(1)
Medical Sciences Division Northern Ontario School of Medicine West Campus, Lakehead University, Thunder Bay, Ontario, Canada
Key Topics
Molecular pathology of brain tumors.
Circulating glioblastoma (GBM) epigenetic biomarkers.
Circulating GBM miRNA biomarkers.
Serum GBM protein biomarkers.
Circulating GBM cells.
Circulating endothelial and endothelial progenitor cells.
Key Points
GBM, the most common brain cancer is associated with dismal median survival of 12–18 month due to the spread of cancer cells to other parts of the brain and hence are not easily amenable to complete surgical removal.
The molecular pathology of GBM involves epigenetic and genetic changes that alter the RTK/RAS/MAPK, PI3K, cell cycle, and growth factor signaling pathways.
The molecular pathologic changes, especially MGMT methylation, IDH1/IDH2, and EGFRvIII mutations, are detectable in circulation as adjuncts to disease management. Emerging circulating GBM biomarkers include miRNAs, serum proteins, circulating GBM cells, endothelial cells, and extracellular vesicles.
18.1 Introduction
Nervous system (NS) tumors are a conglomerate of neoplastic transformation of cells that constitute the nervous system. While there are numerous types, gliomas are the commonest. Gliomas constitute a group of central NS (CNS) tumors further subdivided based on putative cell of origin into astrocytomas, oligodendrogliomas, and oligoastrocytomas. Globally, ~200,000 people are afflicted annually with a diagnosis of glioma. While not as common as other solid tumors such as colon cancer, the prognosis of glioma is very dismal. In the US, 23,770 new cases of nervous system tumors and 16,050 deaths are expected for 2016. The recurrent rate is high, usually with the emergence of high-grade tumors. The median survival is 12–18 months for WHO grade IV glioblastoma (GBM). Even supposedly early stage (grade II) patients have just 5–10 years, while grade III oligodendroglioma and astrocytoma patients survive for 3–10 years and 2 years, respectively. The need for early detection and importantly predictive biomarkers for this tumor is thus urgently needed.
For personalized medicine, accurate classification of CNS tumors is needed. Thus, biomarkers that inform tumor behavior or biology ushers in this paradigm shift in management of brain tumors. As is established, co-deletion of chromosomes 1p and 19q in grade III anaplastic oligodendrogliomas is associated with a better prognosis than other glioma subtypes. Similarly, isocitrate dehydrogenase 1 (NADP+) IDH1 mutations in grade III glioblastoma are associated with better prognosis than patients with IDH1 wild-type tumors. There are currently emerging biomarkers, such as noncoding RNAs, proteomic, and metabolomic biomarkers, which should enhance our knowledge and management of gliomas. Being able to assay these biomarkers noninvasively and longitudinally will definitely improve glioma early detection and optimal patient care.
18.2 Screening Recommendations for Brain Tumors
There are no recommended screening guidelines for brain cancer for a number of reasons. First, because of the low incidence, the harms from screening outweigh any benefit. Second, the risk factors for the general population are obscure, such that no defined high-risk population is currently available to be targeted for screening. The only exceptions are individuals with heritable familial mutations such as neurofibromatosis types 1 and 2, tuberous sclerosis, VHL disease, and Li–Fraumeni syndromes. These families have elevated risk for many cancers including brain cancer and hence require close surveillance. Similarly, people exposed to toxic radiations or those with impaired immune systems such as AIDS patients need close monitoring. While noninvasive tests are being developed for this high-risk group, current screening involves a variety of imaging modalities, including functional MRI and PET scans.
18.3 Molecular Pathology of Brain Tumors
The most common adult primary brain tumor with an estimated global incidence of 3.5/100,000 and one of the deadliest cancers is GBM. It is very lethal because, first, tumor cells infiltrate surrounding brain tissue making complete surgical removal impossible, and second, these tumors are often resistant to chemotherapy and radiation. Thus, the mean survival is 12–18 months.
