Abstract
Only approximately 10% of breast cancers are caused by identifiable mutations in major breast cancer susceptibility genes. Nevertheless, for families affected by these mutations, genetic testing is a powerful tool for determining which relatives are at high risk and which are not. The list of breast cancer predisposition genes is growing longer with the advent of massive parallel sequencing (next-generation sequencing). BRCA1 and BRCA2 are still the most frequent cause of hereditary breast cancer predisposition with CHEK2, ATM, and PALB2 being the next most common. Most patients with deleterious mutations should consider enhanced surveillance with magnetic resonance imaging, and for some, breast cancer risk will be great enough to consider risk-reducing mastectomy. There are several recently described genes in the Fanconi anemia pathway that are related to BRCA1 and BRCA2 . Recognizing mutation carriers at the time of a new breast cancer diagnosis is important for making decisions about surgery, radiation, and systemic therapies.
Keywords
breast neoplasms, cancer genetics, genetic testing, familial breast cancer, multigene panels
Not long after the birth of modern medical science at the turn of the 19th century, two prominent physician scientists observed the hereditary nature of cancer and articulated concepts in cancer genetics that hold true to this day. In 1851 Hermann Lebert suggested “children come into this world carrying within them the seeds of a cancerous disease which remains latent for thirty to fifty years, but which, once developed, is fatal in the space of a few years.” He recognized the importance of identifying individuals with a cancer predisposition and suggested that these individuals might reduce their risk by relocating to regions with a low cancer incidence. In 1866 Lebert’s contemporary, the famed neuroanatomist Pierre Paul Broca, described four generations of a family afflicted with breast and gastrointestinal cancers. Through detailed pedigree analysis, these scientists concluded that major inherited predisposition syndromes account for only 5% to 10% of cases of breast cancer, that women are disproportionately affected with hereditary cancer compared with men, and that identification of high-risk families presents an opportunity to intervene to reduce risk.
The majority of breast cancers are sporadic rather than inherited. It is therefore environmental factors and chance, not germline mutations, that account for a greater number of breast cancers. Twin studies estimate that 12% to 30% of breast cancers have a heritable genetic component. However, only 5% to 10% of breast cancers are related to inheritance of major autosomal dominant predisposition genes. Of these, the causative mutation is identifiable in only 35%. Even with whole genome sequencing, we have nearly exhausted the list of identifiable germline mutations responsible for familial breast cancer. BRCA1 and BRCA2 still account for the majority of mutations identified in genetic high-risk families. More than a dozen other well-described gene mutations predispose to breast cancer, and the list of rare genes is increasing rapidly ( Fig. 17.1 ). Although genetic predisposition accounts for only a fraction of all breast cancers, identification of gene mutation carriers has great value for breast cancer prevention, diagnosis, and treatment.
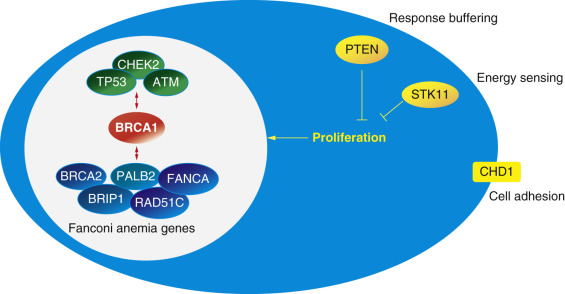
The Value of Genetic Testing
For individuals not yet diagnosed with cancer, genetic testing can be the most accurate tool available for cancer risk stratification. Lifetime breast cancer risk is 57% to 81% for women with pathogenic BRCA1 mutations and 45% to 85% for BRCA2 mutations. Ovarian cancer risk is 39% by age 70 for BRCA1 and 17% for BRCA2. Mutations in other genes may point to increased risk for gastric, thyroid, endometrial, or other cancers ( Table 17.1 ).
