Fig. 1
ER-localized interactions and activities of vIL-6 in latently infected PEL cells. In the context of PEL latency, vIL-6 is expressed at low but functional levels and is largely sequestered in the ER compartment. Latently expressed vIL-6 supports PEL cell growth and viability. In vIL-6-depleted PEL cells, these activities can be complemented by ER-restricted (KDEL motif-tagged) transduced vIL-6, demonstrating sufficiency of ER-localized vIL-6 activity. The vIL-6 signal transducer, gp130, and a novel splice-variant protein, VKORC1v2, each bind vIL-6 within the ER, and depletion of each inhibits PEL cell growth and viability. Available evidence indicates that vIL-6 activity via each of these ER receptors occurs independently. While gp130–mediated activation of STAT and MAPK signaling has been detected in PEL cells, gp130 signaling (in contrast to effects on growth and survival) is not affected by VKORC1v2 depletion or by peptide-mediated disruption of vIL-6:VKORC1v2 interaction. Expression and pro-growth and pro-survival activities of vIL-6 in latently infected PEL cells suggest that the viral cytokine may contribute to latency maintenance in B cells and to PEL disease
2.5 Viral Interferon Regulatory Factor-3 (vIRF-3) in PEL
HHV-8 specifies four viral interferon regulatory factor homologues, vIRFs 1-4 (Cunningham et al. 2003; Lee et al. 2009a), which serve to counter the effects of cellular IRFs and to inhibit innate responses of the cell to virus infection and productive replication (see below). While all of the vIRFs are expressed during lytic replication, consistent with their presumed primary functions in evasion of antiviral host cell defenses, vIRF-3 is expressed as a bona fide latent product in PEL cells (Jenner et al. 2001; Paulose-Murphy et al. 2001; Rivas et al. 2001). As such, vIRF-3 has been referred to by some investigators as latency-associated nuclear antigen-2 (LANA2), despite its partial localization in the cytoplasm (Munoz-Fontela et al. 2005) and the absence of demonstrable latent expression in any other cell type examined. Nonetheless, in the context of PEL/B cells, vIRF-3 has the potential to impact cellular pathways that may be of biological relevance to viral latency and virus-associated pathogenesis. In common with other vIRFs, vIRF-3 can inhibit cellular IRF function. vIRF-3 does so by interfering with the transcriptional activities of IRFs 3, 5, and 7 in addition to inhibiting PKR, a pro-apoptotic kinase that is activated by interferon and dsRNA (Esteban et al. 2003; Joo et al. 2007; Lubyova and Pitha 2000; Wies et al. 2009). Importantly, vIRF-3 has also been found to interact directly with p53 to inhibit the tumor suppressor and to induce c-Myc-directed transcription (Lubyova et al. 2007; Rivas et al. 2001). vIRF-3 can also interact with and inhibit FOXO3a, a transcription factor targeting pro-apoptotic genes (Munoz-Fontela et al. 2007). These activities indicate that vIRF-3 plays a significant role in promoting cell survival and proliferation in the context of PEL and potentially in general B-cell latency. Indeed, vIRF-3 has been demonstrated to be critically important for PEL cell viability in culture, as its depletion triggers apoptosis (Wies et al. 2008). Pro-survival effects of vIRF-3 may in part be the result of its inhibition of PML-mediated repression of survivin (Marcos-Villar et al. 2009). Furthermore, vIRF-3 has recently been reported to repress CIITA transcription factor-directed expression of interferon-γ and class-II major histocompatibility complex in PEL cells (Schmidt et al. 2011). This immune evasion activity of vIRF-3 is probably vital for the long-term survival of these cells in vivo. Therefore, vIRF-3 activities are likely to be important for latency persistence in at least some cell types (where vIRF-3 is latently expressed) and are probably significant contributors to PEL malignancy.
