Fig 4.1
Biomarker-relevant biologic pathways in renal cell carcinoma (RCC). In VHL-/- tumor cells, the absence of pVHL results in the accumulation of hypoxia-inducible factor alpha (HIF-α). HIF accumulation could also be secondary to the activation of the PI3K/AKT/mammalian target of rapamycin (mTOR) pathway. mTOR phosphorylates and activates pS6K, which leads to increasing translation of downstream target proteins, including cyclin D, Myc, and HIF. Activated mTOR also phosphorylates 4E-BP1, disrupts this complex, and allows eIF-4E to stimulate the mRNA translation as well. Activated HIF translocates into the nucleus and results in the transcription of multiple HIF-target genes, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). These proteins bind to their receptors and cause cell migration, proliferation, and permeability. RCC biomarkers could be derived from cell components of tumor cell itself, including DNA, RNA, protein, and metabolites. The soluble cell components could also migrate from the cell into the blood vessels and be detected in blood and urine of RCC patients
4.3 The Importance of RCC Biomarker Development
The early detection and diagnosis of RCC remains a challenge to oncologists and presents a significant barrier to reducing mortality due to this cancer. Roughly 30 % of RCC cases present with metastatic disease at the time of initial diagnosis [3]. Although this percentage has declined in recent years due to increased incidental detection of small renal masses, the mortality rate from RCC has remained steadfastly unchanged [4]. This suggests that RCC with lethal potential are not being identified sufficiently early to prevent metastatic spread, and this presents the single most significant opportunity to reduce death due to RCC. Patients with metastatic RCC have a much poorer prognosis compared with patients with early-stage disease, with a 5-year survival rate of 23 % for stage IV disease as compared to a 5-year survival rate of 96 % for stage I presentation [5].
The use of early detection biomarkers remains in development, but interesting tools are on the horizon. New generations of biomarkers that examine novel substrates such as microRNA (miRNA, miR), proteomic, and metabolomic profiles, with the potential to measure hundreds or more elements simultaneously as a biomarker “profile,” are being investigated intensely as tools for RCC early detection and diagnosis. The results have been encouraging [6, 7], but await full clinical validation.
Several early detection serum and urinary biomarkers have been reported as a first step toward a clinically relevant RCC detection assay. Noninvasive detection methods are promising given the increased frequency of detection of RCC from incidental findings on imaging. In one recent study, analysis of RCC cases revealed elevated plasma levels of N-methyltransferase (NNMT), L-plastin (LCP1), and nonmetastatic cells 1 protein (NM23A) [8]. A three-marker assay was developed with good positive and negative predictive value for RCC, although results of this study remain unvalidated. Examination of urinary samples from newly diagnosed RCC patients and matched controls identified 86 peptides more frequently found in RCC, most of which were fragments of collagen chains. An assay using these peptides was developed and then validated using an independent set of patients, enabling differentiation of RCC from control with excellent discriminative accuracy (AUC of 0.92) [9]. These assays may help indicate the presence of a kidney primary malignancy, although they need to be further validated and studied in a diagnostic capacity.
Metastatic RCC consists of a heterogeneous group of cancers. This creates incredible challenges to prediction of prognosis and response to different therapeutics. Biomarkers have their most immediate potential in RCC to demystify the heterogeneity and classify RCC into meaningful subgroups. Ultimately, having a rational biological signature from which to draw prognostic or predictive information, yet with low cost and minimal specimens from patients, would be invaluable. In the last decade, the emergence of multiple FDA-approved targeted therapies gives promise to patients with advanced RCC; however, it also adds complexity in the effort of tailoring each agent to different individuals in appropriate sequence. Despite increased understanding of the underlying tumor biology of RCC and its variant histologies (which arguably comprise highly distinct disease entities), the current TNM staging and subtyping of RCC give inadequate insight to refine current algorithms for treatment selection, disease monitoring, and management. The identification and utilization of novel biomarkers for prognosis and prediction of response are important approaches for personalized RCC treatment.