Gliomas are classified based on cellular origins as determined by histology and immunohistochemistry, and graded (I–IV) based on aggressive features including mitotic activity, microvascular proliferation, and necrosis. GBM, the most aggressive form of gliomas (grade IV) originates via two mechanisms. Primary or de novo GBM, which is the more common type, develops quite rapidly usually following a short clinical history. GBM can also develop from low-grade gliomas such as diffuse astrocytomas (WHO grade II) or anaplastic astrocytomas (WHO grade III), and these are referred to as secondary GBMs. As expected, the two are molecularly distinct as well. Primary GBMs are characterized by PTEN and EGFR alterations, while TP53 loss of function is more common in secondary GBMs.
18.3.1 Epigenetic Alterations in Brain Tumors
Gene promoter hypermethylation is a recognized feature of GBM. Of interest, methylation of MGMT is an established predictive biomarker in GBM. MGMT methylation, which occurs in ~45 % of adult GBMs, results in loss of gene function and hence reduced efficiency of DNA repair. The fragile state of such tumors is exploited for prediction of patients who will respond to temozolomide (TMZ). Thus, adult GBM patients with MGMT methylation benefit from TMZ chemotherapy, while those without do not. Similarly, the effect of this therapy in childhood high-grade glioma (HGG) is yet to be resolved. Methylation also affects a number of genes such as TP53, RB1, CDKN2A, PTEN, PDGFβ, epithelial membrane protein 3 (EMP3), protocadherin γ subfamily A, member II (PCDHGAII), and suppressor of cytokine signaling 1 (SOCS1) that have established roles in glioblastomagenesis.
18.3.2 Chromosomal Alterations in Brain Tumors
Glioblastoma is characterized by a wide spectrum of gene copy number alterations, as well as LOH. Deletions of CDKN2A and CDKN2B occur in ~53 % of adult GBMs and as many as 20 % of pediatric HGGs. EGFR amplification occurs in ~43 % of adult GBMs and <5 % of pediatric HGGs. These high frequency copy number changes often occur concurrently with EGFR deletions and/or point mutations. In adult GBM, high-level amplifications are found in CDK4 (~13 %), PDGFRA (~11 %), MDM2 (~8 %), MDM4 (~7 %), MET (~2 %), and CDK6 (~1.5 %); amplifications occur in the following genes: MYCN, MYC, CCND2, CCNE1, and SOX2. Additionally, homozygous deletion of RB1, CDKN2C, PTEN, FAF1, NF1, QKI, TP53, and NPAS3 characterizes adult GBM. Amplifications of PDGFRA, MYC, and MYCN are often more associated with pediatric cases.
Glioblastoma is a genetic disease with well-characterized genomic alterations. Multiple chromosomal structural abnormalities underlie GBM pathogenesis. The majority (83–85 %) of adult tumors harbor gains at chromosome 7 and loses at chromosome 10. These two alterations often occur concurrently in the same tumors and are therefore referred to as 7+/10– tumors. They are more common in tumors from people ≥70 than those ≤40. Other frequent chromosomal changes in adult GBM are gains at chromosomes 19 and 20 (occurs in 35–40 % of cases) and losses at chromosomes 9p (~38 %), 13q (~33 %), 22q (~33 %), 14q (27 %), and 6q (22 %). In general, pediatric HGG harbors less frequent chromosomal aberrations, except gains at chromosome 1q that occurs at a frequency of ~20 %.
In addition to the chromosomal gains and losses, a vast majority of GBM (~69 %) displays intergenic, intragenic, and interchromosomal rearrangements, and these alterations are commonly associated with copy number changes at the breakpoints. A classic example is the intragenic deletion of exons 2–7 in EGFR that encodes for the extracellular domain of the receptor (EGFRvIII), and these aberrations often coexist with amplifications and increased expression of the wild-type allele. Similarly, the increased expression of PDGFR is accounted for by amplifications and age-dependent intragenic deletion rearrangements. A few cases of GBMs also display gene fusions. EGFR activation can also occur as a result of recurrent translocation and fusion to SEPT14 or PSPH, and interestingly, this often occurs at amplified regions of the fusion partner. Constitutive kinase activity of FGFR1 in GBM may be caused by inversion and in-frame fusion to TACC1 (similarly FGFR3 can fuse to TACC3 as in non-muscle invasive bladder cancer).
