate immune system that have shown promise as cancer therapies.
The cellular components of the innate immune response
The immune system is divided into two branches, the innate immune system (nonrestricted rapid response) and the adaptive immune system (acquired cellular and humoral response). Innate immune cells comprise macrophages, monocytes, dendritic cells, and other myeloid- and lymphocyte-derived subsets that initiate and/or regulate host immune responses so that the adaptive arm of the immune system can eventually be triggered. Innate immune cells are not restricted by antigen and are functionally attenuated through signaling via immune receptors located on the surface or cytoplasm of the cells. These receptors recognize chemokine and cytokine signals, lipids and other signaling molecules, pathogen-associated molecular patterns (PAMPs), and dead and dying cells (damage-associated molecular patterns or DAMPs). To understand how the innate immune system functions in response to cancer, we will first define elements of innate immunity that are relevant to cancer, with an emphasis on myeloid subsets, given their significant contribution to tumor initiation, growth, immune evasion, and metastasis. Understanding the origins and functional potential of myeloid cells in the context of homeostatic or acute inflammatory settings will provide insight into the role of myeloid cells during cancer.
Monocytes
Monocytes are mononuclear-derived cell subsets that reside within the bone marrow, patrolling the blood and tissues for pathogens or abnormal cells. Monocytes were originally considered a short-lived precursor to tissue macrophages ; however, with the advent of more robust single-cell analysis technologies, fate mapping, and novel investigational model systems to study myeloid ontogeny and function, the origins and contribution of monocytes to tissue inflammation and homeostasis have become more nuanced. Indeed, monocytes do give rise to macrophages during specific inflammatory contexts. However, unlike macrophages, monocytes do not have the capacity for self-renewal, nor are they long-lived, like tissue macrophages derived from embryonic precursors that are maintained into adulthood. Studies have indicated that monocytes can give rise to dendritic cells , and demonstrated that monocytes are able to function as an individual and transient effector populations. For example, monocytes recruited to the liver by the proangiogenic factor vascular endothelial growth factor (VEGF) were educated to remodel the tissue vasculature without differentiating into macrophages. Monocytes recruited into skin and lungs did not terminally differentiate into macrophages or dendritic cells but instead participated in steady-state tissue surveillance and transport of antigen to draining lymph nodes via the lymphatics. Another study highlighted a critical role for monocytes in the expansion of brown adipose tissue. These studies have challenged the dogma that monocytes solely function to repopulate macrophage and dendritic cell pools but instead demonstrate that in multiple inflammatory contexts monocytes contribute to angiogenesis, tissue remodeling, and antigen trafficking to lymph nodes, all of which are pathways involved in cancer initiation and progression. The relevance and contribution of monocytes to cancer progression will be highlighted in the next section.
Monocyte ontogeny
In mice, monocytes consist of at least two functionally and phenotypically distinct subsets. Classic monocytes, or inflammatory monocytes, are Ly6C high (GR-1 + ) with proinflammatory properties, migrating to injured , or infected tissues from the blood or bone marrow via signaling through chemokine receptor CCR2. Nonclassical Ly6C neg (GR-1 neg ), or patrolling, monocytes reside in the vasculature and crawl along the luminal vessel surfaces of endothelial cells to surveil tissues during the resolution of inflammation. Ly6C neg monocytes enter into tissues during the latter phase of infection, via CX3CR1 signaling, exerting a tissue repair immune regulatory program. Ly6C hi monocytes have a short-lived half-life of 20 hours but constitute the steady-state precursors to the blood-residing Ly6C neg migratory pool such that the abundance of Ly6C hi monocytes dictates the pool of Ly6C neg progeny. Monocytes are thought to be derived from the common monocyte progenitor (cMoP), a myeloid precursor population found in the bone marrow in mice, and phenotypically defined as CLEC12A hi CD64 hi subsets in humans. Progenitor populations were originally thought to derive in a hierarchical progression from common myeloid progenitor (CMP)- granulocyte-monocyte progenitor (GMP)- monocyte-macrophage dendritic cell progenitor (MDP)-cMoP-monocyte. However, it has been demonstrated that under specific inflammatory contexts a bifurcation in the pathway occurred whereby CMP arose independently from MDP into GMP-derived neutrophil-like monocytes whereas MDP differentiated into monocyte-derived DCs. Thus, distinct ontogenies likely underlie observed functional heterogeneity in monocyte populations. Besides originating from the bone marrow, large pools of monocytes reside in the spleen and lungs, where they are maintained and are recruited en masse in response to inflammation, injury, or other systemic insults.
Most studies investigating monocyte origins and deployment throughout the body have used murine models. Human counterparts exist and have both similar and distinct functional attributes compared with their murine counterparts, which are outlined in Table 4.1 . In humans, monocytes represent 10% of the nucleated cells in the blood, whereas in mice monocytes represent 4% of nucleated blood cells. In humans, monocytes are defined based on CD14 and CD16 expression. CD14 ++ CD16 − monocytes are more similar in function and phenotype to Ly6C hi monocytes in mice, including having high expression of CCR2 and CD115 while also having lower CD16 expression levels. On the other hand, CD14 + CD16 + monocytes also appear to have similar attributes to Ly6C neg monocytes, including having elevated CX3CR1 expression. Despite these similarities, important differences between murine and human monocytes have been identified. For example, comprehensive transcriptional and flow cytometric profiling of murine monocytes and their human equivalents revealed that murine monocytes had high PPARγ expression signatures, whereas human monocytes had opposed patterns of surface receptors responsible for recognition and uptake of apoptotic cells and phagocytosis.
| Monocyte Subset | Species | Classic Markers | Recruitment | Phenotype | References |
|---|---|---|---|---|---|
| Classic monocyte (inflammatory) | Mouse | CD115 + CD11b + Ly6Chi CCR2 + CX3CR1lo |
|
| , , , , |
| Human | CD14 ++ CD16 − CD115 + CCR2 + CX3CR1lo | Proinflammatory | , , | ||
| Nonclassic (patrolling) | Mouse | CD11b + Ly6Clo CCR2 − CX3CRhi |
|
| , , , , |
| Human | CD14 + CD16 + CD115 + CCR2 − CX3CR1hi |
| , |
Monocyte function: Differentiation during homeostasis and inflammation
Monocytes are recruited in waves, where short-lived Ly6C hi monocytes contribute to early inflammation and Ly6C neg monocytes specialize in tissue repair and immune regulation. Ly6C neg monocytes are longer-lived, with a half-life of up to 2 weeks. Upon entering tissues, and depending on the signals present within a given tissue environment, monocytes will either act as terminally differentiated effectors, as outlined previously, or differentiate into tissue macrophage and dendritic cell populations.
In steady-state conditions, little evidence exists to indicate that classic Ly6C hi monocytes contribute significantly to the pool of tissue-resident macrophages. , , In the context of pathologic inflammation, Ly6C hi monocytes repopulate both tissue-resident macrophage and dendritic cell pools (for in-depth review, see Wynn et al. ). One interesting exception to this paradigm is the lamina propria myeloid cells residing in the intestines. Using macrophage depleted conditions, monocyte fate mapping, adoptive transfer of monocytes, and parabiosis experiments, Varol et al. and Bogunovic et al. both demonstrated that circulating inflammatory Ly6C hi monocytes replenish the pool of immune-regulatory CX3CR1 + macrophages but not of CD103 + lamina propria migratory dendritic cell subsets. , This process occurs during homeostasis and is thought to differ from other steady-state macrophage pools because of the low-level tonic signaling by the commensal microbiota. Follow-up studies demonstrated that signals from the commensal microbiota were essential for monocyte-driven repopulation of luminal macrophages, because antibiotic treatment significantly depleted monocyte recruitment into the intestines, whereas germ-free mice exhibited a significant reduction in luminal macrophage levels. Thus, the chronic signals derived by the gut microbiota were central for continued recruitment of Ly6C hi monocytes into the lamina propria. During acute inflammation, such as during colitis, Ly6C hi monocytes respond to bacterial products and give rise to proinflammatory effectors that eventually differentiate into migratory antigen-presenting dendritic cell subsets. On the other hand, noninflammatory patrolling monocytes in both mice and humans directly give rise to migratory dendritic cell subsets in homeostatic settings. This occurs in the absence of acute inflammation, where Ly6C hi monocytes recirculate back to the bone marrow and are converted into Ly6C neg populations that replenish depleted dendritic cell populations in mucosal, but not splenic, tissues.
Monocytes are versatile cell types and, similar to other myeloid-derived counterparts, exhibit sensitivity to the environment in which they reside. Functionally, monocytes exhibit plasticity and have a broad functional repertoire and differentiation potential that is dictated by the signals present in the tissues that they are recruited into. These properties endow monocytes with the potential to elicit robust inflammatory responses or participate in tissue remodeling and repair. Understanding the basis for monocyte fate and function in specific inflammatory contexts will be important for targeting these functionally diverse subsets for the treatment of various diseases, including cancer.
Macrophages
Macrophages, in Greek meaning “the big eater,” were originally identified as large phagocytic cells of the myeloid lineage having a critical role in maintaining tissue homeostasis and protection during sterile insults or disease. Macrophages are highly plastic phagocytic cells that have tremendous sensitivity to signals within their microenvironment, endowing them with broad functional diversity in terms of cytokines, chemokines, and other effector molecules. Macrophages occupy all tissues of the body, many of which are seeded into tissues during embryogenesis from the yolk sac and fetal liver. In most circumstances, tissue macrophages are long-lived and self-renew to maintain tissue residency throughout adulthood. Because of these properties, there is a significant effort to define the signaling pathways associated with macrophage recruitment and function within tumor-associated tissues and the tumor itself. To appreciate how macrophages function in the context of tumor initiation, metastasis, antitumor immunity and therapy response, we will first review macrophage ontogeny and function during homeostasis and acute inflammation, summarized in Fig. 4.1 .