4.4 Understanding the VHL Pathway for RCC Biomarker Development
4.4.1 VHL
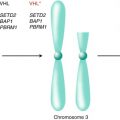
Before embarking on an inventory of biomarkers for RCC, it is essential to understand the biology and molecular pathways which are known in this disease and from which the majority of biomarkers are derived (Fig. 4.1). A key event in the pathogenesis of clear cell RCC (ccRCC) appears to be the inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene, which is a biallelic event in over 90 % of sporadic ccRCC [10]. The mechanisms that lead to the loss of VHL functionality include large-scale and small-scale deletions, missense mutations, early stop codons, truncations, and silencing of the locus by hypermethylation. The VHL gene is located on the short arm of chromosome 3. Large deletions of 3p are commonly identified in ccRCC. This causes a loss of heterozygosity in a majority of ccRCCs, leaving cells susceptible to loss of the remaining allele and full inactivation of VHL [11]. Overall, the disengagement of VHL in this unique tumor type is likely a critical and common event in ccRCC development.
4.4.2 pVHL
The VHL protein (pVHL) performs a critical cellular function in regulating the cellular response to low oxygen. In the presence of sufficient oxygen, pVHL binds to a family of proteins called the hypoxia-inducible factor alpha (HIF-α) subunits, recruiting them to an E3 ubiquitin ligase complex which polyubiquitinates the HIF-α subunits, thus targeting them for proteasome-mediated proteolysis [12]. The loss of pVHL activity, therefore, permits the constitutive stabilization of HIF-α factors, and high-level expression of HIF-α factors has been a widely recognized feature of ccRCC tumor biology. About 90 % of all ccRCC display HIF-α stabilization apparently as a consequence of VHL loss or inactivation [13]. Recent evidence has accrued to indicate that pVHL has functions other than regulation of HIF-related pathways, such as regulation of apoptosis, control of cell senescence, and maintenance of the primary cilium [14].
4.4.3 HIF
HIF is a heterodimeric transcription factor complex consisting of an unstable alpha (α) subunit and a stable beta (β) subunit. Three HIF-α genes (HIF-1α, HIF-2α, and HIF-3α) have been identified in the human genome [15]. Both HIF-1α and HIF-2α function as classical transcription factors, although they can also cooperate with additional factors to maximize activity [16]. The role for HIF-3α, which does not clearly act as a transcriptional regulator and exists with many splice-variant isoforms, is poorly understood [17].
Despite many similarities, HIF-1α and HIF-2α are not fully redundant in function. The global gene expression changes induced by HIF-1α and HIF-2α show that they produce overlapping yet distinct gene expression profiles in both cells and in mice [18].
HIF plays a critical role in tumorigenesis. Indeed, there are several lines of evidence that implicate HIF-α and in particular HIF-2α as playing an active role in VHL-/- renal cell carcinogenesis. First, RCC-associated pVHL mutants are at least partially defective with respect to HIF-2α polyubiquitination [19, 20]. Genetic manipulation of HIF expression in human tumor cell line xenografts has clearly demonstrated a growth advantage for cells expressing HIF-2α but not HIF-1α [12, 21]. Examination of human ccRCC tissues provided the ultimate demonstration of a dependence on HIF-2α stabilization, showing that all VHL-defective RCCs either stabilize dually both HIF-1α and HIF-2α or solely HIF-2α [13]. This observation provides an alternative way of classifying pVHL-deficient tumors based on this distinction of HIF expression. The VHL genotype and the protein expression of HIF-1α and HIF-2α proteins were analyzed in 160 primary tumors. The tumors were examined by immunohistochemistry (IHC) for HIF-1α and HIF-2α and messenger RNA profiling. VHL-deficient tumors that exclusively express HIF-2α (H2) tumors displayed greater c-Myc activity and higher rates of proliferation relative to those of VHL-deficient tumors expressing both HIF-1α and HIF-2α (H1H2), regardless of tumor stage. H2 tumors also demonstrated increased expression of genes involved in DNA repair, decreased levels of endogenous DNA damage, and fewer genomic copy-number changes. Moreover, those VHL-deficient H1H2 tumors and VHL wild-type tumors displayed increased activation of AKT/mTOR and ERK/MAPK1 growth factor signaling pathways and increased expression of glycolytic genes. Thus, there may be two biologically distinct types of VHL-deficient ccRCC: those that produce HIF-1α and those that do not. The relevance of this distinction as a biomarker remains to be demonstrated, although consistent with expectations, H2 tumors were of a higher T stage than their H1H2 counterparts.