| Syndrome | Gene Locus | Neoplasms | Lifetime Breast Cancer Risk | Frequency Among High-Risk Families | Reference |
|---|---|---|---|---|---|
| Hereditary breast/ovarian | BRCA1 (17q12–21) | Female breast, ovarian | 57%–81% | 5% | |
| Hereditary breast/ovarian | BRCA2 (13q12–13) | Male and female breast, ovarian, prostate, pancreatic | 45%–85% | 4% | |
| CHEK2 -related | CHEK2 (22q12.1) | Breast, colorectal, bladder | 25%–37% | 1.3% | |
| PALB2 -related | PALB2 (16p12.1) | Breast, pancreatic, male breast | 20%–79% | 0.7% | |
| ATM -related | ATM (11q22.3) | Breast and ovarian | 15%–20% | 0.7% | |
| Li-Fraumeni | TP53 (17p13.1) | Breast, sarcomas, leukemia, brain tumors, adrenocortical carcinoma, lung | 56%–90% | 0.2% | |
| Moderate risk breast/ovarian cancer | BARD1 (2q34-q35) | Breast/ovarian | Unknown | 0.2% | |
| BRIP1 -related | BRIP1 (17q22-q24) | Breast, ovarian | unknown | 0.2% | |
| Hereditary diffuse gastric cancer | CDH1 (16q22.1) | gastric, lobular breast, colorectal | 60% | 0.1% | |
| Cowden | PTEN (10q23.3) | Breast, thyroid, endometrial | 67%–85% | <0.1% | |
| Other: benign hamartomas (of skin, mucosa, GI, GU, CNS, and bones) macrocephaly | |||||
| Peutz-Jeghers | STK11 (19p13.3) | Breast, ovarian, cervical, uterine, testicular, small bowel, and colon | 32%–54% | <0.1% | |
| Other: hamartomatous polyps of small bowel and mucocutaneous pigmentation | |||||
| RAD51C | RAD51C (17q25.1) | Breast, ovarian | Unknown | <0.1% |
Quantitative risk information can guide decisions about enhanced surveillance to diagnose cancers earlier, or prophylactic surgery or chemoprevention to reduce the risk of ever developing cancer. Pathogenic mutations in most of the genes commonly tested today will be associated with at least a 20% lifetime breast cancer risk. These women all meet the risk threshold for consideration of enhanced surveillance with breast magnetic resonance imaging (MRI).
Pathogenic mutations in breast cancer predisposition genes can influence decisions about surgery, radiation therapy, and systemic treatments in the newly diagnosed breast cancer patient. With respect to surgery, second primary breast cancer risk is often elevated in gene mutations carriers. This information is critical for deciding between breast conservation and unilateral or bilateral mastectomy. Most of the known breast cancer predisposition genes serve a role in DNA maintenance and repair. This knowledge can influence radiation therapy decisions. Although therapeutic whole breast radiation is not contraindicated in any other than homozygous ATM carriers, it should be used with caution in TP53 mutation carriers. Partial breast irradiation is classified as “unsuitable outside of a clinical trial” for BRCA1/BRCA2 gene mutation carrier and is likely ill advised for others with mutations in related DNA repair genes. Pathogenic BRCA1/BRCA2 gene mutations are increasingly recognized as markers of reduced sensitivity to taxol-based chemotherapy and increased sensitivity to platins and poly(ADP-ribose) polymerase (PARP) inhibitors. Although germline genetic tests are not yet recognized as “predictive tests” for systemic therapy decisions, they can gain patients access to clinical trials that are exploiting the unique biology of BRCA1/BRCA2 –associated breast cancer.
Role of the Cancer Genetics Counselor
Receiving the results of a genetic test can be emotionally challenging and set in to motion a chain of events with significant quality-of-life implications. Some patients simply prefer not to know. Decisions about whether to undergo testing, what test to perform, and then what to do with the results are complex. This complexity is amplified in the era of multigene panel testing, making the involvement of a genetics professional essential. Some have advocated genetic testing by primary care providers, or by those who diagnose and treat breast cancer. The ABOUT study, a survey of 11,159 women who underwent BRCA1/BRCA2 testing in 2012, found that only 37% received genetic counseling. Those who received genetic counseling had greater knowledge, understanding, and satisfaction than those who did not. In 2012 the American College of Surgeons Commission on Cancer accreditation program mandated that cancer risk assessment, genetic counseling, and genetic testing services be provided to patients by a qualified genetic professional either on site or by referral. This standard was reiterated in 2016 ( http://www.facs.org/cancerprogram/index.html ). The US Preventive Services Task Force recently completed a systematic review of the literature and concluded that genetic counseling reduces distress, improves risk perception, and reduces intention for testing.