Macrophage ontogeny
Macrophages populate tissues throughout the body in distinct waves during embryogenesis, with inputs from the yolk sac, fetal liver hematopoietic stem cells, or bone marrow. The first wave originates from the yolk sac, where erythroid–myeloid precursors either populate tissues directly or establish a niche of embryonic hematopoietic stem cells in the fetal liver. Monocytes originating from the fetal liver constitute the second wave of macrophage seeding into embryonic tissues. With the exception of the intestinal and cardiac macrophages, the latter of which are renewed by adult monocytes during aging, tissue macrophages are able to self-renew into adulthood. Brain microglia are the only population of tissue macrophages seeded exclusively by yolk sac embryonic precursors, whereas heart macrophages, liver Kupfer cells, and skin Langerhans cells are seeded by both the yolk sac and fetal liver. , Alveolar macrophages, kidney macrophages, and red-pulp macrophages are seeded only by fetal liver monocytes. ,
Macrophage cellular functions: Tissue-specific functions and the spectrum model of activation
Macrophages are considered professional phagocytes, a cell that specializes in phagocytosis, deriving from the Greek words phago (to devour) and cytos (cell). Phagocytosis is a cellular process in which innate immune cells engulf and ingest large materials (greater than 0.5 μm in size) such as dead and dying cells, pathogens, or other particulate materials. Phagocytosis by macrophages and other professional phagocytes is essential for immune homeostasis and host defense, a process that was first described by Elie Metchnikoff, who received a Nobel Prize for his work in 1908 (for in-depth review, see Gordon ). Phagocytosis occurs in multiple phases beginning with sensing of the particle to be ingested through receptors on the cell surface, , internalization of the particle through the plasma membrane and into a distinct cellular compartment termed the phagosome , and fusion of the phagosome with lysosomes, an organelle responsible for degradation of phagocytosed macromolecules.
Tissue-resident macrophages exhibit multiple specialized functions for distinct tissue compartments and are identified by specific transcription factors that are induced in response to the tissue microenvironment. Several functionally nonredundant macrophage populations reside in the spleen. Macrophages residing in the red pulp are regulated by the transcription factor PU.1, enabling localization to red pulp for phagocytosis of damaged and dying red blood cells scavenging released iron to maintain iron homeostasis. On the other hand, marginal zone splenic macrophages are dependent on LXRa signaling, where they specialize in the clearance of apoptotic cells while also regulating selective uptake of apoptotic cells by CD8a dendritic cells, a subset in the spleen responsible for regulating autoreactivity to self-antigens. Alveolar macrophages express the transcription factor B lymphoid transcription repressor BTB and CNC homology 2 (Bach2), which enables lipid handling and clearance of surfactant in the alveolar spaces of the lungs. They also facilitate gas exchange and prevent against hypoxia during infection.
Although there are multiple additional examples of tissue specification for macrophages, these studies underscore that macrophage function and phenotype are dictated by the signals present within the tissues that they reside. The advent of single-cell technologies to measure differences in gene expression, protein production, and chromatin regulation have enabled discovery of specific signaling pathways contributing to macrophage heterogeneity during health and disease. This concept is central to the heterogeneity of macrophages observed in disease settings such as cancer. Through transcriptional and epigenetic profiling of tissue-resident macrophages, Lavin et al. identified that aside from lineage-specific transcription factors, tissue-specific microenvironmental signals regulated macrophage identity and function through the induction of core transcription factors and specific chromatin modifications. Tissue signals were so robust that transplanted macrophage precursors or mature macrophages were effectively epigenetically “reprogrammed” when transferred into new tissue environments, exemplifying macrophage functional and transcriptional plasticity. Macrophage plasticity and the ability to be reprogrammed in response to changes in the microenvironment are key considerations when developing anticancer therapies, as will be outlined in the following.
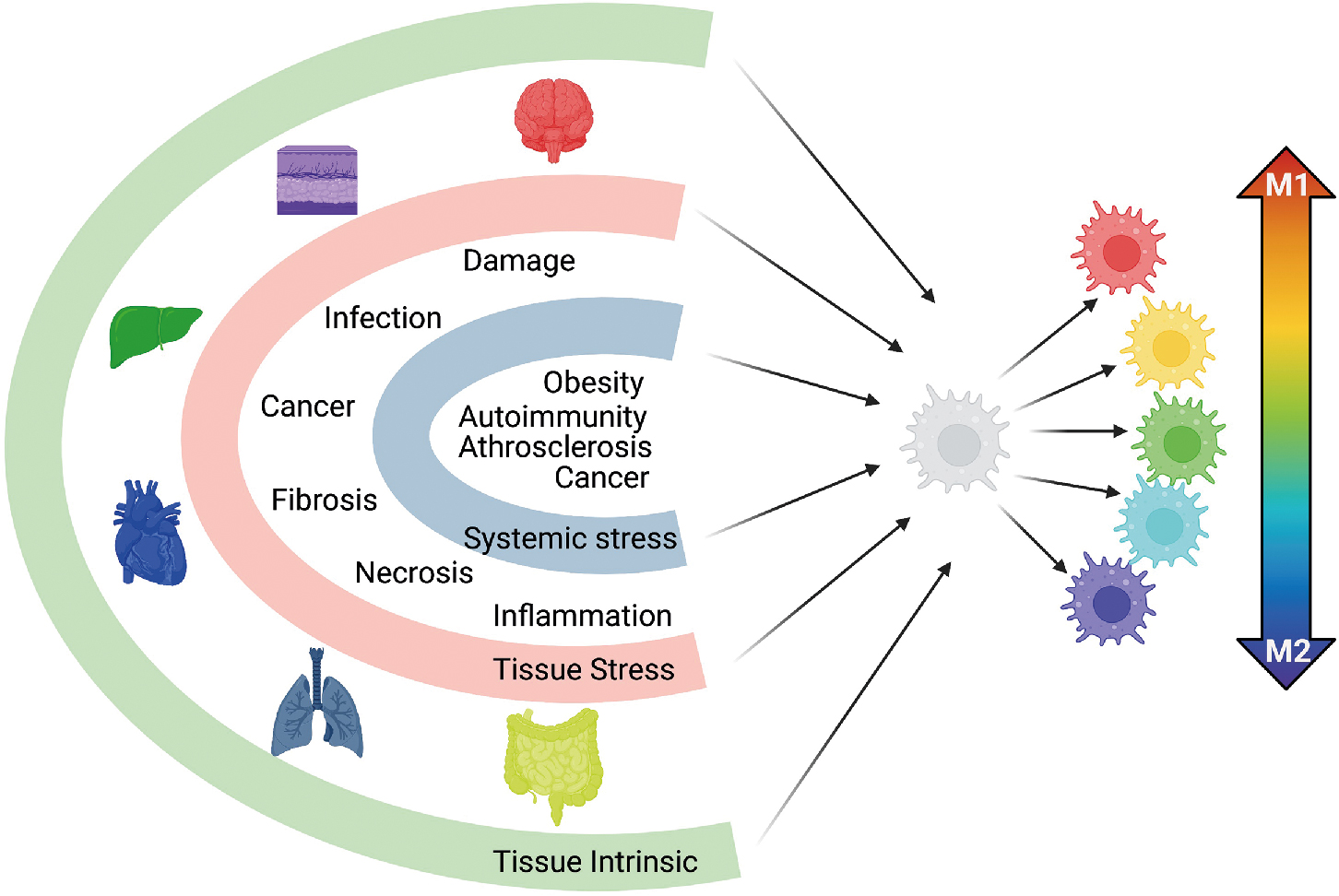
The conceptual framework of macrophage activation has long maintained that macrophages can be polarized into two opposing states: M1, or classically activated inflammatory macrophages, and M2, or alternatively activated immune regulatory macrophages. Studies evaluating macrophage response to acute infection, allergies, obesity, and asthma supported the paradigm that macrophages existed in one of two polarized states. However, this model did not account for the specialized functions of tissue macrophages, macrophages during sterile inflammation and tissue repair, and resolution of inflammation, nor did it encompass macrophage functional changes during chronic and complex disease states such as autoimmunity, chronic infection, and cancer. This paradigm has evolved coincident with the work related to macrophage ontogeny, with elegant studies demonstrating that macrophages have a broad functional repertoire with multiple, distinct, activation programs. Using ex vivo–derived human macrophages, Xue et al. differentiated macrophages in 29 distinct conditions ranging from stimuli associated with M1 and M2 activation axis to stimuli associated with free fatty acids, high-density lipoprotein, or combinations associated with chronic inflammation. Using transcriptomics and network analysis, it was found that in conditions diverging from the classical M1 and M2 paradigm, macrophages had a broad spectrum of activation signatures. These studies support the framework that macrophage activation is multidimensional, where macrophages receive input from tissue environments and external stimuli such as sterile insult, pathogens, or cancer, culminating in a broad range of activation phenotypes. The multispectral model is highlighted in Fig. 4.1 .
Dendritic cells
Dendritic cells were first discovered by Ralph Steinman and Zanvil Cohn in 1973, with Ralph Steinman receiving the Nobel Prize in Physiology or Medicine for this discovery in 2011. Dendritic cells were first identified as a unique cell subset, distinct from macrophages, based upon their extensively branched dendrites, multiple mitochondria, motility, lack of robust phagocytic activity compared with macrophages, and a robust ability to activate T cells. , After years of skepticism, it is now well accepted that dendritic cells are instrumental for inducing adaptive immune responses to self- and foreign antigens, highlighting their critical role in the initiation of tolerance and protective immunity. Macrophages are professional phagocytes, and dendritic cells are considered professional antigen-presenting cells, a sentinel of immune activation and host memory responses. Given these attributes, much work has gone toward understanding dendritic cells to therapeutically enhance adaptive immune responses against pathogens and tumors and to suppress responses during autoimmunity.