2.6 Micro-RNAs (miRNAs)
Micro-RNAs (miRNAs) are ~22 nt non-coding RNAs that regulate the expression of mRNAs via cleavage or by inhibiting translation. miRNAs are encoded as primary miRNAs (pri-mRNAs), synthesized by RNA Polymerase II, and processed to pre-miRNAs (stem-loop structures) by the RNAse III domain of DROSHA prior to nuclear export by exportin 5 (Lee et al. 2003b; Lund et al. 2004). In the cytoplasm, pre-miRNAs are cleaved into 21–24 nt double-stranded RNAs by an RNAse III domain of DICER, and one strand of the miRNA duplex is then incorporated into RISC (RNA-induced silencing complex) (Bartel 2004; Lee et al. 2003b). The incorporated miRNA guides the loaded RISC to the mRNA target (Schwarz et al. 2003). Generally, the mRNA target is degraded if the miRNA is perfectly complementary to the targeted sequence. Alternatively, binding of a miRNA lacking perfect complementarity inhibits translation of the mRNA (Zeng et al. 2003).
While miRNAs have been detected in all metazoans, virus-encoded miRNAs were discovered only recently (Pfeffer et al. 2004). To date, 12 HHV-8-encoded miRNAs have been identified, termed miR-K12-1 to miR-K12-12. A total of ten of the 12 miRNAs are located between latently expressed ORFs 71 and K12 in the HHV-8 genome; miR-K12-10 is located within the ORF K12, and miR-K12-12 is within the 3′ UTR of K12 (Pfeffer et al. 2005; Samols et al. 2005). All 12 of the HHV-8 miRNAs are oriented “in sense” with ORFs 71 to K12 and are expressed during latency (Cai et al. 2005). Many of the miRNAs can be detected (and some are even induced) during lytic infection (Cai et al. 2005; Umbach and Cullen 2010). Bioinformatic approaches have utilized miRNA seed sequences (nt 2-7/8 of the miRNA) to search for miRNA gene targets (Gottwein and Cullen 2010; Lu et al. 2010b; Nachmani et al. 2009; Qin et al. 2010), but these approaches yield large numbers of potential candidates. Additionally, targets with less than perfect seed sequence complementarity may be overlooked, and experimental validation of identified candidates is necessary to assess authenticity. Functional approaches involving transduction of recombinant viruses containing single or a combination of HHV-8 miRNAs and monitoring potential changes in mRNA levels via microarray have also been used (Ziegelbauer et al. 2009). More recently, immunoprecipitation of RISC and/or associated Argonaute proteins has been conducted, and microarray analysis of co-precipitated mRNAs (RIP-ChIP) has been employed (Dolken et al. 2010). These methods have identified multiple mRNA targets within the host cell. From these data, the virally-encoded miRNAs have been deduced to: (1) limit NK cell recognition of virally infected cells (Nachmani et al. 2009), (2) repress RTA expression to inhibit latent-lytic switching (Bellare and Ganem 2009; Lin et al. 2011; Lu et al. 2010a), (3) interfere with TGFβ signaling (Liu et al. 2012), (4) alter NF-κB signaling pathways (Lei et al. 2010), (5) modulate cytokine production (Abend et al. 2010), and (6) modify cell cycle progression (Gottwein and Cullen 2010). These targets and pathways are summarized in Table 1 and are discussed further below.
Table 1
HHV-8-encoded microRNAs and their functions
Functional Class | Viral microRNA | Target | Activity/Function | References |
|---|---|---|---|---|
Cell Cycle Regulation | miR-K12-1 | p21 | Inhibits cell cycle arrest | Gottwein and Cullen (2010) |
Apoptosis | miR-K12-10a | TWEAKR | Reduced caspase activation | Abend et al. (2010) |
miR-K12-5, 9, 10 | BCLAF1 | Inhibits apoptosis | Ziegelbauer et al. (2009) | |