4.4.4 HIF-Responsive Genes
HIF is a potent transcriptional activator of the cellular hypoxia response and more than 100 direct HIF-responsive genes have been described, with a number of these genes active in carcinogenesis [22]. Although some of these genes and their products are being studied in RCC, two deserve special attention: vascular endothelial growth factor (VEGF) and carbonic anhydrase IX (CAIX, CA9).
4.4.4.1 VEGF
VHL-/- ccRCCs are notoriously angiogenic and overproduce a variety of proangiogenic molecules including the HIF-responsive VEGF. VEGF stimulates endothelial cell proliferation, migration, maturation, and survival and is among the most potent endothelial mitogens. Furthermore, the VEGF receptor, kinase insert domain-containing receptor (KDR), may be present on renal carcinoma cells, suggesting the possibility of an autocrine feedback loop, although receptor activity on tumor cells remains to be demonstrated [23, 24].
VEGF and VEGF receptors (VEGFR) have been thrust into the spotlight as a result of substantial activity of targeted therapies, which engage these factors. Bevacizumab is an antibody that binds circulating VEGF protein and has activity in metastatic RCC [25]. In addition, potent tyrosine kinase inhibitors, such as sunitinib, sorafenib, and pazopanib, target the intracellular signaling pathways of multiple members of the VEGF receptor family of proteins. Multiple phase III trials have demonstrated substantial clinical benefit from blocking VEGFRs with sunitinib, sorafenib, and pazopanib [26, 27, 28]. Below, we will discuss the potential utility of biomarkers of VEGF activity in the context of therapeutics that directly target this signaling pathway, either via tumor cells directly or via supporting cells of the endothelium.
4.4.4.2 CAIX
CAIX is a transmembrane protein that may play a role in the regulation of cell proliferation, oncogenesis, and tumor progression. CAIX is a HIF-responsive, hypoxia-induced protein that accumulates in VHL-defective RCCs [29]. A study of CAIX expression in 317 primary and 42 metastatic renal neoplasms showed correlation between CAIX expression with ccRCC histology as well as histologic grade, suggesting that this HIF-dependent protein may provide an effective surrogate for HIF stabilization with the potential to independently serve as a biomarker [30].
4.4.5 AKT/mTOR/HIF Pathway
A better understanding of the molecular biology underlying RCC will lead to the development of biomarkers reflecting aberrant signal transduction pathways within these tumors. Mammalian target of rapamycin (mTOR) is a kinase that activates substrates critical for protein synthesis. It directly phosphorylates the ribosomal subunit S6 kinase (S6K) as well as eukaryotic initiation factor 4E (eIF-4E), which is released from its inhibitory binding partner 4E-BP1 upon its phosphorylation by mTOR. Loss of function mutations of the PTEN tumor suppressor gene result in increased mTOR activity via AKT-dependent inactivation of the tuberous sclerosis complex (TSC1 and TSC2), and key members of this pathway have been identified to have non-overlapping mutations in a substantial percentage of tumors [31]. Inhibitors of mTOR decrease global translation of proteins including HIF, cyclin D1, and Myc [32]. There are now two FDA-approved mTOR inhibitors used in the clinic for advanced RCC: temsirolimus [33] and everolimus [34], which have led to both improved progression free survival (PFS) and overall survival. The detection of the effector molecules (phospho S6, phospho 4EBP1, and phospho AKT) has been linked with response to VEGF-targeted therapy [35] and is both prognostic for overall survival and predictive of response to mTOR therapy [36–38].
4.4.6 Other 3p Genes Involved in RCC
In addition to VHL, several other genes located on chromosome 3p have been recently shown via massively parallel sequencing to be commonly mutated in RCC. These genes are important in histone modification and chromatin remodeling. The most commonly mutated gene is PBRM1 (polybromo 1) [39]. Histone methyltransferase SETD2 (SET domain-containing protein) and the histone deubiquitinase BAP1 (BRCA1 associated protein–1) have also recently been described, in addition to several other less commonly mutated genes [40, 41]. There are both positive and negative genetic interactions among these genes, with PBRM1 mutations and SETD2 mutations commonly occurring in the same tumor and PBRM1 and BAP1 rarely occurring together [42, 43]. These genes, similar to VHL, are tumor suppressor genes in which one allele is typically inactivated by mutation or hypermethylation and the second is inactivated through a large deletion in chromosome 3p, resulting in loss of heterozygosity [44].