Most subsets of dendritic cells, with the exception of Langerhans cells, are constantly replenished from hematopoietic precursors in the bone marrow. Dendritic cells are subdivided into three subsets based upon lineage precursors, expression of core transcription factors, and functional attributes (summarized in Fig. 4.2 ). Subsets of dendritic cells are as follows: classic dendritic cells (cDCs), plasmacytoid DC (pDCs), and monocytic DCs (moDC).
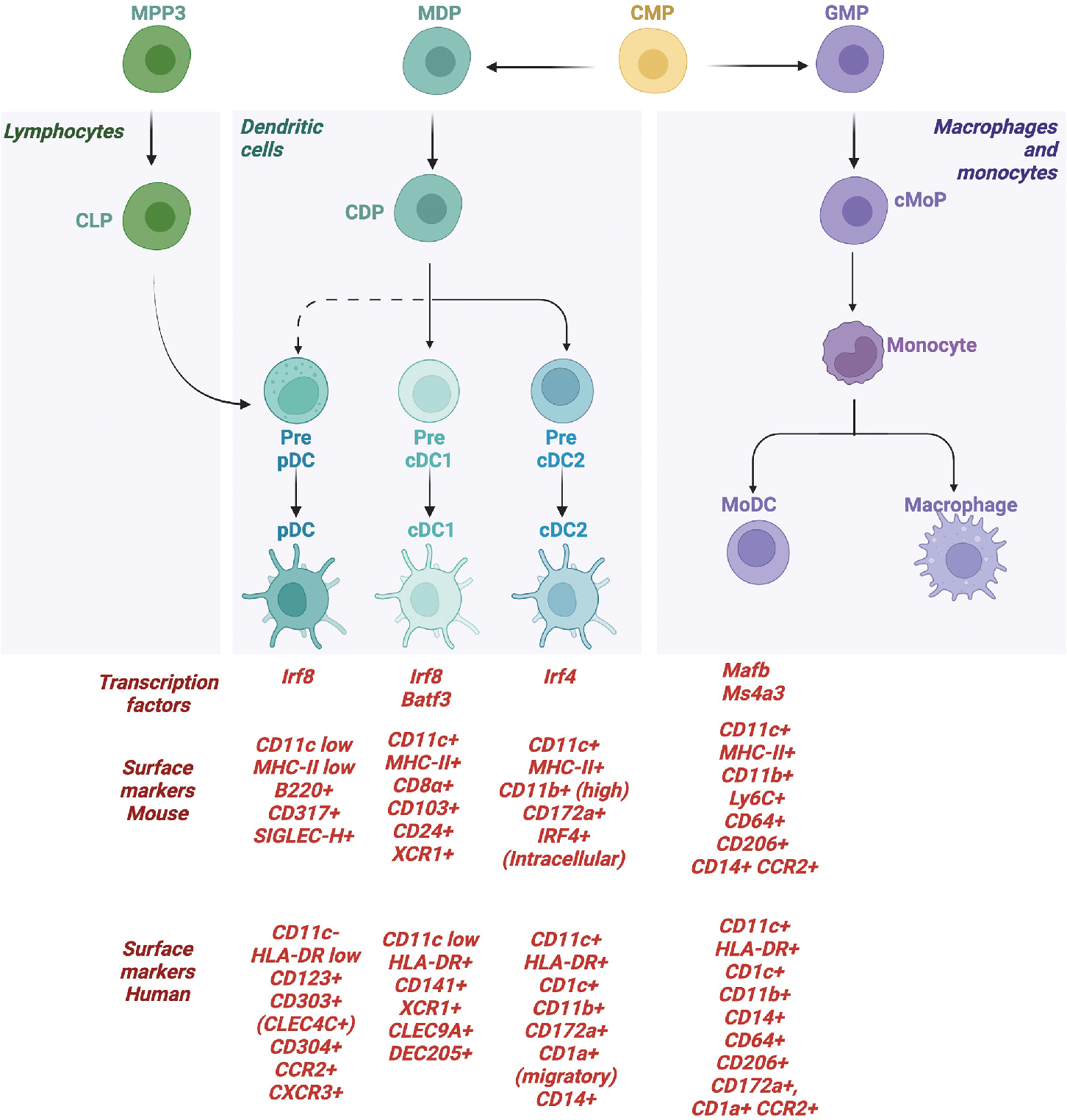
cDCs are the most well-characterized subsets, given their ability to activate CD4 and CD8 T cells. cDCs derive from the common dendritic cell progenitor (CDP), with precursors entering into the lymph nodes to disperse via high endothelial venules where they form an integrated network of DCs. cDCs are subdivided further based upon distinct developmental pathways, expression of core transcription factors, and functional attributes. cDCs are considered a heterogenous population, but the two most well-characterized subsets are cDC1 and cDC2. cDC1 express XCR1 and CLEC9A and can cross-present antigen to CD8 T cells, having an outsized role during antitumor immunity in both mice and humans, as detailed later. cDC2 are more heterogenous and have been identified to represent multiple distinct and heterogenous transcriptional programs (for in-depth review, see Chen et al. ). cDC2 typically activate CD4 T cells and, depending on the subset, can be proinflammatory or immune regulatory. A third cDC subset has been identified in humans, termed cDC3, which primes a subset of tissue-homing CD8+ T cells expressing CD103. ,
Plasmacytoid dendritic cells (pDCs) produce large amounts of type I interferon in response to viral infection or inflammation. The origins of pDCs are not as well-defined compared with their classical counterparts. Original studies demonstrated that pDCs can be derived from CDP. However, studies tracking progenitor subsets from the lymphoid lineage using the expression of yellow fluorescent protein (YFP) discovered that a majority of pDCs in the thymus and spleen were marked by YFP expression, indicating that the YFP-expressing pDCs derived from lymphoid progeintors. Dress et al. explored this paradigm further by combining single-cell mRNA sequencing with in vivo fate mapping and in vitro single cell–based clonal assays to demonstrate that pDCs arose from a subset of common lymphoid-like progenitors, indicating that pDCs are a distinct lymphoid-derived dendritic cell subset. Monocyte-derived DCs also do not arise from CDP but instead form monocytic precursors in inflamed settings.
Other innate immune cell subsets
Natural killer cells
Natural killer (NK) cells are central for host defense against virally infected or transformed cells. NK cells are widely distributed throughout the body, present within secondary lymphoid tissues such as lymph nodes, spleen, and mucosal associated lymphoid tissue in addition to tissues such as the lung, uterus, and liver. In the blood and lymph nodes, NK cells represent approximately 5% to 15% of total lymphocytes, whereas in the liver, lung, and uterus they are represented in higher frequencies. NK cells derive from common lymphocyte progenitors in the bone marrow. However, unlike B- and T-cell lymphocytes, NK cells do not require sensitization to kill target cells because of the lack of expression of antigen-specific receptors. Instead, NK cells express a series of germline-encoded activating and inhibitory receptors that enable distinction between self and nonself, a process referred to as education or licensing . Although NK cells are generally considered an innate lymphocyte subset, NK cells are also distinct from innate myeloid cells because they do not have the ability to phagocytose cells. Instead, NK cells possess effector capabilities resembling that of T cells, including the secretion of perforin, granzymes, and interferon gamma (IFN-γ). NK cells also express FasL and TRAIL on the cell surface, both of which bind to death receptors on recipient cells, leading to the induction of apoptosis. NK cells also produce a multitude of effector cytokines and chemokines, including tumor necrosis factor alpha (TNF-α), interleukin (IL)-10, GM-CSF, G-CSF, IL-3, CCL2, CCL3, CCL4, CCL5, XCL1, and CXCL8.
NK cells are derived from common lymphoid precursors in the bone marrow prior to populating peripheral lymphoid and nonlymphoid tissues. Commitment to the NK lineage coincides with expression of CD122, a receptor for IL-15, followed by the induction of transcription factors such as STAT5, EOMES, Nfil3, Ets, T-bet, Id2, Tox, and Tcf1. As NK cells mature, they upregulate expression of receptors such as NKp46 and CD49b, a point at which they are considered to be mature because of their potential to exert cytotoxic functions. NK cells continue to progress through a four-stage process of maturation, consisting of transcriptional and functional changes and defined in mice by upregulation of CD11b and gradual loss of CD27, with terminally differentiated CD11b + CD27 − effectors having potent cytolytic capabilities and reduced potential for expansion. In humans, NK-cell maturation is thought to associate with diminishing levels of surface CD56 expression. Using single-cell RNA sequencing, CD56 bright populations were found to have gene expression similar to that of the less differentiated CD11b − CD27 + subset, whereas CD56 dim populations resembled CD11b + CD27 − populations. However, it remains unknown whether CD56 expression truly identifies human NK subsets from the same lineage, progressing through distinct maturation states, or whether its expression defines subpopulations with unique developmental trajectories.
Central to NK cell function is the expression of a broad array of membrane proteins that calibrate NK cell interactions between other cells, resulting in inhibition or induction of cell cytotoxicity. NK receptors are broadly categorized into activating, inhibitory, and costimulatory receptors. Licensed NK cells express inhibitory receptors that contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in their cytoplasmic domains. Upon recognition of self-MHCI, ITIM domains are phosphorylated by SH2 domain–containing protein tyrosine phosphatase 1 (SHP1), effectively inhibiting intracellular signaling pathways controlling NK cell activation. Many NK-activating receptors contain immunoreceptor tyrosine-based activation motif (ITAM)-signaling molecules that, upon receptor crosslinking, lead to activation of the ITAM motif and downstream signaling events associated with NK cytotoxicity and effector function (for in-depth review, see Yokoyama and Plougastel ).