miR-K12-1, 3, 4-3p | Caspase 3 | Inhibits apoptosis | Suffert et al. (2011) | |
miR-K12-11 | BACH1 | Pro-survival | ||
Latency Maintenance | miR-K12-3 | NFIB | Inhibits RTA, Stabilizes latency | |
miR-K12-5, 7-5p, 9 | RTA | Stabilizes latency | ||
Growth Signaling | miR-K12-11 | SMAD5 | Limits TGFß signaling | Liu et al. (2012) |
miR-K12-6, 11 | MAF | Cell fate reprogramming | Hansen et al. (2010) | |
Immune Evasion | miR-K12-7 | MICB | Evasion from NK cells | Nachmani et al. (2009) |
Angiogenesis | miR-K12-1, 3, 6, 11 | THBS1 | Increases angiogenesis | Samols et al. (2007) |
Viral Replication | miR-K12-1 | IkBalpha | Limits viral replication | Lei et al. (2010) |
Chromatin Modification | miR-K12-4-5p | Rbl2 | Global epigenetic modification | |
Unvalidated Targets | miR-K12-3 | NHP2L1 | Splicesome assembly? | Dolken et al. (2010) |
mIR-K12 cluster | LRRC8D | Unknown | Dolken et al. (2010) | |
miR-K12 cluster | SEC15L | Vesicle trafficking? | Dolken et al. (2010) | |
miR-K12-4-3 | GEMIN8 | mRNA splicing? | Dolken et al. (2010) | |
miR-K12 cluster | ZNF684 | Transcriptional regulation? | Dolken et al. (2010) | |
miR-K12 cluster | CDK5RAP1 | CDK5 inhibitor? | Dolken et al. (2010) |
HHV-8 employs many strategies to remain undetected by the host immune response, and viral miRNAs are believed to play a vital role. HHV-8-encoded miR-K12-7 targets MICB, a viral infection-induced cell-surface marker that functions to induce natural killer (NK) cell recognition and killing via engagement with the NK-expressed NKG2D receptor (Glas et al. 2000; Nachmani et al. 2009). MICB targeting by virus-encoded microRNAs is conserved in human cytomegalovirus (Stern-Ginossar et al. 2007; Stern-Ginossar et al. 2008) and EBV infection (Nachmani et al. 2009). In addition to immune evasion, several HHV-8 miRNAs target RTA, thereby functioning to maintain viral latency and inhibit viral replication. Several research groups have identified miR-K12-3, miR-K12-5, miR-K12-9, and miR-K12-11 as directly or indirectly targeting RTA expression (Bellare and Ganem 2009; Lin et al. 2011; Lu et al. 2010b). Viral replication is also limited through inhibition of the NF-κB inhibitor IκBα by miR-K12-1; upregulation of NF-κB signaling abrogates lytic replication of HHV-8 (Lei et al. 2010). By reducing virion production, the virus is able to further circumvent the host immune response.
In addition to immune evasion, the virus counteracts pro-apoptotic pathways induced by the host cell upon viral infection. Caspase 3 is a target of multiple viral miRNAs (miR-K12-1, miR-K12-3, and miR-K12-4-3p), and its inhibition desensitizes HHV-8-infected cells to caspase-induced apoptosis (Suffert et al. 2011). Similarly, miR-K12-10a suppresses TWEAK receptor (TWEAKR) expression, which limits TWEAK-induced caspase activation and apoptosis (Abend et al. 2010). TWEAKR inactivation also reduces production of pro-inflammatory cytokines IL-8/CXCL8 and MCP-1/CCL2 (Abend et al. 2010). BCLAF1, a transcriptional repressor, can be targeted by several of the HHV-8 microRNAs (miR-K12-5, miR-K12-9, and miR-K12-10a/b), leading to decreased etoposide-induced apoptosis in PEL cells (Ziegelbauer et al. 2009). Finally, miR-K12-11, a homologue to cellular miR-155, alters expression of BACH1, which leads to a variety of phenotypic changes including upregulation of HMOX1 (increased cell survival), upregulation of xCT (increased infection permissivity in macrophages and endothelial cells), and protection against apoptosis mediated by reactive nitrogen species (Gottwein et al. 2007; Qin et al. 2010; Skalsky et al. 2007).