BAP1 is a nuclear deubiquitinase and tumor suppressor gene mutated in about 9–15 % of ccRCCs [41, 45–48] (and commonly in other cancers, most notably metastatic uveal melanoma) [49]. It causes expression of a specific gene expression signature and is associated with increased mTOR activation. BAP1 mediates deubiquitination of histone H2A and binds to host cell factor-1 (HCF-1), a component of the chromatin-remodeling complex, and binding is required for suppression of cell proliferation. Therefore, loss or mutation of BAP1 is thought to result in loss of tumor suppression [41, 45, 50–52]. A missense variant of BAP1 has also been found as a germline mutation in familial RCC, although it rarely occurs [53].
SETD2 is a two-hit tumor suppressor gene located in the region of chromosome 3p that is deleted in a majority of ccRCCs. It is present in about 7–16 % of ccRCCs [45, 46]. SETD2 functions as a histone modifier and methyltransferase and is responsible for trimethylation of lysine 36 of histone H3, causing decreased H3K36 levels in some tested ccRCC cell lines [54] and thereby possibly influencing gene expression and transcription activation.
PBRM1 encodes a chromatin/nucleosome remodeling complex protein BAF180 and is mutated in 30–45 % of RCC [39, 46]. It is thought to work as a tumor suppressor gene through regulation of DNA accessibility and gene expression and therefore regulate cell proliferation, although the full mechanism of tumorigenesis due to loss of PBRM1 is not fully understood [42]. Nearly all PBRM1-mutated tumors exhibit a hypoxia signature, suggesting a loss of VHL even though not all cases are associated with a detectable VHL mutation [39]. Overall, the recently identified mutations of BAP1, PBRM1, and SETD2 represent novel genetic contributors to the pathogenesis of ccRCC, a finding that may reveal important prognostic classification groups and potentially inform therapeutic decisions in the future.
4.5 The Development of RCC Biomarkers in Clinical Decision-Making
While biomarkers for early detection and diagnosis remain at an early stage of development, more advances have been made for prognostic and predictive biomarkers of RCC. Here we will focus our discussion on these markers.
4.5.1 Prognostic Biomarkers
Prognostic biomarkers have been studied in parallel with advances in the tumorigenesis of this cancer. A summary of the potential molecular prognostic biomarkers that have been investigated for RCC is provided (Table 4.1). We will focus the following discussion on the broad spectrum of prognostic biomarkers.
Table 4.1
Potential individual molecular prognostic biomarkers for renal cell carcinoma (RCC)
Biomarker | Type | Source | No. of patients | Reference | Results |
|---|---|---|---|---|---|
Circulating tumor cells (CTCs) | Cells | Blood | 154 | Bluemke et al. [124] | Detection of CTCs was correlated with poor overall survival (RR 2.3; p = 0.048) |
miRNA-106b | RNA | Tumor | 38 | Slaby et al. [125] | miR-106b is a potential predictive marker of early metastasis after nephrectomy in RCC patients (p = 0.032) |
miR-23b/27b | RNA | Tumor | Ishihara et al. [126] | Lower expression of miR-23b/27b was associated with shorter OS (p = 0.018 and p = 0.025, respectively) | |
Serum amyloid A protein (SAA) | Protein | Blood | 119 | Wood et al. [127] | Total SAA protein was of independent prognostic significance (p = 0.017) |
Angiotensin receptor type 2 (AR2) | Protein | Tumor | 84 | Dolley-Hitze et al. [128] | AR2 was overexpressed in the most aggressive forms of RCC and correlates with PFS (p = 0.006) and cancer stage (p < 0.001) |
CAIX | Protein | Tumor | 357 | Klatte et al. [83] | CAIX expression was a strong independent prognostic factor for patients with metastatic cc RCC (p < 0.05) |
C-reactive protein | Protein | Blood | 282 | Jagdev et al. [129] | C-reactive protein was highly significant for cancer-specific survival (P < 0.0001) and OS (p < 0.002) |
CXCR4, CXCR7 | Protein | Tumor | 223 | D’Alterio et al. [130] | High CXCR4 expression (p = 0.0061), high CXCR7 (p = 0.0194) expression, and the concomitant high expression of CXCR4 and CXCR7 (p = 0.0235) are independent prognostic factors |