The National Society of Genetic Counselors has outlined the essential functions of the genetic counselor which include (1) estimating cancer risk based on personal and family medical history before genetic testing, (2) offering genetic testing when certain criteria are met, (3) fully informing the individual about the test and the possible results, and (4) disclosing test results in conjunction with a reestimation of cancer risk and a discussion about options for reducing risk. Psychosocial assessment and intervention, when required, is a key function throughout the genetic testing process.
Identifying Mutation Carriers
Mutations in BRCA1 and BRCA2 are the most frequently identified cause of hereditary breast cancer predisposition. The prevalence (allelic frequency) of pathogenic BRCA1/BRCA2 gene mutations is estimated at 0.13% to 0.26% for the general population and 1.3% to 2.7% for Ashkenazi Jewish populations. Therefore, considering other genes in addition to BRCA1 and BRCA2, there are 350,000 to more than 500,000 individuals in the United States who carry pathogenic mutations in breast cancer predisposition genes.
Current genetic testing guidelines nearly ensure that genetic high-risk families are only recognized after one or more individuals have already been diagnosed with cancer. Ideally, mutation carriers would be recognized before they develop cancer so they have the opportunity to act to avoid cancer. Nevertheless, current guidelines are based on patterns of personal and family cancer history. Third party payors usually adopt these guidelines, so that financial constraints often limit testing outside of these guidelines. The American Society of Clinical Oncology has recommended genetic testing when the following criteria are met: (1) there is a personal or family history suggesting genetic cancer susceptibility, (2) the test can be adequately interpreted, and (3) the results will aid in the diagnosis or influence the medical or surgical management of the patient or family members at hereditary risk for cancer. A fourth criteria must be considered as well: there must be some provision for paying for the test.
Genetic testing guidelines are directed at recognizing individuals who are reasonably likely to carry a pathogenic mutation. This is currently largely determined by personal and family cancer history unless there is a known gene mutation in the family. National Comprehensive Cancer Network (NCCN) guidelines for recommending genetic evaluation are quite liberal. BRCA1/BRCA2 testing is recommended, even in the absence of any family history, for women diagnosed with breast cancer at age 50 or younger, triple negative breast cancer at age 60 or younger, bilateral or ipsilateral second primary breast cancer, ovarian cancer, breast or pancreatic cancer at any age in an Ashkenazi Jewish person, or male breast cancer. Combinations of personal or family history of breast, prostate, pancreas, ovarian or other syndrome-associated cancers are also sufficient for pursuing genetic evaluation ( Table 17.2 ). NCCN guidelines include testing criteria for TP53 (Li-Fraumeni syndrome) and PTEN (Cowden syndrome).
| Personal History | Required Family History a |
|---|---|
| Breast cancer ≤50 b Triple-negative breast cancer ≤60 Two primary breast cancers (same or opposite side) Ovarian cancer Breast cancer in Ashkenazi Jewish Pancreatic cancer in Ashkenazi Jewish Male breast cancer | None required |
| Breast cancer (any age) | Breast cancer ≤50 Two or more breast cancers (any age) Ovarian cancer Two or more pancreatic cancer and/or prostate cancer (Gleason score ≥7) Male breast cancer |
| None required | Two or more primary breast cancers in a single individual Breast cancer ≤45 in a first- or second-degree relative Two or more relatives with breast cancer, one age ≤50 Breast cancer at age ≤50 Ovarian cancer Male breast cancer |
a First-, second-, or third-degree relative in the same lineage.
There are more than a dozen genes firmly linked to inherited breast cancer predisposition. Additionally, a step-wise approach to genetic testing may be cost-prohibitive. Generating and maintaining detailed testing criteria for each one may not be practical. In general, hereditary predisposition can be suspected based on early age at breast cancer diagnosis, multiple breast cancers in the same lineage, and the presence of associated cancers such as ovarian, pancreas, or prostate for hereditary breast/ovarian cancer syndrome, and gastric, melanoma, sarcoma, endometrial, or others for the other high penetrance syndromes (see Table 17.1 ).