Mast cells
Mast cells are evolutionarily conserved innate immune cells found in connective tissues throughout the body, such as perivascular sites, brain, smooth muscle, peritoneal cavity, gastrointestinal tract, and respiratory tract. Mast cells are known for initiating inflammatory responses during allergic diseases. Although mast cells are often neglected in cancer research because of their low abundance in tissue and tumor microenvironments, it has become clear that in cancer and noncancer settings, mast cells are pivotal regulators of immune function.
Non-tissue-resident mast cells develop from hematopoietic progenitor populations in the bone marrow where precursor populations are recruited to distal sites via chemokine gradients into the tissue and undergo division, , followed by maturation and activation by tissue microenvironmental factors. On the other hand, tissue-resident mast cells are derived from yolk sac progenitors. Tissue mast cells are long-lived and can be replenished from precursors residing in the tissue. , However, very little is known how progenitor tissue-resident mast cells are matured. For bone marrow–derived mast cells, progenitors differentiate from common myeloid progenitors (CMPs), a process that is tightly controlled by CCAAT/enhancer binding protein α (C/EBPα), GATA-binding factor 2 (GATA-2), GATA-binding factor 3 (GATA-3), Hes-1, and melanocyte-inducing transcription factor (MITF). The downregulation of C/EBPα in GMP along with upregulation of GATA-2, GATA-3, and Hes-1 allows GMP to differentiate into bipotent progenitors, which have the potential to become mast cells and basophils. The subsequent upregulation of MITF and further downregulation of C/EBPα lead to the commitment of progenitors into mast cells.
A tremendous amount of work has been done to identify subtypes of mast cells in animals and humans, where the classification of mast cell subsets is based on the anatomic location and/or the protein contents in the secretory granules. In mice, the most well-characterized subtypes of mast cells are connective tissue mast cells (CTMCs) and mucosal mast cells (MMCs). CTMCs are found in skin, the peritoneal cavity, and the gastrointestinal tract, whereas MMCs usually populate at mucosal sites. CTMCs have granules that contain heparin, histamine, tryptase, and chymase, whereas the granule contents of MMCs mainly comprise chymase and have low levels of histamine and undetectable levels of heparin. In humans, mast cells are classified as MC T (mast cells containing mainly tryptase) and MC TC (mast cells containing both tryptase and chymase). MC T reside in the external mucosa of the gastrointestinal and respiratory tracts, whereas MC TC are usually found in the submucosa and perivascular tissues. , Distinct tissue tropisms give rise to distinct functional attributes, with MC T critically regulating the immune responses and MC TC being involved in tissue repair. , Although human and murine mast cells share multiple protein signatures, mast cells from each species also produce a distinct repertoire of proteases and respond differently in the presence of the mast cell stabilizer cromolyn. Host-intrinsic and/or tissue microenvironment–driven variations can also contribute to phenotypical differences in mast cells. ,
Neutrophils
Neutrophils, or polymorphonuclear leukocytes, are the most abundant cell type in the peripheral blood, approximating 50% to 70% of cells in human blood and 10% to 25% of cells in murine blood. Neutrophils are phagocytic and granulocytic cells considered one of the first lines of defense against pathogens. On the other hand, neutrophils also have a central role during immune homeostasis and tissue repair. The neutrophil life cycle is still an intense area of debate, owing to a lack of reliable techniques to accurately trace the path of neutrophil differentiation, circulation, tissue seeding, and eventual death in vivo. Compared with other innate immune cell populations, it is generally accepted that neutrophils are short-lived, surviving only hours to days once in the periphery. Given their critical role for innate immune defense, the numbers of neutrophils circulating in the periphery, and their short half-life, there is a significant requirement for de novo production of progenitors from the bone marrow daily. It is estimated that approximately 60% of all leukocytes produced daily in the bone marrow are granulocytic precursors, with 10 neutrophils produced and released from the bone marrow in humans and 10 neutrophils produced in mice daily. During inflammation, as many as 10 billion neutrophils can be produced in humans.
Neutrophils are derived from the multipotent progenitors in the bone marrow, undergoing commitment prior to entrance into the blood. Prior to the work by Evrard et al., it was unknown what factors contributed to the phenotypic and functional heterogeneity of peripheral neutrophils. Evrard et al. demonstrated that neutrophils arise from CMPs, transitioning into GMPs and eventually into a previously unrecognized precursor population they termed preneutrophils . In homeostatic conditions, commitment of GMP into preneutrophils is driven by expression of the transcription factor C/EBPε and expression of cKit and CXCR4. Preneutrophils are highly proliferative and reside in the bone marrow and spleen. During inflammatory stress, such as sepsis or cancer, preneutrophils rapidly expand to meet the increased demand, giving rise to two distinct neutrophil subsets: nonproliferative immature Ly6G lo/+ CXCR2 − CD101 − neutrophils and Ly6G + CXCR2 + CD101 + mature neutrophils within the blood. Other studies have identified that multipotent progenitors could act as a source of neutrophils in response to hematopoietic demands, whereas Cugurra et al. identified that the skull and vertebral bone marrow can supply the meninges with neutrophils under pathologic conditions such as spinal cord injury and neuroinflammation. Altogether, these studies have begun to establish the underlying mechanisms of neutrophil heterogeneity, underscoring that neutrophil phenotype and function are dependent on systemic inflammatory factors in addition to the tissue of origin.
The production, trafficking, and egress of mature neutrophils from the blood was demonstrated to occur in a diurnal pattern, where the exit of CD62L low CXCR4 high “aged” neutrophils from the blood back into the bone marrow and other tissues peaked during resting phases and was followed by emergence of “fresh” neutrophils during awake hours. Casanova-Acebes et al. went on to demonstrate that the circadian clock gene Artnl1 (BMAL1) was responsible for the diurnal regulation of neutrophil migration. BMAL1 was found to be a cell-intrinsic regulator of CXCL2 production and subsequent signaling through the ligand CXCR2, initiating transcriptional and migratory changes in neutrophils that trigger the aging cascade. On the other hand, the chemokine receptor CXCR4 antagonized aging via CXCR2 signaling. Although immature neutrophils express high levels of CXCR4 while being retained in the bone marrow, CXCR4 is paradoxically expressed at high levels in aged neutrophils, a process that is hypothesized to signify a return into the bone marrow and other sterile tissues to bolster to tissue immunity.
Functionally, neutrophils exhibit a broad range of capabilities that are dictated by the tissue environments into which they migrate, by the signals in the tissue or systemic environment they are exposed to, and by a culmination of physiologic factors that differently prime neutrophils in their functional response. For example, neutrophils in an obese setting can metabolize lipids and affect energy expenditure, whereas neutrophils in the spleen provide help to B cells. During inflammation, neutrophil longevity is increased upon activation, which can occur via recognition of tissue insults through expression of multiple innate signaling receptors. Neutrophils are highly phagocytotic cells; they can release potent amounts of reactive oxygen species and proteases from their granule contents and can release neutrophil extracellular traps, or NETs. NETs are an extracellular release of neutrophil DNA into a net-like configuration, effectively trapping microorganisms. NETs contain a high concentration of antimicrobial molecules and other molecules that are toxic to the captured microbes. As discussed below, the heterogeneity of neutrophil effector function and sensitivity to systemic signals result in significant heterogeneity in neutrophil phenotypes and functions in cancer-bearing individuals.
Innate immune networks in the promotion of tumor growth, immune suppression, and metastasis
In the previous section, the origins, differentiation pathways, and diverse functions of innate myeloid cells were covered. During cancer, myeloid cells have the potential to provide robust and protective antitumor immune control, acting as sentinels against tumor growth. Here, we will focus on the mechanisms whereby inflammation and innate immune signaling increase cancer risk or promotion of tumor growth. Enhanced tumor progression is coordinated by innate immune cells, leading to immune evasion, increased angiogenesis, and metastasis. Tumors and the tumor microenvironment act on terminally differentiated myeloid cells or on immature precursors recently recruited into the tumor microenvironment, both of which transform myeloid cells into potent immune suppressive and protumorigenic innate immune subsets. Here, we will define key innate immune pathways that initiate cancer formation or that contribute to tumor progression and metastasis.
Inflammation is a key modulator of tumor initiation
In the context of typical immune function, inflammation often is associated with host response against pathogens or sterile insults. Tumor promoting on the other hand, indicates nonspecific and unabated inflammation. In this context, inflammation itself can result in cancer development and progression. Inflammation is a hallmark of cancer, , with tumor-promoting inflammation affecting all aspects of tumorigenesis: driving oncogenic transformation, enhancing proliferation and survival of premalignant cells, facilitating tumor escape from the immune system, driving therapy failure and/or resistance, enhancing angiogenesis, and promoting metastasis. How inflammation is involved in cancer initiation and progression is summarized in Fig. 4.3 .