Cathepsin D | Protein | Urine | 239 | Vasudev et al. [131] | Cathepsin D showed evidence of independent prognostic value for OS (p = 0.056) |
E-selectin | Protein | Tumor | 559 | Tran et al. [107] | Higher baseline E-selectin levels associated with better PFS (p = 0.002) |
EZH2 | Protein | Tumor | 520 | Wagener et al. [132] | High nuclear EZH2 expression was an independent predictor of poor cancer-specific survival (HR 2.72, p = 0.025) |
Global histone acetylation | Protein | Tumor | 193 | Mosashvilli et al. [133] | Global histone modification level was a universal cancer prognosis marker (p < 0.05) |
HGF | Protein | Blood | 559 | Tran et al. [107] | High baseline level of HGF was associated with worse PFS (p = 0.013) |
HIF-1α | Protein | Tumor | 357 | Klatte et al. [83] | Patients with high HIF-1α expression (>35 %) had significantly worse survival than patients with low expression (< or =35 %); median survival, 13.5 vs. 24.4 months, respectively (p = 0.005) |
HuR | Protein | Tumor | 152 | Ronkainen et al. [134] | HuR expression was associated with reduced RCC-specific survival (HR 2.18; p = 0.015) |
IL-6, IL-8 | Protein | Blood | 559 | Tran et al. [107] | High baseline levels of IL-6 and IL-8 were associated with worse PFS (p = 0.021 and 0.013, respectively) |
IMP3 | Protein | Tumor | 716 | Hoffman et al. [135] | IMP3 expression was associated with a 42 % increase in death from RCC (p = 0.024) |
MMP-9 | Protein | Tumor | 120 | Kawata et al. [136] | MMP-9 was associated with high nuclear grade and was an independent prognostic factor (p = 0.003) |
Osteopontin | Protein | Blood | 559 | Tran et al. [107] | High baseline levels of osteopontin were associated with worse PFS (p = 0.041) |
p-AKT | Protein | Tumor | 40 | Jonasch et al. [35] | Higher levels of p-AKT were associated with increased OS (HR 1.15, 95 % CI 1.02–1.29) |
PI3K | Protein | Tumor | 176 | Merseburger et al. [137] | Increased PI3K expression was associated with lower survival (p = 0.030) |
p-mTOR | Protein | Tumor | 132 | Abou Youssif et al. [138] | Cytoplasmic p-mTOR showed independent prognostic significance (p = 0.029) and fidelity between primary RCCs and their matched metastases (p = 0.004) |
PAI-1 | Protein | Tumor | 167 | Zubac et al. [139] | PAI-1 was a significant prognostic factor of cancer-specific survival (p < 0.001) |
S100A4 | Protein | Tumor | 32 | Bandiera et al. [140] | Five-year survival was lower in patients with high S100A4 expression than weak expression (41 % vs. 78 %; p < 0.05) |
TIMP-3 | Protein | Blood | 903 | Pena et al. [102] | TIMP-3 was the only biomarker prognostic for overall survival in TARGET trial (p = 0.002) |
TS-1 | Protein | Tumor | 172 | Thrombospondin-1 expression was associated with high nuclear grade, advanced stage (p < 0.001), and tumor progression (p = 0.006) | |
VEGF-A | Protein | Blood | 559 | Tran et al. [107] | High baseline VEGF levels were associated with worse OS (p-0.004) |
4.5.1.1 Clinical Biomarkers
Historically, multiple clinical algorithms were used to estimate prognosis, including the UCLA Integrated Staging System (UISS) to predict risk for disease recurrence or disease-associated death [55] and the Memorial Sloan Kettering Cancer Center (MSKCC) risk criteria for estimating survival for patients with metastatic disease [56]. The UISS incorporates the TNM staging systems, performance status, and the Fuhrman grade of the tumor and is heavily weighted based on tumor stage. While valuable, this staging system does little to risk stratify those patients with nonmetastatic but sizeable primary tumors. For patients with metastatic disease, which remains incurable with current therapeutic options, the MSKCC algorithm is a valuable clinical tool to establish prognostic intervals for a disease that can range from indolent to rapidly lethal. This system also takes into account the Karnofsky performance status (which can be highly subjective and variable), time from diagnosis to treatment, and laboratory values of hemoglobin, lactate dehydrogenase, and corrected serum calcium. With the widespread clinical use of targeted therapies in RCC, it is necessary for those criteria, which were validated in the era of cytokine therapies, to recruit new biomarkers to match deregulated pathways with effective inhibitors.