MicroRNAs encoded by HHV-8 also play significant roles in growth signaling and angiogenesis. Transforming growth factor beta (TGFβ) signaling can be downregulated via the direct targeting of SMAD5 by miR-K12-11 (Liu et al. 2012); downregulation of TGFβ signaling induces cell proliferation. It has been noted that HHV-8+ B-cell lines have decreased levels of miR-155, the cellular homologue of miR-K12-11; miR-K12-11 may compensate for the limited levels of miR-155 in these cells (Skalsky et al. 2007). Inhibition of miR-K12-11 was found to derepress TGFβ signaling in HHV-8+ B cells (Liu et al. 2012). TGFβ signaling can be modulated by thrombospondin 1 (THBS1), a target of HHV-8 miRNAs miR-K12-1, miR-K12-3, miR-K12-6, and miR-K12-11 (Samols et al. 2007). Thrombospondin 1 is an antiangiogenic factor, and its downregulation leads to repressed TGFβ signaling (Samols et al. 2005). Thus, multiple viral microRNAs are capable of altering growth signaling and increasing angiogenesis to support the establishment of HHV-8-associated neoplasias. Additionally, viral miRNAs can alter the transcriptomes of endothelial cells and assist in cellular reprogramming. Both blood vessel endothelial cell (BEC) and lymphatic endothelial cell (LEC) expression markers are found in KS tissue, and the tissue can be reprogrammed toward the LEC or BEC fate under the appropriate stimuli. Specifically, miR-K12-6 and miR-K12-11 target the cellular transcription factor MAF (musculoaponeurotic fibrosarcoma oncogene homologue) (Hansen et al. 2010), which is found in LECs but not in BECs. Silencing of MAF by the microRNAs increases expression of BEC marker genes within the KS tissue. Targeting of MAF by miR-155 in CD4+ T cells has also been described (Rodriguez et al. 2007).
Several other studies have identified potential targets of viral microRNAs, though the validation of these targets is still ongoing. For example, Rbl2 (retinoblastoma-like protein 2) has been identified as a target of miR-K12-4-5p (Lu et al. 2010b). Rbl2 is an inhibitor of specific DNA methlytransferases (DNMT3a and 3b). Epigenetic changes have been observed following inhibition of Rbl2, but the consequences of these alterations are unclear (Lu et al. 2010b). Lastly, a large-scale study utilizing a luciferase assay approach has identified several potential targets of various HHV-8 miRNAs. These targets include proteins with roles in vesicle trafficking, spliceosome assembly, transcription regulation, and cell cycle progression (Dolken et al. 2010). Validation of these protein targets is needed and will further enhance the understanding of the roles of miRNAs in HHV-8 biology and pathogenesis.
3 Novel Virus–Host Interactions via Lytic Activities
3.1 Viral Interleukin-6 (vIL-6)
In contrast to its direct autocrine role in PEL pathogenesis, vIL-6 is believed to contribute to KS and MCD predominantly via paracrine signaling. Newly infected cells and those undergoing lytic replication express vIL-6 as an early gene product, and vIL-6 is rapidly induced following RTA expression in these cells (Sun et al. 1999). The majority of HHV-8-infected cells in the KS lesion remain latently infected, but small subsets of cells are lytically active. This minority of cells expresses lytic proteins, including vIL-6, vGPCR, and K1; these proteins ultimately enhance the expression of cellular inflammatory and angiogenic cytokines (Mesri et al. 2010). For example, vIL-6 can induce expression of VEGF (Aoki and Tosato 1999), considered to be a key contributor to KS development. VEGF and cytokines such as IL-6, CXCL8, and bFGF are secreted from lytically active cells and can further activate nearby latently infected and uninfected cells in a paracrine fashion. The secreted proteins modulate survival of HHV-8-infected cells, angiogenesis (predominantly through VEGF), and recruitment of uninfected cells to the lesion site. Inflammatory cytokines (including IL-6) and angiogenic factors have been proposed to play a role in the initial development of KS (Ensoli and Sturzl 1998).
In MCD patients, levels of IL-6 are substantially higher in affected lymph nodes when compared to unaffected nodes of the same patient (Yoshizaki et al. 1989). Additionally, disease severity correlates with IL-6 levels, and when affected lymph nodes are resected, IL-6 levels decrease (Yoshizaki et al. 1989). Similarly, cell lines derived from HIV positive KS patients express IL-6, and KS tissue produces increased amounts of IL-6 compared to normal tissue (Miles et al. 1990). IL-6 antisense oligonucleotides were found to suppress the growth of KS cells from HIV positive patients as well as the production of IL-6, and addition of exogenous, recombinant IL-6 was able to restore growth and proliferation (Miles et al. 1990). An additional study observed substantially elevated levels of IL-6 produced by malignant plasma cells from MCD patients though not from other B-cell tumors (Burger et al. 1994). At the time of these discoveries, the mechanism of disease-associated IL-6 dysregulation was not understood. More recent reports have demonstrated that vIL-6 can induce the expression of IL-6 and VEGF in some cell types (Aoki et al. 1999; Mori et al. 2000). It is likely that vIL-6 also contributes directly to MCD. Additionally, vIL-6 downregulates CCL2 and inhibits the infiltration of neutrophils during acute infection of B cells (Fielding et al. 2005).