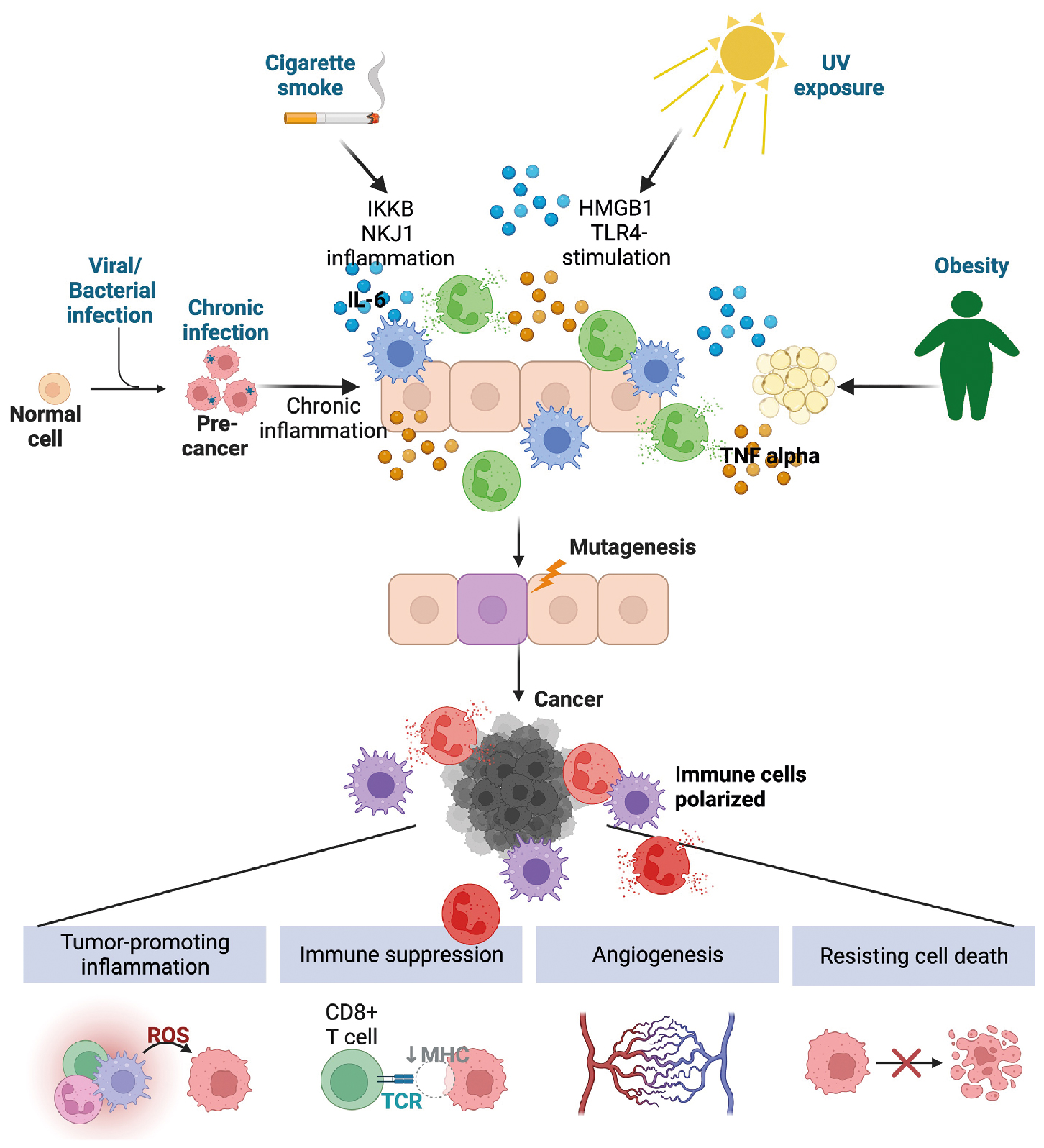
The association between inflammation and cancer was first obtained by experimental evidence from preclinical animal models in addition to large-scale epidemiologic studies that identified a strong association between the use of nonsteroidal antiinflammatory drugs (NSAIDs) and reduced cancer incidence and/or cancer-associated mortality for multiple types of cancer. , However, caution should be observed when considering prescribing NSAIDs for prevention of or treatment of certain cancers, because the dose and cancer type could have negative effects. For example, a randomized clinical trial called ASPREE found that in individuals over 70 years of age, compared with the use of a placebo, low-dose aspirin was associated with an increased risk of advanced metastatic cancer. On the other hand, there is evidence that chronic inflammatory diseases, such as inflammatory bowel disease and systemic lupus erythematosus, increase the risk of colorectal and gastric cancer, respectively.
A majority of cancers are caused by environmental factors, many of which are associated with inflammation. Cancer-causing inflammation can occur as a result of chronic exposure to sunlight, , smoking, and obesity, the known inflammatory mechanisms of which are summarized in Table 4.2 . Chronic infection can also lead to cancer. In the estimation of global burden of cancers attributable to infectious disease from the year 2018, 13% of all identified cancers arose because of infectious disease, with increased incidence in underdeveloped and developing countries. Strikingly, 90% of the infection-associated cases worldwide were a result of infection with four pathogens: Helicobacter pylori , human papillomavirus (HPV), hepatitis B virus (HBV), and hepatitis C virus (HCV), all of which are either vaccine preventable (HPV, HBV) or treatable ( H. pylori , HCV). Infection with Schistosoma is linked with bladder cancer and certain Bacteroides species are associated with colorectal cancer. Unlike acute infection, where a robust inflammatory response leads to clearance of each pathogen, in these scenarios, the ability of the microorganism to evade the immune system results in a low level and chronic inflammation.
| Stimulus | Type of Cancer | Inflammatory Mechanism | References |
|---|---|---|---|
| Smoking | Lung cancer | Pulmonary inflammation marked by IKKB and JNK1-induced inflammation. Increased IL-6 and TNF-α, accumulation of macrophages and neutrophils | |
| Intermittent intense ultraviolet exposure | Metastatic melanoma | Ultraviolet-damaged keratinocytes release HMGB1, TLR4-dependent recruitment of neutrophils. Stimulation of angiogenesis | , |
| Obesity | Pancreatic | Increased mitochondrial fatty acid B-oxidation, increased TNF-α signaling, adipose-derived IL-6 and TNF | |
| Colorectal cancer | Microbiome dysbiosis, reduced DC-mediated tissue homeostasis. Obesity and chemical carcinogens increase IL-6 and repolarize tissue macrophages | ||
| Liver cancer | High-fat diet combined with fermented foods increased dysbiosis, cholestasis, hepatocyte death, and neutrophil influx |
Depending on the tissue context, chronic inflammation can lead to cellular transformation because of recruitment of myeloid cells producing reactive oxygen species (ROS) and reactive nitrogen intermediates (RNIs) or the elevated production of proliferative and growth factors. For example, cytokines can activate STAT3 signaling, which in some instances can cause differientated cells to loose their identity acquiring a non-differientated stem cell phenotype capable of self-renewal. Chronic signaling of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) can suppress cell death, activate genes related to cell proliferation, and enhance the expression of growth factors and protumorigenic cytokines in the tissue environment. Tumor-promoting cytokines and growth factors, such as TNF-α, IL-1α, IL-1β, IL-6, IL-11, IL-23, and IL-17, initiate proinflammatory signaling networks resulting in enhanced cell proliferation and cell survival. One of the most mutated pathways in cancer is loss of or mutation of Tp53 , which encodes for the p53 protein, a critical regulator of cell homeostasis. Another critical role for p53 is control of NF-κB-mediated inflammation. In multiple contexts, loss of p53 leads to dysregulation of NF-κB expression, increasing the production of inflammatory cytokines and chemokines from mutant cells. , This signaling pathway further contributes to tumor growth via the establishment of a supportive immunologic niche that enables tumor growth and eventual metastasis.
Inflammation-mediated reprogramming of myeloid cells during cancer
Multiple cytokine and chemokine networks have also been implicated as essential for the orchestration of tumor progression through the establishment of an immune suppressive environment that promotes angiogenesis and facilitates metastatic spread. The mechanisms outlined here are by no means comprehensive but are intended to provide general mechanistic insight into relevant inflammatory pathways and their effects on individual immune cells and the effects of these signaling pathways on tumor progression. Myeloid cells are exquisitely responsive to the multitude of inflammatory signals within the tumor microenvironment, and as tumors progress, both lineage-committed , and immature myeloid precursors , are polarized into protumorigenic and suppressive phenotypes.
Monocytes
One of the major bottlenecks in defining how monocytes affect cancer is the lack of markers that differentiate monocyte-derived macrophages and dendritic cells from monocytes. This is because monocytes upregulate macrophage and dendritic cell markers such as F4/80, MHCII, and CD11c once they enter the tissue. These attributes, along with progenitor pathways that are distinct from those of other myeloid subsets during conditions of inflammation, make tracking the trajectory that monocytes take from the bone marrow into the tumor environment more challenging. Despite these challenges, lineage tracing, specialized transgenic mice, and single-cell analysis technology have provided insight into the pro- and antitumor activity of monocytes during cancer.
Tumor- and stroma-associated expression of the chemokine CCL2 is one of the main signaling pathways involved in the recruitment of inflammatory monocytes into the tumor microenvironment. Once monocytes enter the tumor microenvironment, they are coopted to enhance tumor growth, enable immune evasion, or promote metastatic dissemination. Qian et al. demonstrated CCL2 signaling recruited Ly6C hi CCR2-expressing monocytes into metastatic sites enhancing extravasation, tumor seeding, and growth in the lungs. Once in the tumor microenvironment, inflammatory CCR2-expressing monocytes contribute to the pool of tumor-associated macrophages. Other relevant signaling pathways associated with polarizing monocytes to acquire tumor-associated macrophage functions include CSF1 signaling. In a tumor setting, CSF1 enhances recruitment and polarization of monocytes into macrophages. However, other pathways have been identified, indicating that there are multiple tissue and microenvironmental contexts in which monocytes can become polarized to become part of the tumor-associated macrophage pool. For example, expression of the receptor for IL-4 and IL-13 (IL-4R) results in polarization and enhanced tumor-promoting functions of monocytes recruited in the bone marrow, culminating in enhanced recruitment to and outgrowth of breast tumor cells in the bone marrow. Another study identified that IL-6 and GM-CSF produced by cancer-associated fibroblasts induce polarization of monocytes into tumor-associated macrophages. Inflammatory monocytes can also directly promote metastasis. In certain tumor models, inflammatory monocytes recruited into the tumor microenvironment via CCL2 express factor XIIIA, a transglutaminase that covalently crosslinks fibrin, creating a scaffold for tumor cells to invade and metastasize. Monocytes also modulate the tumor microenvironment via production of IL-1β, IL-6, TNF-α, and CCL3, resulting in immune suppression and enhanced angiogenesis (see review by Olingy et al. ).