The probability that an individual carries a mutation in BRCA1 or BRCA2 can be estimated using family history models such as BRCAPRO, Tyrer-Cusick, or BOADICEA. There is no specific mutation probability that is sensitive and specific enough to use as the sole criterion for offering genetic testing. One study found that 6% to 8% of BRCA1 / BRCA2 mutation carriers are missed when the testing criterion is a 10% mutation probability estimated by genetic counselors or the computer model BRCAPRO.
The US Preventive Services Taskforce recommends that primary care providers screen women for personal and family cancer histories that may suggest an inherited predisposition to breast cancer. Numerous screening tools are available. The simplest tool is shown in Table 17.3 . Despite these recommendations, detailed cancer family history screening is not routinely practiced. One approach for increasing genetic counseling referrals has been to provide physicians with education and tools to facilitate screening, but this approach has not been particularly successful. Many organizations have articulated guidelines for genetics referrals, but providers are often unaware of these guidelines. For all of these reasons, there is interest in moving family history screening out of primary care practices into other venues. A modified Bellcross tool was recently used to screen 96,055 women having mammograms. Five percent of the women met criteria for genetic counseling referral, and 29 mutation carriers identified. This is only 22% of the expected mutation prevalence for a population of this size and composition.
| Breast cancer at age ≥50 | 3 |
| Breast cancer at age <50 | 4 |
| Ovarian cancer at any age | 5 |
| Male breast cancer at any age | 8 |
| Ashkenazi Jewish heritage | 4 |
There is reason to question whether self-reported family history screening is the best approach for identifying mutation carriers. In the first place, there are issues with the accuracy of self-reported history, and in the second place, limited family size and structure can contribute to false negatives. A recent study found that half of 306 women with breast cancer diagnosed before age 50, who were seen for genetic risk assessment, had fewer than two first- or second-degree female relatives living past the age of 45. Nevertheless, BRCA1/BRCA2 mutations were identified in 14% of these women.
These concerns have prompted some to suggest universal mutation testing irrespective of personal or family history. One criticism of this approach is that we may not know how to interpret cancer risks in individuals ascertained in this fashion, and another is that the cost of the testing would need to be less than $250 to get the cost per quality life year gained down to $53,000. A demonstration project among 8195 healthy Ashkenazi Jewish men identified BRCA1/BRCA2 mutations in 2.2% resulting in the identification of 211 mutation carriers among 629 female relatives. Among mutation carriers identified in this fashion, breast cancer risk to age 80 was 60% for BRCA1 and 40% for BRCA2. Ovarian cancer risk was 53% for BRCA1 and 62% for BRCA2. This has not yet been evaluated in the general (non–Ashkenazi Jewish) population.
The pathology laboratory may also have a role in the identification of gene mutation carriers. Multigene panel testing was performed in 1824 triple-negative breast cancer patients unselected for family history. Pathogenic BRCA1 or BRCA2 mutations were identified in 11.2% and mutations in other genes in 3.7%.
In summary, in the United States alone there are hundreds of thousands of individuals with unrecognized mutations in breast cancer predisposition genes. At a minimum systematic family history screening should be incorporated into primary care, breast care, and cancer screening activities. The limitations of this approach are well recognized. Novel approaches based on histologic and biomarker features of primary tumors may engage pathology laboratories for genetic risk screening in the future. Universal testing generates new social, economic, and political concerns but does offer the opportunity to identify mutation carriers before they develop cancer.
Genetic Testing Technology
Oswald Avery demonstrated that nucleic acids are the medium of genetic transmission in the 1940s. How a mixture of phospho-sugars and nucleotides could encode information could not even be guessed at until Watson and Crick worked out the structure of DNA in 1953. It took many years after that to realize that messenger RNAs (mRNAs) were working copies of specific nucleotide sequences that could travel to the cytoplasm and direct ribosomes to assemble amino acids into specific proteins. Frederick Sanger devised methods to determine the sequence of nucleotides in DNA molecules in 1977. Once this was worked out, it was quickly discovered that small differences in nucleotide sequences could generate abnormal proteins that were associated with inherited diseases.