In a recent revision of the model, Motzer et al. developed a nomogram that includes both statistically significant and insignificant factors as biomarkers to create a non-biased prognostic model for patients receiving sunitinib [56]. The additional factors included were the number of metastatic sites (p < 0.01), the presence of hepatic metastases (p < 0.1), thrombocytosis (p < 0.01), prior nephrectomy (p = 0.37), the presence of lung metastases (p = 0.74), and serum alkaline phosphatase levels (p = 0.82) [56].
4.5.1.2 Histological Biomarkers
Tumor stage is widely considered by many clinicians as the most important prognostic factor. Historically, effort has focused on identifying critical features in addition to tumor size, such as extracapsular extension, renal vein invasion, inferior venous cava invasion, lymph node involvement, and presence or absence of adrenal gland metastases. It is only recently that the histologic subtyping of RCC into clear cell, papillary, and chromophobe variants gained its long-deserved attention. Aggregation of data has shown that each tumor subtype is associated with different pathophysiology and clinical behavior. In the largest and most comprehensive retrospective review to date, a group of 3,062 cases was identified between 1970 and 2003, among them 2,466 patients (80.5 %) with clear cell, 438 (14.3 %) with papillary, and 158 (5.2 %) with chromophobe RCC. A significant difference in metastasis-free and cancer-specific survival existed between patients with ccRCC and the two other dominant subtypes. Even after multivariate adjustment, the ccRCC subtype remained a significant predictor of metastasis and cancer-specific death [57].
In an effort to estimate prognosis within the ccRCC group, the Fuhrman grading system has been used to further categorize tumors according to tumor cell morphology and correlates tumor grade to mortality [58]. Other histologic features, including the presence of alveolar features, lymphovascular invasion [59], and sarcomatoid dedifferentiation [60] play pivotal roles in prognosis as well, although the degree to which each of these affect prognosis is uncertain.
4.5.1.3 Genetic Biomarkers
Traditional cytogenetic karyotyping studies have altered the approach used in classifying RCC subtypes. Characteristic karyotypes have been consistently associated with each of the most common subtypes of RCC (clear cell, papillary, and chromophobe) [61–63]. In ccRCC, the most frequently observed cytogenetic abnormalities were loss of 3p (60 %), gain of 5q (33 %), loss of 14q (28 %), trisomy 7 (26 %), loss of 8p (20 %), loss of 6q (17 %), loss of 9p (16 %), loss of 4p (13 %), and loss of chromosome Y in men (55 %) [64]. It is interesting that tumors with loss of 3p typically presented at lower TNM stages. Loss of 4p, 9p, and 14q were all associated with higher TNM stages, higher grade, and greater tumor size. A deletion of 3p was associated with better prognosis, while loss of 4p, 9p, and 14q were each associated with worse prognosis [64]. With regard to the less common RCC variants, in papillary RCC, trisomies of chromosomes 7 and 17 were found to be specific genetic alterations irrespective of their size, grade, and cellular differentiation [65]. Another study indicated trisomy 16 and chromosome Y were specifically involved in papillary RCC [66]. The rarest subtype of the three, chromophobe RCC, predominantly showed loss of whole chromosomes, such as loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 [67]. A recent evaluation of the somatic mutation spectrum of chromophobe RCC showed these tumors have commonly mutated TP53 and PTEN genes, although less than half of all tumors had one of these mutations [68]. Further analysis revealed frequent TERT promoter genomic rearrangements in chromophobe RCC, as well as alterations in mitochondrial DNA including increased mitochondrial genome copy numbers and electron transport gene complex 1 mutations [68].
Karyotyping provides a piece of the genetic puzzle of RCC tumorigenesis by elucidating some chromosomal changes. However, in order to complete the puzzle and identify the stepwise progression of RCC carcinogenesis, we have to rely on genomic or exomic sequencing, array comparative genomic hybridization (a-CGH), or SNP analysis.
Recent advances in sequencing technology have made large-scale genomic sequencing rapid and cost-effective. As above, several genes located on chromosome 3p (PBRM1, SETD2 and BAP1) have recently been identified as commonly mutated in ccRCC, along with the frequently mutated VHL gene. These results indicate that large-scale gene sequencing is no longer limited by cost and can provide substantial genetic information to identify heterogeneity in ccRCC.