Nonclassical or patrolling monocytes are longer-lived than classic or inflammatory monocytes, constantly surveilling tissues and transporting antigen to draining lymph nodes. These attributes make patrolling monocytes well-suited for antitumor immune functions. Nonclassic monocytes patrol the lung environment for tumor cell debris and can activate NK cells to prevent metastatic seeding of the tissue. Mechanistically, it was subsequently shown that patrolling monocytes are expanded by tumor-produced exosomes and activated to produce IL-15, leading to subsequent activation of IFN-γ-producing NK cells that prevent metastatic colonization of the lungs. Despite the tumor protective functions of patrolling monocytes, they are still susceptible to the signals within the tumor microenvironment and can be modulated or polarized to support tumor growth. For example, tumors treated with anti-VEGFR2 therapy acquire resistance. One of the mechanisms of resistance involves upregulation of CX3CR1 and recruitment of patrolling monocytes into the tumor microenvironment. Once recruited, the monocytes enhance angiogenesis and immune suppression through the production of chemokines that recruit suppressive neutrophils into the tumor microenvironment.
Dendritic cells
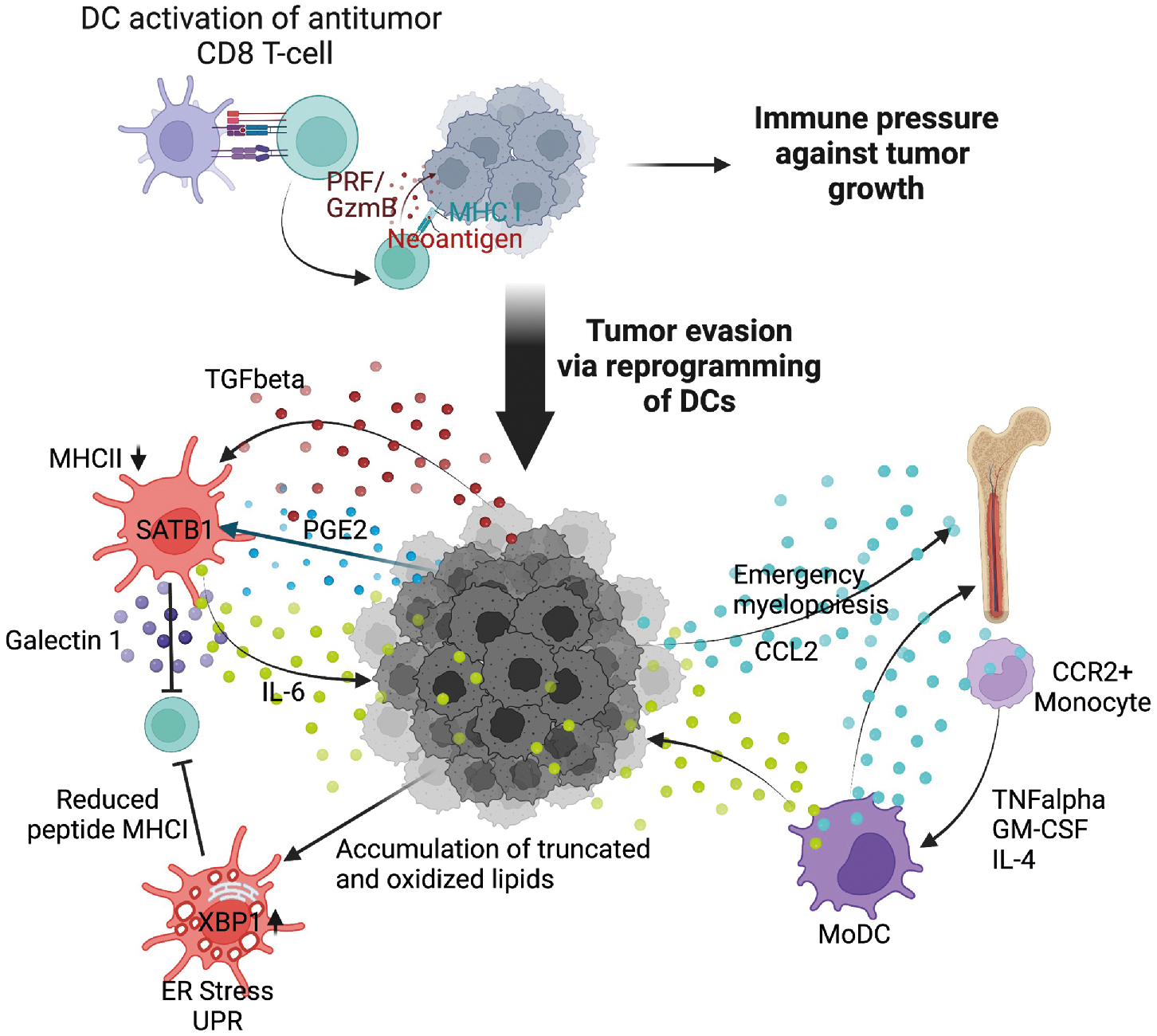
Dendritic cells are critical for the initiation of adaptive tumor immune responses. However, as tumors progress, conventional dendritic cells are coopted by tumors acquiring tolerogenic functions. These dendritic cells are termed tumor-induced regulatory dendritic cells . Although distinguishing dendritic cells from macrophages and other myeloid cells is more challenging under conditions of emergency myelopoiesis, bona fide dendritic cells can be identified by expression of the transcription factor Zbtb46 , and the presence of DNGR1. Using an approach to selectively deplete dendritic cells in an autochthonous model of ovarian cancer, Scarlett et al. demonstrated that during early stages of tumor progression, dendritic cells were instrumental for orchestrating antitumor CD8 T-cell immune responses. However, as tumors continued to evolve and the tumor microenvironment became more suppressive, tumor-produced transforming growth factor beta (TGF-β) and prostaglandin E2 (PGE2) polarized dendritic cells to suppress T-cell responses. Mechanistically, dendritic cells became suppressive in response to sustained and elevated expression of the chromatin regulator Satb1. Although required for differentiation of conventional dendritic cells, unremitting Satb1 expression induces a phenotypic switch in conventional dendritic cells, leading to enhanced secretion of immune suppressive galectin 1 and tumor-promoting IL-6.
Conventional dendritic cells can be polarized via additional suppressive pathways, including interactions with MDSCs, regulatory T cells, and metabolic stress. For an extensive review of mechanisms associated with dendritic cell polarization and suppression, see Veglia and Gabilovich, Wculek et al., and Conejo-Garcia et al. Herber et al. observed that dendritic cells in patients and in murine models of cancer were abnormally laden with oxidized lipids, a state termed dyslipidemia , resulting in endoplasmic reticulum (ER) stress, which is discussed later, and a reduced ability of dendritic cells to activate T cells. Mechanistically, lipid accumulation was driven by expression of scavenger receptor A whereby oxidatively truncated lipids were bound to chaperone HSP70, effectively preventing transport of exogenous peptide-MHC class I complexes to the cell surface, limiting the ability of dendritic cells to stimulate CD8 T cells.
Plasmacytoid dendritic cells represent a rare population within the tumor microenvironment. As such, evaluating the effects of pDCs on cancer progression has been challenging, and it is still unclear whether pDCs conclusively have pro- or antitumor functions. However, given the plasticity in the dendritic cell lineage, it is likely that pDCs have varied function depending on the signals that are present in the tumor microenvironment. Within the tumor microenvironment, pDCs were shown to enhance immune suppression by expanding regulatory T cells in an inducible T-cell costimulatory ligand (ICOSL)-dependent manner while also inducing IL-10 in regulatory T cells or initiating type 2 immune responses in CD4 T cells, both of which are immune suppressive and protumorigenic. However, evidence also exists that plasmacytoid dendritic cells induce cytotoxic T-cell responses via production of IFN-α. Additionally, when activated with ligands for TLR7, pDCs initiate substantial antitumor immune responses.
Abnormal differentiation of myeloid cells and infiltration of tumors with immature dendritic cells are considered primary mediators of myeloid dysfunction within the tumor microenvironment. Myeloid-derived dendritic cells are often referred to as plastic or nonclassical monocytes , rather than bona fide dendritic cells, and are thought to acquire dendritic cell–like attributes once they enter the tumor environment. Myeloid-derived dendritic cells are associated with a negative prognostic outcome. Monocyte-derived dendritic cells arise from the differentiation of CCR2-expressing monocytes in response to inflammation, such as what would be found within the tumor microenvironment. Myeloid-derived dendritic cells promote immune suppression within the ovarian tumor microenvironment and are associated with poor outcome in other models of cancer.
cDC1 dendritic cells, although a rare subpopulation within the tumor microenvironment, express CD103 and are dependent on IRF8 and Batf3 signaling. Importantly, cDC1 are one of the most capable cell subsets that stimulate CD8 T cells for tumor control. In support of this, the abundance of cDC1 in tumors corresponds with positive cancer outcomes. cDC1 can also produce IL-12 to potentiate adaptive antitumor immune responses. In situ expansion of cDC1 within tumors using systemic administration of FLT3L and intratumoral poly I:C resulted in activation and enhanced response to BRAF inhibition in combination with programmed death ligand 1 (PD-L1) blockade. The response was so robust that mice were subsequently protected from tumor challenge, demonstrating the potency of cDC1 in achieving protective antitumor immunity. NK cells recruit cDC1 into the tumor microenvironment via CCL5 and XCL1 expression, identifying a key NK–dendritic cell–chemokine regulatory axis that is currently being explored as a potential cancer therapeutic. Mechanisms of tumor-induced dendritic cell dysfunction are summarized in Fig. 4.4 .