The search for breast cancer genes began in the 1980s with the collection and analysis of family histories. An early modeling study by King, using 1579 pedigrees, concluded there is an autosomal dominant breast cancer predisposition gene with an allelic frequency of 0.0006, that breast cancer risk is 82% with the gene and 8% without, and that the gene accounts for 4% of breast cancers. Using markers for chromosomal regions, the King laboratory eventually determined that this gene was likely located on the long arm of chromosome 17. It was the group at Myriad Genetics that ultimately identified and sequenced BRCA1 in 1994. Knowing which part of the genome to sequence and being able to reliably sequence that part in any individual using the methods developed by Sanger led to the birth of clinical cancer genetics as a medical discipline.
Sanger Sequencing
For more than a decade, Sanger sequencing was the primary laboratory test performed on DNA extracted from white blood cells or oral mucosal cells to determine whether an individual carried a mutation in a known breast cancer predisposition gene. Sanger sequencing involves separating the two strands of DNA and then defining the region to be sequenced by allowing short complementary sequences (primers) to bind to the DNA at either end of the region of interest. Double-strand DNA is then rebuilt by filling in the gap between the primers with new nucleotides (A, C, T, and G). Included in the mix of new nucleotides are low concentrations of specially prepared A’s, C’s, T’s, and G’s that are labeled with a colored probe that will stop the elongation of the new DNA strand at that point. Several cycles of DNA synthesis in a test tube will produce a mixture of DNA strands of various lengths, each capped with a color marker representing the last nucleotide added to that particular strand. After many cycles of DNA synthesis, the mixture is separated by size using electrophoresis. The order that the colored nucleotides exit the electrophoresis gel will establish the original sequence of nucleotides between the two primer pairs. One limitation of this approach is that only predefined, relatively short (300–900 nucleotides) segments of DNA between the primer pairs will be assessed. A gene like BRCA2 has 10,254 coding nucleotides so more than a dozen sequencing reactions need to be set up just to assess the coding region. People decide whether other sequencing reactions need to be designed to also assess portions of the promoter region or introns that may contain disease-causing mutations. The point being that sequencing tests in use around the turn of the 21st century may miss important mutations. Another limitation of this approach is that point mutations are recognized by observing two colors coming off of the electrophoresis gel at the same point (one representing the correct nucleotide and one representing the variant nucleotide). If one copy of a particular section of DNA is simply missing (a deletion), only one color will come off of the gel for that section (the normal sequence), and an important genomic alteration will be missed. This is why additional tests to identify insertions, deletions, or rearrangements of entire sections of the DNA have been gradually added to genetic testing protocols over time.
Next Generation Sequencing
Massive parallel sequencing, or next-generation sequencing (NGS), became commercially available in 2005. There are several iterations of the technology, but in general, DNA is minced up to generate short fragments that are then widely distributed across glass surfaces. Each short fragment is duplicated several times to create bundles of DNA with the same sequence. These appear as spots on an imaging screen. Reagents, which include color-labeled nucleotides, are sequentially added and washed away. Each spot is photographed between each cycle. The color of a given spot after any cycle corresponds to the specific nucleotide most recently bound to the immobilized DNA strands at that location. In this way, a nucleotide sequence is generated for each spot ( Fig. 17.2 ). Each sequencing test will include tens of thousands to millions of spots, each generating 50 to 100 base sequences. Translating this enormous data set into nucleotide sequences for specific regions of the genome is a daunting task that is done by computers. First, each individual sequence is aligned to a standard human genome to determine exactly what part of the genome is represented. Because of the random nature of the initial DNA digestion and spot creation, each nucleotide of interest will occur several times on overlapping sequences. Statistical filters are used to assess the quality of nucleotide calls based on the number of replicates generated by the test and the consistency of the calls between replicates. Comparison of the test genome with the standard genome will reveal tens of thousands of sequence variants for any given individual. This very long variant list can be shortened by ignoring known polymorphisms and by focusing only on specific regions of interest (e.g., the BRCA1 gene). Nevertheless, NGS produces a wealth of sequence variants that pose a problem for interpretation. Technically, NGS is reliable and will identify the same mutations identified by Sanger sequencing. NGS can identify large insertions, deletions and rearrangements missed by Sanger sequencing but may have difficulty recognizing small rearrangements.