3.2 Viral CC-Chemokine Ligands (vCCLs)
The three HHV-8 chemokines, vCCL-1, vCCL-2, and vCCL-3, are encoded by ORFs K6, K4, and K4.1, respectively (Moore et al. 1996; Neipel et al. 1997b; Nicholas et al. 1997; Russo et al. 1996). All are expressed early during the lytic cycle (Jenner et al. 2001; Paulose-Murphy et al. 2001). vCCL-1 and vCCL-2 are most closely related structurally to cellular chemokines CCL3 and CCL4, while vCCL-3 shares significant primary sequence similarity with a number of CC-chemokines. However, the properties of the v-chemokines are distinct from those of their cellular counterparts. With respect to receptor usage, vCCL-1 is an agonist for CCR8; vCCL-2 signals through CCR3, CCR8, and potentially also CCR5; vCCL-3 functionally targets CCR4 and XCR1 (Boshoff et al. 1997; Dairaghi et al. 1999; Luttichau 2008; Luttichau et al. 2007; Nakano et al. 2003; Stine et al. 2000). In addition, vCCL-2 binds several CCR- and CXCR-type chemokine receptors and CX3CR1 as a neutral (non-signaling) ligand and effectively inhibits cellular chemokine activity through these receptors (Chen et al. 1998; Crump et al. 2001; Dairaghi et al. 1999; Kledal et al. 1997; Luttichau et al. 2001). The nature of the v-chemokine-targeted receptors suggests that they may mediate immune evasion via Th2 polarization and blocking of leukocyte trafficking, as has been demonstrated for vCCL-2 in in vivo experiments (Chen et al. 1998; Weber et al. 2001).
Apart from these immune evasion properties, each of the v-chemokines has been demonstrated to promote angiogenesis, in part via induction of VEGF (Boshoff et al. 1997; Liu et al. 2001; Stine et al. 2000). Like vIL-6, HHV-8 vCCLs have the potential to promote KS pathogenesis via paracrine signaling and may also play roles in PEL, where VEGF has been implicated as an important factor based on data from murine studies (Aoki and Tosato 1999; Ensoli and Sturzl 1998; Haddad et al. 2008). Additional contributions of vCCL-1 and vCCL-2 to pathogenesis may include pro-survival signaling via CCR8, demonstrated in uninfected and HHV-8-infected endothelial cells (Choi and Nicholas 2008). vCCL-1 and vCCL-2 were also found to promote survival of PEL and murine cell lines (Liu et al. 2001; Louahed et al. 2003). Unlike most (non-secreted) viral proteins, the v-chemokines have the potential to function in a paracrine manner. Therefore, these chemokines may promote cell survival of latently infected and uninfected cells surrounding those supporting lytic replication, thus contributing to viral pathogenesis. Nonetheless, an important aspect of the pro-survival activities of vCCL-1 and vCCL-2 is the positive contribution to productive replication via autocrine signaling. The endogenously produced v-chemokines inhibit lytic cycle-induced apoptosis and increase virus yields in HHV-8-infected endothelial cultures (Choi and Nicholas 2008). This activity involves CCR8 signaling-dependent suppression of lytic cycle stress-induced pro-apoptotic protein Bim, a powerful inhibitor of productive replication.