Macrophages
In a tumor setting, macrophages are hijacked by tumors to promote malignant growth, invasion, metastasis, and outgrowth of tumors in distal organs. In general, the presence of macrophages associates with poor clinical outcomes for most cancer types. , Phenotypically, tumor-associated macrophages resemble M2-like macrophages, upregulating CD206 and acquiring a range of effector functions that enable the promotion of metastasis, angiogenesis, immune suppression, and tumor escape from therapy.
Aside from the surface expression of CD206, tumor-associated macrophages are functionally diverse and heterogenous in nature. Multiple signaling pathways have been identified that control macrophage polarization within the tumor microenvironment. One major mechanism involved in the polarization of macrophages is tumor hypoxia. Tumor hypoxia occurs because of aberrant growth of tumor cells and abnormal tumor vasculature, outpacing the limited oxygen and nutrient supply. Hypoxia forces tumors to adapt to new metabolic demands, initiating prosurvival, invasion, and inflammatory programs via upregulation of phosphoinositide 3-kinase (PI3K)/protein kinase b (Akt)/the mamalian target of rapamycin (mTOR), mitogen activated protein kinase (MAPK), and NF-κB signaling. Heterogeneity of monocyte-derived macrophages has been demonstrated using murine models and single-cell phenotypic analysis. For example, in a murine model of breast cancer, seven functionally and molecularly distinct macrophage subsets were identified based upon Ly6C and MHC II expression levels. The most suppressive macrophage populations resided adjacent to hypoxic tumor areas and had increased angiogenic and suppressive activities. Tumor-derived lactic acid was also demonstrated to have similar effects on the polarization of macrophages. Another contributor to macrophage heterogeneity is the nature of myeloid precursors that macrophages arise from. Macrophages that differentiate from M-MDSC are transcriptionally distinct and more immune suppressive compared with monocyte-derived tumor macrophages. M-MDSC-derived tumor macrophages are dependent on S100A9 activity within the tumor microenvironment, a signaling pathway that induces C/EBPβ, a transcription factor associated with potent immune suppressive activity of M-MDSCs. On the other hand, prostaglandin E2 ( PGE2), a product of COX-1 or 2 activity, enhances the suppressive function of committed M2 macrophages, , and inhibition of PGE2 using COX-2 inhibitors restores efficacy of PD-L1 blockade in certain tumor models.
Tumor-associated macrophages originating from tissue-resident macrophage pools complicate efforts to target tumor-associated macrophages. Tissue-resident macrophages self-renew locally, in the absence of input from the bone marrow. This establishes a scenario where tissue-resident macrophages can provide early and supportive signals to developing tumors to enhance growth, invasion, and avoidance of immune destruction. In non-small cell lung cancer, tissue-resident macrophages established a niche for developing tumors, facilitating epithelial to mesenchymal transition while also shielding developing tumors from adaptive immune pressure. For pancreatic cancer, CSF1-polarized monocyte-derived macrophages are more immune suppressive, and polarized tissue-resident macrophages have a profibrogenic phenotype.
Depending on the signals present within the tumor microenvironment, a multitude of additional transcriptional and regulatory pathways influence the polarization of tumor-associated macrophages. Signaling through the aryl hydrocarbon receptor (AhR) can polarize multiple immune cell subsets in the tumor microenvironment, including dendritic cells and macrophages. AhR resides in the cytoplasm, and when AhR ligands enter the cytoplasm via passive diffusion, the AhR receptor complex translocates into the nucleus, binding target DNA sequences located in the promoter regions of multiple genes relevant to inflammation and procancer activities. AhR ligands include kynurenine pathway metabolites and arachidonic acid metabolites, both of which have tolerogenic effects on immune cells within the tumor microenvironment. AhR signaling in macrophages and dendritic cells within the tumor microenvironment increases expression of IL-1β, IL-6, TNF-α, IL-10, and NF-κB. Hezaveh et al. added to this body of literature by demonstrating that indoles produced by certain Lactobacilli species from diet-derived tryptophan polarized tumor-associated macrophages overexpressing the AhR ligand in pancreatic cancer. The effects of the gut microbiome on cancer progression and therapy response are covered in Chapter 11 . These studies highlight the systemic effect of cancer and a cancer-affected gut microbiome on innate immune cells. Thus, potential therapies could involve dietary modifications such as consuming low-tryptophan diets. Indeed, for pancreatic cancer, provision of a diet low in tryptophan reversed macrophage-mediated immune suppression.
Transcription factors associated with macrophage polarization into suppressive proangiogenic phenotypes include, but are not limited to, jumonji domain-containing protein 3 (JMJD3) and IRF4 , ; STAT1, 3, and 6 signaling ; and C/EBPβ. Due to the underlying phenotypic plasticity of macrophages, protumor transcriptional changes can be triggered in response to multiple inflammatory cytokines and chemokines, PAMPs and DAMPs, metabolic changes, and changes in the vasculature and in the surrounding stroma. Other immune cells present within the tumor microenvironment can also polarize macrophages. For example, B-cell production of IL-10 leads to subsequent activation of TRIF/STAT1 signaling in macrophages, resulting in decreased proinflammatory functions; production of IL-10, IFN-β, CCL2; and upregulation of M2-like markers Ym1 and Fizz1. Other studies have found that autoantibodies polarized macrophages by binding Fc receptors on macrophages. Regulatory T cells and Th2 CD4 T cells are also able to polarize macrophages, through the production of IL-4, IL-13, and IL-10, as detailed in Gabrilovich et al. Several reviews cover signaling pathways associated with macrophage polarization extensively. ,
Mast cells
Mast cells are potent innate immune cells whose role in inflammation extends beyond allergy and hypersensitivity reactions. As described previously, mast cells possess multiple effector functions that enable modulation of the tumor microenvironment. The effect of mast cells on cancer is variable and dependent on the types of cancer. For ovarian cancer and esophageal cancer, mast cells have been shown to be associated with a positive prognosis, whereas for melanoma, breast cancer, lung cancer, and gastric cancer, associations are either mixed or correspond with poor outcomes. Using a humanized model of melanoma, Somasundaram et al. demonstrated that mast cells are associated with immune suppression and failure of PD-1 blockade.
Mast cells are potent immune effectors and are also highly sensitive to microenvironmental cues present in the tissues in which they are residing. As such, mast cells are functionally and phenotypically modulated by tumor-associated signals while having a reciprocal effect upon the inflammatory milieu of the tumor microenvironment. Mast cells can rearrange the tumor environment through release of metalloproteinases, serine proteases, and other factors that enhance angiogenesis and remodeling of the basement membrane. Cytokines such as IL-6, CCL2, and VEGF enhance angiogenesis and recruit cells into the tumor environment, whereas profibrogenic mediators such as PDGF-β activate fibroblasts. Mast cell–produced IL-6 and CCL2 can also enhance cell proliferation and polarization of other myeloid subsets. The chemokine IL-8 (CXCL1, a proposed homologue in mice) and histamine can chemoattract and induce proliferation of tumor cells. Mast cells can also produce IL-13 and IL-4. Typically associated with an allergic response, these two cytokines have been associated with a fibrotic response, immune suppression, and tumor growth. The alarmin IL-33, which is also produced by gastric tumor cells, has been shown to induce IL-13, CSF-2, CCL3 and IL-6 from mast cells.
Mast cells can accumulate in tumors and the tumor microenvironment through multiple chemotactic factors such as SCF, CCL2, VEGF, angiopoietin 1, IL-8, CXCL1, osteopontin, and CXCL10. Activation and degranulation are also affected by signals in the tumor environment. For example, PGE2 produced by macrophages, other myeloid cells, and tumor cells can activate mast cells to degranulate via signaling through the PGE2 receptor EP3. , On the other hand, stimulation of EP2 and EP4 by PGE2 reduces activation of mast cells. Mast cells are also activated and phenotypically modulated by recognition of PAMPs and DAMPs. TLR4-mediated recognition of LPS can induce GM-CSF, IL-8, and IL-10 release without degranulation, whereas recognition of peptidoglycan via TLR2 signaling can trigger degranulation.
Neutrophils
Neutrophils are often synonymous with antimicrobial defense; however, their recruitment into sterile tissues suggests that neutrophils also have a role in tissue defense or damage, a process with high relevance to cancer. In patients with cancer, cancer-associated neutrophils sediment in the low-density fraction of blood gradients, along with myeloid-derived suppressor cells, which are described later. Low-density neutrophils are derived from TGF-β signaling and have been shown to be highly immune suppressive, protumorigenic, and prometastatic. GM-CSF is also a potent chemokine produced by tumors that triggers emergency myelopoiesis and pathologic expansion of immature neutrophils from the bone marrow and other extramedullary sites. CXCR1 and CXCR2 signaling on neutrophils, the ligands of which are induced by TGF-β and produced by tumor cells, are also involved in neutrophil exit from the bone marrow and into tumor-affected tissue. The ligands for these two pathways are produced in abundance by transformed cells, initiating the release of neutrophils from the bone marrow and into the tumor microenvironment.