Large Rearrangements
All of the DNA sequencing technologies are highly sensitive and specific for recognizing single nucleotide changes and short insertions or deletions. Recognizing rearrangement of larger regions is more problematic. These large rearrangements account for up to 17% of pathogenic BRCA1/BRCA2 gene mutations in individuals of Near East/Middle Eastern ancestry and up to 22% for individuals with Latin American/Caribbean ancestry. In 2012 NCCN guidelines specified that BRCA1/BRCA2 gene mutation testing should routinely include special tests for large rearrangements. Testing before this may not have included this special testing. NextGen sequencing data files generally include enough information to recognize rearrangements if specific bioinformatics algorithms are used to look for them. Some test providers will supplement NGS “allele dose” calculations with additional tests specifically directed at identifying large rearrangements such as microarray comparative genomic hybridization and multiplex ligation-dependent probe amplification analysis. Thus far sensitivity for detecting these rearrangements seems high, though for some platforms, specificity needs to be improved to reduce the false positive rate. Confirmatory testing using one of the classical assays is currently recommended.
Classifying Variants
DNA sequencing of any kind will always identify small differences between individuals or small differences compared with some standard genome. Each variant must be adjudicated on the basis of available evidence. Variants that occur in greater than 1% of the population are generally considered polymorphisms and easily classified as almost certainly benign. Variants that have previously been definitively linked to breast cancer in family studies are more easily classified as almost certainly pathogenic. In the middle, however, is every shade of gray from “variant likely benign” to “variant likely pathogenic.” For some variants there is simply not enough information to make an educated guess. These are reported as “variants of uncertain significance.”
The American College of Medical Genetics has published guidelines for reporting DNA sequence variations. There are five possible classifications based on the availability of published data and the type of DNA change ( Table 17.4 ). Classification is straightforward for variants that have previously been reported and for which there are quite a lot of data available. For everything else, it is people who weigh the evidence and initially assign a classification.
| Classification | Abbreviated Criteria |
|---|---|
| Pathogenic | Variant causes loss of protein function, which is known to cause disease. |
| Likely pathogenic | The bulk of available laboratory and epidemiologic data point to loss of protein function and statistical correlation with disease. |
| Benign | Known polymorphism that is common in the population. Reliable data do not support loss of protein function or segregation with disease. |
| Likely benign | Available data suggest no loss of protein function; variant sometimes seen in patients with known pathogenic variants. |
| Uncertain significance | None of the preceding criteria are met, or available data are contradictory. |
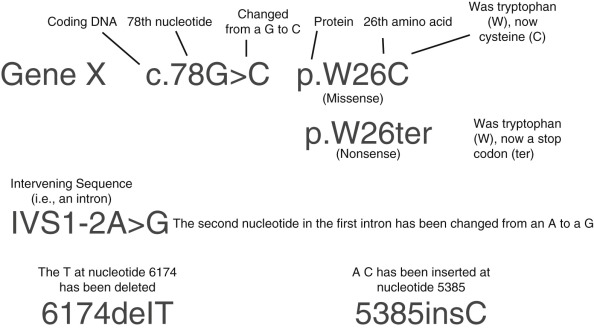
There are thousands of distinct rare variants in BRCA1 and BRCA2, and the list is rapidly growing for many other genes as a result of increasing use of multigene panel testing. Each new variant is initially classified somewhere along the spectrum of benign to pathogenic based on features of the specific nucleotide change. Certain types of mutations are more likely to be pathogenic than others. These include single nucleotide changes that result in an early stop codon and a shorter than normal protein (nonsense or truncating variants) as well as insertion or deletion of some number of nucleotides that is not a multiple of three. This will result in a shift in the reading frame and often an early stop codon (frameshift). Missense variants are changes in one nucleotide that result in a different amino acid being added when the mRNA is read. Most of these variants are not pathogenic, although some are. Several additional lines of evidence can move the classification toward or away from pathogenic. Variants that have previously been observed in individuals that have other, definitely pathogenic variants in the same autosomal dominant gene are more likely neutral. A variant that occurs in a region that is normally coded exactly the same across many species (i.e., conserved) may be more likely to be pathogenic, as is a variant that causes a significant change in the type of amino acid that is added to the growing protein chain (e.g., switching from a hydrophilic to a hydrophobic amino acid). Variants that occur near the intron-exon boundary can affect the way that mRNA is spliced together and may also be pathogenic. Fig. 17.3 illustrates the nomenclature used to describe these different types of variants in laboratory reports.