18.3.3 Mutations in Brain Tumors
The Cancer Genome Atlas (TCGA) data indicate that ~46 % of GBM harbors at least one somatic mutation that affects genes associated with DNA methylation (IDH1), histone modifications (SETD2, HDAC, KDM6A, MLL2–4, and EZH2), or chromatin remodeling (ATRX, CREBBP, CHD5–9, and SWI/SNF-related matrix-associated actin-dependent regulator of chromatin A2—SMARCA2).
The majority (58–84 %) of adult GBMs harbors mutational hotspots in TERT promoter that results in increased telomerase expression and activity. These mutations tend to occur in association with amplifications in EGFR and negatively correlate with mutations in IDH1, IDH2, ATRX, and TP53. Pediatric tumors tend to harbor ATRX loss, PDGFRA mutations, and amplifications, and few (3–11 %) also have TERT mutations.
Other genes frequently mutated in adult GBMs are in order of decreasing frequencies: PTEN (~29 %), TP53 (~29 %), EGFR (~20 %), NF1 (~9 %), SPTA1 (~9 %), RB1 (~8 %), PIK3CA (~7 %), ATRX (~6 %), PIKBR1 (~6 %), IDH1 (~5 %), KEL (~5 %), GABRA6 (~4 %), LZTR1 (~3 %), BCOR (~2 %), BRAF (~2 %), CTNND2 (~2 %), and QK1 (~2 %).
Almost all the genetic lesions in GBM impinge on three main signaling pathways to promote tumor growth and spread. The RTK/RAS/PI3K pathway is activated in ~88 %, p53 pathway in ~87 %, and the RB pathway in as many as 78 % of GBMs.
18.3.4 Molecular Subtypes of GBM
Various subtypes of GBM have been identified based on methylation, gene expression, and genomic aberration data sets. Currently, at least six subtypes are recognized.
First, using unsupervised hierarchical clustering of TCGA expression data set, copy number alterations, and sequencing, Verhaak et al. subclassified GBMs into four molecularly distinct subclasses [1].
Proneural subtype is enriched for PDGFRA, CDK4, CDK6, and MET aberrations and frequent IDH1 mutations.
Classical subtype harbors EGFR amplifications, as well as PTEN and CDKN2A loss.
Mesenchymal subtype features mutations or losses in NF1, TP53, and CDKN2A.
Neural subtype has no unique genetic signature.
Second, Noushmehr et al. similarly used unsupervised hierarchical clustering of DNA methylation profiles from the TCGA samples to delineate subtypes of GBM based on glioma CpG island methylator phenotype (G-CIMP) status [2]. This G-CIMP-positive subclasses are enriched in the proneural subclass of Verkhaak et al., and thus associated with IDH1 mutations that could account for the G-CIMP.
Sturm et al. observed mutations in H3F3A and its chromatin remodeling complexes ATRX and DAXX in 44 % of pediatric cases [3]. Thus, they decided to include these in subclassification. Using epigenetic, genetic, and transcriptomics data from 210 GBM patient samples composed of cases from adults and children, six subtypes of GBM were mapped:
Subtype 1 occurs mostly in young adults and is characterized by G-CIMP-positive, proneural gene expression, and IDH1 and TP53 mutations.
Subtype 2 is a feature of childhood midline and brainstem GBMs, also expresses proneural genes, but has H3F3A (K27) mutation, and strongly positive for OLIG2 staining. The prognosis of this subtype is very poor.
Subtype 3 occurs mostly in teenagers and young adults and is characterized by G-CIMP-positive, TP53, and H3F3A (G34) mutations.
Subtype 4 is similar to adult classical subtype with similar transcription map, EGFR amplification, and CDKN2A loss.
Subtype 5 has profile of adult mesenchymal subtype but with relatively few mutations and copy number changes.
Subtype 6 has adult proneural expression profile, but PDGFR2A amplification, and CDKN2A loss.