The presence of these genetic mutations has been shown to have prognostic and predictive significance. Patients with BAP1-mutated tumors have significantly worse median overall survival with a nearly threefold increased hazard ratio for death than those with PBRM1 mutations [50]. BAP1 is also an independent marker of poor prognosis in patients with low-risk disease and may be able to help risk stratify this group of patients [51]. Presence of BAP1 is also associated with metastatic disease at presentation [45]. The combination of BAP1– and PBRM1-mutated tumors is rare and has been associated with an even worse overall survival than either mutation alone in most studies, although not in one small study [45, 50]. The BAP1 mutation was originally described via genetic sequencing [41], but immunohistochemical testing has now been validated and also correlates with poor overall survival and adverse clinicopathological tumor features [52]. SETD2 mutations are associated with worse cancer-specific survival in a cohort of patients from the Cancer Genome Atlas, but not an MSKCC cohort [46]. The presence of PBRM1 mutation does not seem to be associated with a change in cancer-specific survival [47], although it has been associated with advanced tumor stage in some earlier studies [69]. It therefore has been suggested to play a more prominent role in tumor initiation instead of disease progression [46].
4.5.1.4 Gene Expression Profiles
Multiple studies have used traditional gene profiling using RT-PCR to quantify RNA expression. In 2001, Takahashi et al. studied the expression profile of 29 ccRCC samples and found 51 genes, which could categorize RCC for prognostic purposes [70]. More recently, an analysis of gene expression profiles using machine learning algorithms refined the notion that more than one type of ccRCC was present and used 49 ccRCC samples to define a panel of 120 genes which can accurately define two groups of ccRCC, designated ccA and ccB [71]. This model was refined for application using a NanoString platform using archival renal tumor tissues, demonstrating the feasibility of the approach and showing an advantage of molecular classification using the ClearCode34 biomarker for ccA and ccB integrated with stage and grade over conventional clinical algorithms [72].
Using an RT-PCR platform adapted for fixed tissue analysis, 931 archival formalin-fixed tumor tissues from patients with localized ccRCC were examined across 732 candidate genes [73]. With a median follow-up of 5.6 years, 448 genes were found to be associated with a longer recurrence-free interval (p < 0.05). Sixteen genes had a strong association after consideration of clinical pathologic covariates and false discovery adjustments (HR 0.68–0.80). Among the 16 genes, increased expression of angiogenesis-related genes (EMCN and NOS3) was associated with lower risk of recurrence, as was increased expression of immune-related genes (CCL5 and CXCL9). This profile provides a feature set readily adaptable to validation studies and has additional promise as a potential predictive biomarker as well. Several of the recently discovered 3p genes commonly mutated in ccRCC also have unique gene expression profiles, but they have been thus far indistinguishable from nonmutant tumors using unsupervised hierarchical clustering algorithms and are therefore not ready for clinical use at this time [42].
4.5.1.5 Hybrid Strategies
The current trend is to incorporate multiple complementary approaches for better identification and understanding of cancer-related genes. Cifola et al. performed the first integrated analysis of DNA and RNA profiles of 27 RCC samples [74]. Seventy-one differentially expressed genes (DEGs) were found in aberrant chromosomal regions and 27 upregulated genes in amplified regions. Among them, the transcripts encoding LOX and CXCR4 were found to be upregulated. Both are implicated for cancer metastasis. Such combinations of genomic and transcriptomic profiling may potentially provide us a more powerful tool for prognostic estimation.
Another trend is to combine epigenetic data with gene expression profiling for better understanding of these interactions. In a preliminary study, an 18-gene promoter methylation panel using quantitative methylation-specific PCR (QMSP) for 85 primarily resected RCC was evaluated [75]. Significant differences in methylation among the four subtypes of RCC were found for CDH1 (p = 0.0007), PTGS2 (p = 0.002), and RASSF1A (p = 0.0001). CDH1 and PTGS2 hypermethylation levels were significantly higher in ccRCC compared to non-ccRCC. RASSF1A methylation levels were significantly higher in papillary RCC than in normal tissue (p = 0.035). Further validation of epigenetic data in larger cohorts is needed to explore the true prognostic value.