Various informatics tools are available to help classify variants. ClinVar is a publically available list of sequence variations and their current interpretation that is maintained by the National Institutes of Health. The Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) is an international group devoted to the collection and interpretation of rare variants in BRCA1 and BRCA2. They have developed a software tool to estimate the probability of pathogenicity for any variant using combined evolutionary sequence conservation, family-based segregation and cancer history, tumor pathology, and RNA splicing effects. Some clinical laboratories use personal and family history weighting to assist in the classification of new variants.
Variants of Uncertain Significance
Every new variant identified in a known breast cancer predisposition gene has uncertain significance until it has been investigated as described in the preceding. Many times, at the conclusion of this investigation there are insufficient data to confidently place it anywhere along the benign-to-pathogenic spectrum. These variants are temporarily classified as variants of uncertain significance (VUS). The natural history of most VUS is definitive reclassification over time as new information becomes available. Sometimes this new information is proactively generated by testing affected and unaffected relatives from the same family or by assessing the functional significance of the DNA change in the laboratory. Sometimes new information is passively acquired as the same variant is observed in more informative families. The point is that VUS rates are high when a new test begins identifying new variants, but this rate decreases as experience increases. This was observed with BRCA1 and BRCA2 gene mutation testing where the VUS rate was 7% to 15% in 2002 but had decreased to 2.9% by 2012. VUS rates are somewhat higher for non-Caucasian populations but have declined from 22% to 46% to 2.6% to 7.8% in recent years.
The introduction of multigene panel testing has generated a torrent of new variants. VUS rates are currently 20% to 40% for genes that are less well studied than BRCA1 and BRCA2. One study that evaluated a 42-gene panel in 175 patients reported an average of 2.1 VUS calls per patient. This degree of uncertainty is understandably unsettling. It will take time to accumulate sufficient clinical and functional data to definitively reclassify these variants. To this end, the Prospective Registry of Multiplex Testing (PROMPT) was established. This is an online registry that collects variant and clinical information from patients who have had multigene panel testing.
Patients whose genetic test result returns a VUS are said to have had a noninformative result. In this case the test was not helpful, and the patient is managed based on the personal and family history. Variant classifications can change over time as more data become available. It is necessary to establish and maintain procedures for recontacting patients if their variant is reclassified.
Multigene Panels
NGS has made it possible to screen numerous genes simultaneously at a significantly reduced cost per nucleotide compared with Sanger sequencing. The first clinical application of multigene panels was in patients with cardiomyopathy. Next-generation multigene panels for hereditary breast cancer first became commercially available around 2010. Early panels did not include BRCA1 or BRCA2 because of the Myriad Genetics, Inc. patent on these sequences. On June 13, 2013, the Supreme Court overturned this patent, and subsequent panels included these genes. One effect of this Supreme Court ruling was the introduction of competition into the genetic testing space, which led to multiple choices and significantly reduced costs for patients. Dozens of companies offer hereditary breast cancer panel testing, including several university laboratories. Important questions for the consumer are the following: (1) Are all of these tests equally accurate for mutation detection? (2) Apart from cost, are there any important differences between test providers? (3) To what extent do multigene panels improve the identification of high-risk families?
To measure the sensitivity and specificity of NGS for variant detection, Myriad Genetics, Inc. assessed BRCA1 and BRCA2 by NGS for 1864 samples that had previously undergone clinical testing by Sanger sequencing. NGS identified 15,877 variants in these samples, one fewer than had been identified by Sanger sequencing. One polymorphism was missed because a primer for that region happened to overlap the variant. Analytic sensitivity was estimated at greater than 99.96% and specificity at greater than 99.99%. Further validation of an NGS panel that included 25 genes reported 100% concordance with variants identified by Sanger sequencing. In another validation study that included 198 individuals who had undergone standard BRCA1 and BRCA2 mutation testing, the InVitae NGS test identified 58 of 59 known pathogenic mutations. One large insertion was missed because no specific deletion-duplication assay was performed at that time and one variant previously classified as pathogenic was considered by InVitae to be a variant of uncertain significance. Other validation studies have reported excellent concordance between NGS and Sanger sequencing or between NGS assays performed at different centers.