3.3 Viral G Protein-Coupled Receptor (vGPCR)
While not unique to HHV-8, the vGPCR encoded by this virus is structurally and functionally diverged from other gamma-2 herpesvirus vGPCRs and is strongly implicated as a paracrine contributor to KS development (Cannon 2007; Nicholas 2005; Rosenkilde et al. 2001; Verzijl et al. 2004). HHV-8 vGPCR is unusual in its promiscuous functional association with three classes of Gα proteins (i, q, and 13) in addition to its direct association with and activation of the signaling protein SHP2 (Couty et al. 2001; Liu et al. 2004; Philpott et al. 2011; Shepard et al. 2001). Initial reports that vGPCR can function as a classical “autocrine” oncogene in in vitro and in vivo experimental systems employing vGPCR-transduced cell lines (Bais et al. 1998) implied that vGPCR may be expressed as a latent protein (enabling it to contribute directly to HHV-8 oncogenesis). However, no evidence of vGPCR latent expression has been forthcoming. Nonetheless, vGPCR can induce KS-like tumors in receptor-transduced mice, and this phenotype is supported despite only a minority of cells expressing vGPCR (Montaner et al. 2003; Yang et al. 2000). This can be explained by angiogenic, mitogenic, and inflammatory cellular cytokine induction via vGPCR signaling, leading to local endothelial activation, proliferation, and tumorigenesis (Montaner et al. 2004). As stated above, cytokine dysregulation is considered to be the principle driver of KS disease. vGPCR is known to activate key factors, such as VEGF, bFGF, CXCL8, and IL-6, that are found in KS lesions and believed to promote and be required for KS development and progression (Bais et al. 1998; Cannon et al. 2003; Pati et al. 2001; Schwarz and Murphy 2001). Thus, vGPCR produced in a small minority of spontaneously reactivating endothelial cells may induce levels of cellular cytokines sufficient to promote KS. It should be noted that in animal models, vGPCR-expressing cells are able to cooperate with cells expressing latency genes v-cyclin and/or vFLIP to increase the frequency of KS achieved with inoculated vGPCR+ cells alone (Montaner et al. 2003). This supports the notion that both autocrine latent and paracrine lytic activities can function together in HHV-8-associated neoplasia.
3.4 Viral Interferon Regulatory Factors (vIRFs)
HHV-8 vIRFs 1-4 are specified by the genomic region encompassing ORFs K9 to K11; an equivalent locus (encoding 8 vIRFs) has been identified only in the closely related rhesus rhadinovirus (Alexander et al. 2000; Cunningham et al. 2003; Moore et al. 1996; Searles et al. 1999). All four of the vIRFs are expressed during lytic replication, but vIRF-1 and vIRF-3 are also expressed in PEL latency (Pozharskaya et al. 2004; Rivas et al. 2001). Additionally, vIRF-1 transcripts have been detected in KS cells using reverse transcription-polymerase chain reaction techniques (Dittmer 2003). As outlined above (Sect. 2.5), vIRF-3 is required for PEL cell viability (Wies et al. 2008); however, vIRF-1 depletion has no detectable influence on PEL cell growth or survival in culture (Y. Choi and J. Nicholas, unpublished data). The vIRFs appear to function primarily to evade host cell defenses against de novo infection and virus productive replication, which trigger cellular IRF and interferon signaling cascades leading to cell cycle arrest and pro-apoptotic signaling (Lee et al. 2009a; Offermann 2007). The vIRFs counter these cellular signals in several ways. IRF5 and IRF7 functional dimerization and promoter association are antagonized by direct binding of vIRF-3 to these cellular factors, and vIRF-3 also inhibits IRF3 activity (Joo et al. 2007; Wies et al. 2009). vIRF-1 mediates transcriptional repression of IRF-targeted genes by blocking IRF-directed promoter recruitment of p300/CBP via competitive binding to the transcriptional coactivators (Burysek et al. 1999a; Li et al. 2000; Lin et al. 2001; Seo et al. 2000; Zimring et al. 1998). Transcriptional repression of IRF1-, IRF3-, and ISGF3-targeted genes is mediated by vIRF-2, in part by activation of caspase 3-mediated destabilization of IRF3 (Areste et al. 2009; Burysek et al. 1999b; Fuld et al. 2006). In addition, vIRF-2 and vIRF-3 directly target and/or inhibit dsRNA-activated PKR kinase activity; this suppresses PKR promotion of protein translation and inhibits interferon signaling (Burysek and Pitha 2001; Esteban et al. 2003). Interestingly, vIRF-3 associates with and stabilizes the pro-angiogenic transcription factor HIF-1α (Shin et al. 2008), and this could serve not only to promote endothelial cell survival but may also contribute to KS and PEL pathogenesis via induction of cytokines such as VEGF. vIRFs 1, 3, and 4 have been shown to inhibit p53 activity via (1) direct binding to the tumor suppressor (vIRF-1 and vIRF-3), (2) through interaction with p53-phosphorylating and p53-activating ATM kinase (vIRF-1), or (3) via stabilization of MDM2 (vIRF-4), which promotes ubiquitination and proteasomal degradation of p53 (Lee et al. 2009b; Nakamura et al. 2001; Rivas et al. 2001; Seo et al. 2001; Shin et al. 2006). Inhibitory interactions of vIRF-4 with deubiquitinase HAUSP have been reported to be involved in p53 destabilization (Lee et al. 2011). Therefore, p53 represents a major vIRF target that is important to control for efficient virus productive replication to occur. vIRF-4 also binds to CSL, the target of Notch, but the significance of this interaction is unclear (Heinzelmann et al. 2010).