Signaling through CXCR2 can culminate in the release of NETs into the tumor microenvironment. Unlike their host-protective role during infectious disease, NETs in the tumor microenvironment protected tumors from T cell– and NK cell–mediated cytotoxicity. NETs also have tumor metastasis effects, with studies demonstrating that NETs enhance extravasation from the primary tumor site , and recruit tumors into the premetastatic niche to facilitate metastatic colonization. Thus, inhibitors limiting NET formation or neutrophil egress from the bone marrow have the potential to improve adaptive immune responses and reduce tumor growth and invasion. Several studies have identified multiple heterogenous neutrophil populations in cancer-bearing individuals, suggesting that host-intrinsic factors also affect neutrophil phenotype and abundance. Indeed, in a comprehensive outlook of neutrophils in cancer biology, Quail et al. outlined multiple factors, including but not limited to age, sex, circadian clocks, obesity, the microbiome, and smoking.
Inflammation and tumor vasculature
To sustain rapid growth and increased metabolic demand, tumors need to establish a blood supply. Cancer cells can initiate formation of tumor-supportive blood vessels from existing vessels, which is termed sprouting angiogenesis . Angiogenesis is facilitated via the coordination of multiple proangiogenic factors. Although the innate immune system is integral to this process, a consequence of angiogenesis is subsequently increased inflammation, highlighting the reciprocal relationship between angiogenesis and a developing tumor.
Proinflammatory cytokines such as IL-1 family cytokines, IL-6, IL-17, TGF-β, TNF-α, and VEGF are upregulated in the evolving tumor microenvironment, leading to activation of NF-κB and STAT3 signaling in endothelial cells and pericytes. STAT3 upregulation in endothelial cells upregulates expression of adhesion molecules on endothelial cells, such as ICAM-1 and VCAM-1, to facilitate accumulation of immune cells into the tumor microenvironment. STAT3 acts upstream of HIF1 and VEGF, two potent angiogenic factors, while also increasing the expression of additional angiogenic factors such as FGF and Ang-2. STAT3 signaling also enhances synthesis of matrix metalloproteinases, which remodel the basement membrane and extracellular matrix, all part of the angiogenic pathway to support exponential tumor growth. NF-κB activation also contributes to angiogenesis of multiple tumor types through upregulation of VEGF, enhancing expression of vascular adhesion molecules, subsequently amplifying the expression of multiple proangiogenic and inflammatory cytokines and chemokines. Despite the known dependence of tumors on angiogenic signaling pathways, antiangiogenic agents have not yielded significant improvements in progression-free survival and overall survival for pancreatic adenocarcinoma, prostate cancer, breast cancer, or melanoma, with more mixed results for non-small cell lung cancer and glioblastoma. Reasons for failures of antiangiogenic therapies are varied but could arise from tumors using nonangiogenic mechanisms to acquire vasculature, or vessel cooption. Vessel co-option is a process whereby tumor cells use existing tissue vasculature to meet their metabolic and growth needs.
The mechanisms of vessel cooption are not as well understood as angiogenesis. However, evidence has emerged to suggest that VEGF receptor blockade can initiate vessel cooption as a mechanism of tumor escape from antiangiogenic therapies. , Using a model of VEGF blockade–induced vessel cooption, single-cell RNA sequencing identified that coopted vessels were enriched in a population of macrophages with inflammatory potential—having an inflammatory M1-like phenotype, characterized by having high expression levels of IL-1β and IL-6, TNF-α, and IFN-γ —all of which are capable of normalizing the abnormal tumor vasculature that arises during angiogenesis. The question arises as to whether vessel cooption is an attempt to normalize the vasculature, once tumor cells co-opt it, or whether inflammatory macrophages are sufficient to drive the process. Several questions remain and more work is needed to develop models of tumor vessel cooption that would enable mechanistic understanding of the underlying process of vessel cooption and what role, if any, inflammation has during this process.
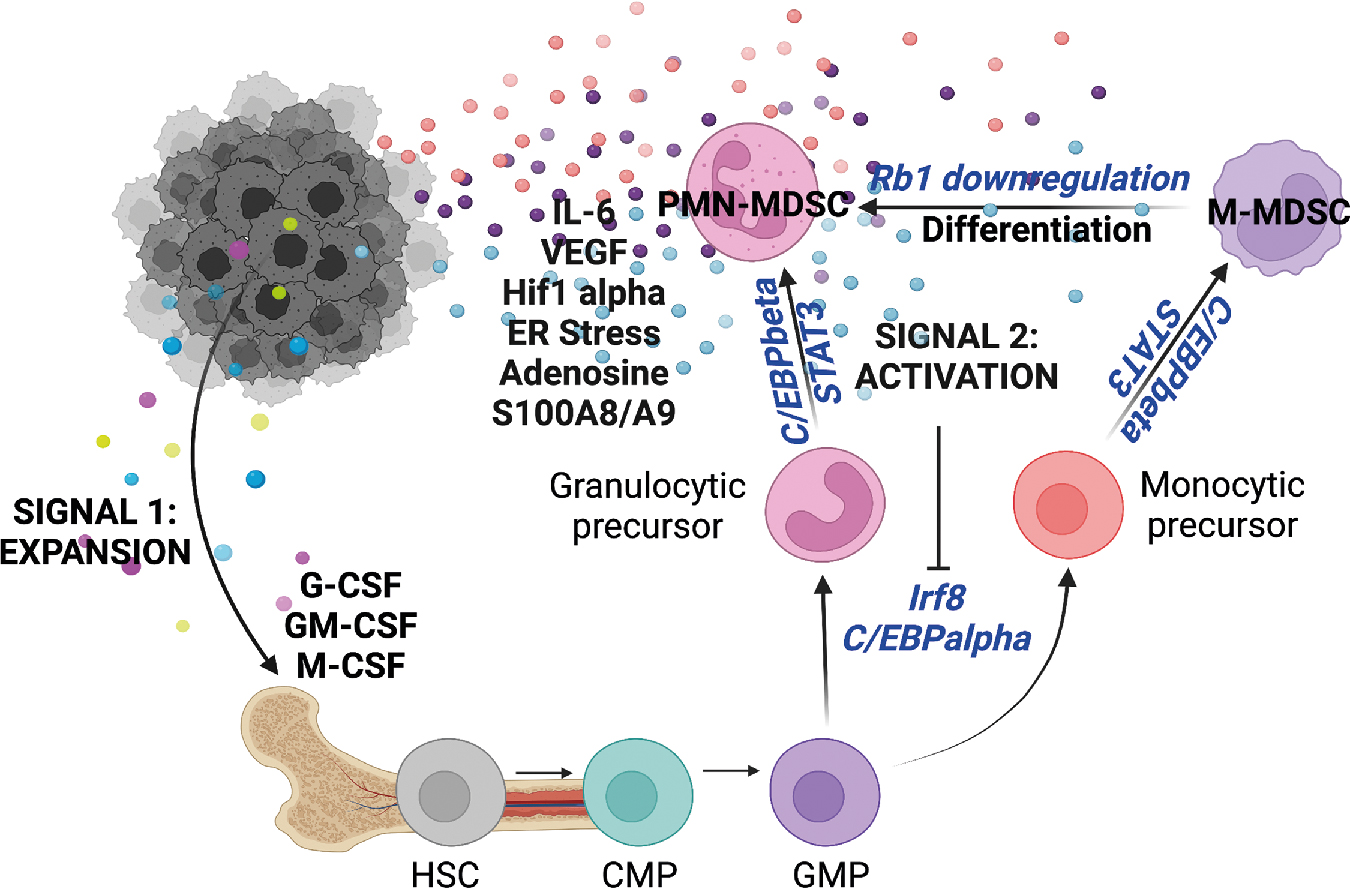
Inflammation-mediated emergency myelopoiesis: Myeloid-derived suppressor cells
The hematopoietic system is capable of rapidly deploying immune cells from the bone marrow in response to systemic inflammation, referred to as demand-adapted hematopoiesis . This can occur locally, in response to tissue-restricted infections or in response to systemic inflammatory signals. Emergency myelopoiesis is a state of pathologic immune activation, characterized by prolonged stimulation through release of cytokines, growth factors, and other tumor-associated mechanisms associated with chronic inflammation. Emergency myelopoiesis results in the systemic emergence of immature neutrophils and myelomonocytic cells from the bone marrow, where they become pathologically activated to become myeloid-derived suppressor cells (MDSCs, covered in the latter half of this chapter). MDSCs accumulate in distal sites of tumor spread and are programmed into suppressive cells in the presence of tumor-associated inflammatory factors. Myeloid cells entering the tumor microenvironment are conditioned by the tumor to facilitate angiogenesis, promote metastasis, and aide tumor evasion from the immune system, all of which are hallmarks of cancer. , Thus, coordination between tumors and the hematopoietic compartment underlies a critical process of cancer-mediated cooption of host innate immune cells to support growth and invasion. Emergency myelopoiesis is a major node of this process, culminating in extensive efforts to define relevant pathways involved in tumor-associated myelopoiesis.
It is now well established that MDSCs become pathologically activated in two phases ( Fig. 4.5 ) (for in-depth review, see Condamine et al. ). During phase 1, the bone marrow and spleen are conditioned by chronic low-level stimulation by cytokines and growth factors such GM-CSF, G-CSF, and M-CSF, resulting in expansion of granulocytic and monocytic precursor cells. During phase 2, pathologically expanded myeloid progenitors are exposed to inflammatory signals in the periphery, such as IL-6, IL-1β, VEGF, ER stress, adenosine signaling, and HIF1α. Myeloid cells derived in these conditions become unable to differentiate, have reduced phagocytic activity, produce antiinflammatory cytokines and signaling molecules, and produce ROS, NOS, MPO, and other suppressive factors, culminating in a significant inhibition of CD4 and CD8 T-cell effector activity in the tumor microenvironment.