18.4 Circulating Brain Tumor Biomarkers
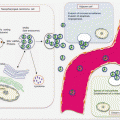
Circulating biomarkers complement GBM management especially with the development and deployment of targeted therapies. Thus, the molecular pathologic alterations including MGMT methylation, IDH1/IDH2 mutations, and EGFRvIII are detectable in circulation. MiRNAs, serum proteins, circulating GBM, and endothelial cells (CECs), as well as extracellular vesicles, are all potential noninvasive GBM biomarkers (Fig. 18.1).
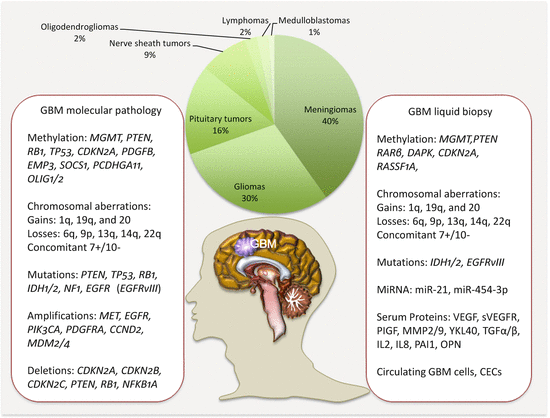
Fig. 18.1
Frequencies of brain tumors, GBM molecular pathology, and some liquid biopsy biomarkers
18.4.1 Circulating Brain Tumor Epigenetic Biomarkers
The epigenome has been studied in circulation of glioma patients. Using bisulfite sequencing, hypomethylation of ALU repeats in glioma tissues and matched serum samples from patients revealed high correlation (r = 0.882) between the two sample sets [4]. As a diagnostic biomarker, serum ALU hypomethylation achieved an AUROCC of 0.861 in glioma detection. Also, in tissue and serum samples, high methylation of ALU sequences correlated with longer survival. Methylation of multiple genes including CDKN2A/P16, MGMT, RARβ, and TP73 in glioma tissues and plasma from patients was studied for the first time as a proof of principle [5]. Methylation in at least one gene was present in 90 % of the tumor samples, of which 67 % plasma samples harbored identical alteration in this small sample set. Another multi-marker methylation analysis targeted MGMT, RASSF1A, CDKN2A/p16, DAPK, and TMS-1 in tumor tissue and matched serum samples from patients with GBMs [6]. The frequencies of methylation in tissue samples were MGMT (38.1 %), CDKN2A/p16 (66.7 %), DAPK (52.4 %), and RASSF1A (57.1 %). Corresponding gene methylation frequencies in serum samples were 39.3 %, 53.6 %, 34.3 %, and 50 %. There was strong correlation between methylation in tissue and serum samples. CDKN2A/p16 methylation in diffuse gliomas (astrocytomas and oligodendrogliomas) from the brain and brainstem as well as matched serum samples from patients and controls revealed methylation in 60 % of astrocytomas but in only one oligodendroglioma [7]. Similar methylations were detected in 75 % of serum samples from patients with astrocytomas.
A predictive study of response to 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and temozolomide treatment of patients with GBM targeted methylation of multiple genes [8]. Both tumor and serum samples were assayed for methylation in MGMT, CDKN2A/p16, DAPK, and RASSF1A. MGMT methylation and silencing enhanced the response to BCNU treatment. Methylation in all the genes assayed correlated with serum methylation patterns. But only MGMT methylation predicted response to BCNU treatment. Overall survival and stability of disease were observed in 90.9 % of patients with MGMT methylation compared to only 35.7 % of patients without MGMT methylation. Similarly, time to progression was 29.9 weeks for patients with MGMT methylation compared to 15.7 weeks for patients without methylation.
18.4.2 Circulating Brain Tumor Genetic Biomarkers
MYC-N gene amplification in neuroblastoma is detectable in plasma and serum samples from patients, and may be useful in treatment monitoring. Tumor-specific epigenetic and genetic markers were assayed in serum from patients with gliomas (astrocytomas and oligodendromas) [9]. LOH on chromosomes 1p, 19q, and 10q, as well as promoter methylation of MGMT and PTEN were assessed in serum samples. While the specificities were high, the sensitivities were only 51 % for LOH and 55 % for methylation targets in serum. Tumor-serum concordance was statistically significant for MGMT methylation (for both types of gliomas) and LOH at 1p and 10q (for oligodendromas).