4.5.1.6 Copy-Number Analysis
Array comparative genomic hybridization (a-CGH) has been used to identify the specific copy number changes associated with RCC. A comprehensive analysis incorporated a-CGH and gene expression profiles from 90 tumors in order to identify new therapeutic targets in ccRCC [76]. There were 14 regions of nonrandom copy-number change, including seven regions of amplification (1q, 2q, 5q, 7q, 8q, 12p, and 20q) and seven regions of deletion (1p, 3p, 4q, 6q, 8p, 9p, and 14q). An analysis aimed at identifying the relevant genes revealed VHL as one of three genes in the 3p deletion peak, CDKN2A and CDKN2B as the only genes in the 9p deletion peak, and MYC as the only gene in the 8q amplification peak. An integrated analysis to identify genes in amplification peaks that are consistently overexpressed among amplified samples confirmed MYC as a potential target of 8q amplification and identified candidate oncogenes in the other regions.
a-CGH may also improve the diagnostic accuracy for RCC. A recent study examined a-CGH on ex vivo fine-needle aspiration (FNA) biopsies and tumor fragments of 75 RCC patients. The pattern of genomic changes identified by a-CGH was used blindly to classify the renal tumors and the genetic findings were subsequently compared with the histopathologic diagnosis. a-CGH was successful in 82.7 % of FNA biopsies and in 96 % of tumor fragments. The genetic pattern correctly recognized 93.5 % of ccRCC, 61.5 % of chromophobe RCC, 100 % of papillary RCC, and 14.3 % of oncocytoma, with the negative predictive value being above 90 % [77]. As RCC histology is an independent predictor of prognosis, one could postulate that a-CGH will have powerful prognostic value as well.
4.5.1.7 SNP Genotyping
Single nucleotide polymorphism (SNP) genotyping has been used to detect cytokine gene polymorphisms in RCC patients to determine its prognostic significance. A panel of 21 SNPs within the promoter regions of 13 cytokine genes were analyzed in a single-center study of 80 metastatic RCC patients [78]. IL4 genotype -589T-33T/-589C-33C was identified as an independent prognostic risk factor in metastatic RCC patients with a median overall survival decreased 3.5-fold (3.78 months, p < 0.05) compared with patients homozygous for IL4 haplotype -589C-33C (13.44 months). An association was also found between three SNPs (−2578C/A, −1154G/A, and −634C/G) in the VEGF gene and survival of 213 RCC patients [79]. A more recent study found an SNP in IL-8 was associated with survival in patients treated with pazopanib, and these results were validated using data from the COMPARZ trial in sunitinib-treated patients [80, 81]. Multiple VEGF SNPs have also been associated with response and survival as well [80, 82]. These studies contribute evidence that SNP genotyping could be used to develop prognosis algorithms in patients with metastatic RCC.
4.5.1.8 VHL and HIF as Prognostic Biomarkers
Based on the extensive discussion of the derangement of this pathway as a result of VHL mutation, it is not surprising then that VHL loss or HIF stabilization might provide a prognostic resource. Perhaps owing to the high prevalence of VHL mutation among ccRCCs, numerous efforts to demonstrate that VHL mutation is a prognostic indicator have been unfruitful. Klatte and colleagues showed preliminary evidence that HIF-1α expression can provide an independent prognostic factor for patients with ccRCC. Patients with high (>35 %) tumor immunostaining of HIF-1α had shorter survival than patients with low (≤35 %) immunostaining of HIF-1α [83]. However, more recent studies have suggested that higher expression of HIF-1α and HIF-2α are associated with improved prognosis [84, 85]. Whether tumor expression of HIF-1α provides substantial prognostic information with respect to the natural history of ccRCC remains to be determined, as does the role of HIF-2α in this setting.
4.5.1.9 Circulating Cells
Levels of circulating endothelial cells and circulating tumor cells have been recently gaining attention as prognostic biomarkers. Several studies have shown that higher levels of circulating endothelial cells or circulating endothelial progenitor cells during the first cycle of VEGF-targeted therapy were associated with improved PFS [86, 87]. However, this technology remains investigational for assessing disease at this time.
4.5.2 Predictive Biomarkers
With the abundance of approved therapies for RCC, oncologists now have the luxury to choose individualized therapy for each patient. Traditional immunotherapy should be retailored to fit selected patients better. Targeted therapies not only have invigorated RCC oncologic practice but also have changed the approaches used to predict response to therapy and to measure clinical outcome. In the next section, we differentiate and discuss biomarkers according to different therapies (Table 4.2).
Table 4.2
Potential predictive biomarkers of response to targeted therapies for renal cell carcinoma (RCC)