NGS technology is robust for the identification of single base pair substitutions and small insertions or deletions and can also reliably detect large rearrangements depending on the specific assay design and bioinformatics. Another potential source of variability between test providers is the approach used to manage the very long list of variants generated by NGS to exclude the trivial and recognize the pathogenic. After filtering out polymorphisms, this becomes a very human endeavor that is often more art than science. It is important to understand how each laboratory approaches this process. Cost is one straightforward metric for comparing genetic test providers apart from any quality considerations. The cost for hereditary breast cancer predisposition testing can range from less than $500 to nearly $5000 depending on the provider used and the tests performed. On a very practical level, test providers may also be assessed based on the level of service they provide to their clients. This includes legwork for insurance preauthorizations, accurate estimation and constraint of out-of-pocket expenses for the patient, provision for providing testing to the uninsured and underinsured, and vigor with which they pursue VUS reclassification including no-charge testing for family members.
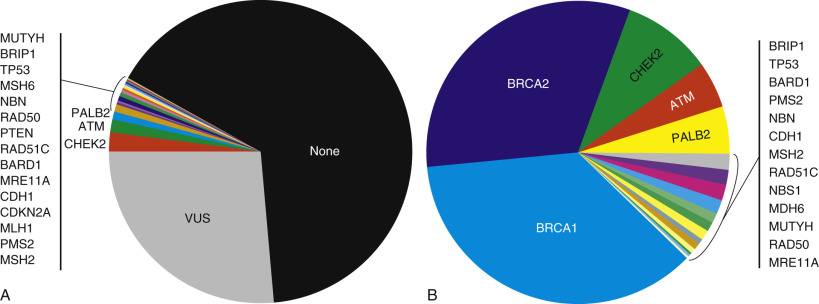
It was anticipated that most apparent familial breast cancer would be explained when testing panels were expanded to dozens of genes beyond BRCA1 and BRCA2. This has not been the case. Multigene panel testing will identify a pathogenic mutation in 4% to 12% of familial high-risk patients who have tested negative for BRCA1 and BRCA2 mutation. BRCA1 and BRCA2 mutations still account for 65% to 81% of pathogenic mutations identified on multigene panel testing, with CHEK2, ATM, and PALB2 the most frequently affected non– BRCA1/BRCA2 genes. Even with multigene panel testing, most patients with personal and family histories of breast and ovarian cancer will have no identifiable pathogenic mutation, and testing will identify one or more variants of uncertain significance in 19% to 42% ( Fig. 17.4 ).
Managing Cancer Risk
The goal of risk management in mutation carriers is to reduce the probability of developing a breast cancer or to diagnose a breast cancer early when it can be treated with the best outcome and least morbidity. The primary options for managing cancer risk include enhanced surveillance, chemoprevention, and prophylactic surgery. Certain reproductive and lifestyle factors may also modify breast cancer risk in gene mutation carriers. Most available data are derived from studies in BRCA1 and BRCA2 gene mutation carriers and may not be relevant to every inherited predisposition syndrome. Table 17.5 lists current recommendations by gene.
| Breast MRI | Discuss RRM | Consider RRSO |
|---|---|---|
| ATM BRCA1 BRCA2 CDH1 CHEK2 PALB2 PTEN STK11 TP53 | BRCA1 BRCA2 CDH1 PTEN TP53 PALB2 | BRCA1 BRCA2 Lynch syndrome BRIP1 RAD51C RAD51D |
Related posts:
 Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
 Therapeutic Strategies for Breast Cancer
Therapeutic Strategies for Breast Cancer
 Breast Conservation Therapy for Invasive Breast Cancer
Breast Conservation Therapy for Invasive Breast Cancer
 Radiation Complications and Their Management
Radiation Complications and Their Management
 Immunologic Approaches to Breast Cancer Therapy
Immunologic Approaches to Breast Cancer Therapy
 Delayed Diagnosis of Symptomatic Breast Cancer
Delayed Diagnosis of Symptomatic Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