18.4.3 Clinical Utility of Liquid Biopsy in GBM
The molecular pathology of glioma has provided lead biomarkers for disease management. The detection or measurement of MGMT methylation, 1p/19q co-deletion, IDH1 and 2 mutations, EGFR amplification, and EGFRvIII expression offers clinical insights into various diseases. They serve as biomarkers for diagnosis, prognosis, treatment prediction, and monitoring. For example, 1p/19q deletion serves as a diagnostic, prognostic, and predictive biomarker of diffuse glioma. Additionally, these molecular alterations serve as targets for drug development. As examples, bevacizumab targets VEGF, the inhibitor AGI-5198 targets IDH1/2, and rindopepimut targets EGFRvIII. While traditionally tissue biopsies are used for these genetic analyses, liquid biopsy is paving the way for the noninvasive and longitudinal assessment of these molecular alterations in ctDNA. As demonstrated examples, promoter methylation of MGMT is both of prognostic and predictive relevance, EGFRvIII alterations are present in extracellular vesicles, and mutations in IDH1/IDH2 are detectable in ctDNA.
18.4.4 Circulating GBM Noncoding RNA Biomarkers
MiRNA alterations in circulation of GBM patients have been addressed with some promise. Plasma miR-21 levels are altered in a number of neoplastic conditions. In GBM, Ilhan-Mutlu et al. observed a four-fold increase in a particular patient samples years before the clinical diagnosis of GBM (control group had significantly decreased circulating miR-21) [10]. In another study, miR-21 levels were significantly elevated in GBM patients (p = 0.02), and the levels dropped significantly (p = 0.05) after surgery. Increasing miR-21 levels were associated with a patient who developed recurrent disease [11, 12]. Li et al. identified miR-21 in exosomes derived from a GBM cell line (U251) [13]. Indeed, over 28 miRNAs were enriched in these exosomes, with higher levels of nine miRNA*s including miR-93*, miR-106a*, miR-181a*, and miR-452* than their matured counterparts. However, serum miR-21 has also been shown to be a diagnostic and prognostic biomarker of primary CNS lymphoma (PCNSL) [14]. The levels were significantly increased in PCNSL compared to other brain tumors and healthy controls, and this achieved a diagnostic accuracy with AUROCC of 0.930 and 0.916 in the test and validation data sets. Indeed, miR-21 could separate patients with PCNSL from GBM with an AUROCC of 0.883 in the test set and 0.851 in the validation cohort. MiR-21 emerged in both Kaplan–Meier and multivariate Cox regression analysis as a strong independent predictor of OS of PCNSL.
MiR-21, miR-128, and miR-342-3p show differential plasma levels between GBM patients and healthy controls [15]. However, these miRNAs were not specific to GBM as they were associated with other brain tumors (meningiomas, pituitary adenomas) as well. Their levels correlated with histopathologic grades of glioma and returned to normal levels following surgery. This group subsequently uncovered miR-454-3p to be significantly elevated in plasma samples from glioma patients compared to healthy controls [16]. MiR-454-3p levels were higher in patients with high-grade than low-grade tumors. The diagnostic performance AUROCC was 0.9063. The levels decreased significantly following surgery, and high plasma miR-454-3p was significantly associated with poor prognosis. Serum miR-125b levels were significantly lower in glioma patients than healthy controls, and the levels correlated with WHO tumor grade [17]. As a diagnostic biomarker of glioma, serum miR-125b had an AUROCC of 0.839. This diagnostic accuracy improved with increasing tumor grade (grade II AUROCC was 0.868 and grade III–IV was 0.959 compared to grade I with an AUROCC of 0.691). But circulating miR-125b is not specific to glioma, as it performed equally well in detection of other cancers. Similar to miR-125b, circulating levels of miR-29 family were more useful at detecting advanced stage gliomas (AUROCC of 0.81) compared to early stage disease (AUROCC of 0.66) [18]. Again, this miRNA family is associated with other cancers as well, achieving an AUROCC of 0.83 in meta-analysis of other cancers.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