vIRF-1 associates with and inhibits the activities of other cellular proteins involved in innate cellular responses to infection and the promotion of apoptosis. These targets include retinoic acid/interferon induced protein GRIM19 and TGFβ receptor-activated transcription factors Smad3 (tumor suppressor) and Smad4 (co-Smad) (Angell et al. 2000; Ma et al. 2007; Seo et al. 2002, 2005). More recently, vIRF-1 has been found to bind directly to members of the so-called BH3-only protein (BOP) family (Choi and Nicholas 2010; Choi et al. 2012). BOPs are Bcl-2-related proteins that function to promote apoptosis either via inhibitory interactions with pro-survival members of the Bcl-2 family or by direct activation of apoptotic executioners Bax and Bak (Kuwana et al. 2005; Willis and Adams 2005). A region of vIRF-1 comprising residues 170-184 (BOP-binding domain, BBD) interacts with BOP BH3 domains, required for pro-apoptotic activities of these proteins, thereby mediating BOP inhibition (Choi and Nicholas 2010; Choi et al. 2012). vIRF-1 BBD, a predicted amphipathic α-helix, resembles the Bid BH3-inhibitory BH3-B domain of Bid and represents only the second example of a BH3-B-type BH3-inhibitory domain and therefore a novel viral mechanism of apoptotic inhibition. BBD-mediated interactions with BOPs are functionally important, as indicated by the following findings: (1) BBD-mutated vIRF-1 is less active than wild-type vIRF-1 in promoting productive replication and inhibiting apoptosis in lytically infected endothelial cells; (2) vIRF-1:BOP-disrupting BBD peptide causes significant inhibition of virus production in these cells; and (3) depletion of vIRF-1-targeted BOPs Bim or Bid leads to substantial increases in replicative titers (Choi and Nicholas 2010; Choi et al. 2012).
In summary, a wealth of published data indicates that the vIRFs of HHV-8 represent an effective panel of antiapoptotic proteins that promote productive replication via interactions with an important group of cellular proteins involved in host cell defenses against infection (summarized in Fig. 2).
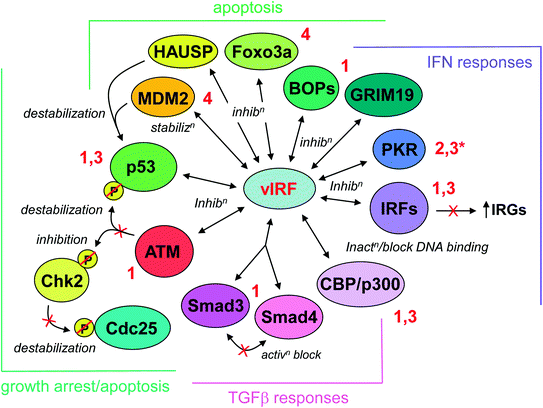
Fig. 2
Summary of inhibitory interactions of vIRFs with cellular proteins. The particular vIRFs interacting with each target are indicated by the adjacent numbering (red lettering), and the effect of each interaction is indicated (italics). The activities of vIRF interactions are grouped into four general, overlapping biological categories, as indicated by the brackets and color coding. The vIRF:protein interactions and their significance are discussed fully in the text. (*Functional inhibition; no physical interaction detected)
3.5 K7-Encoded Viral Inhibitor of Apoptosis (vIAP)
In common with other gammaherpesviruses, HHV-8 specifies a homologue of cellular Bcl-2 proteins. However, HHV-8 encodes an additional BH-like domain-containing antiapoptotic protein that is a homologue of cellular survivin. This HHV-8 protein is referred to as K7 (corresponding to the encoding ORF) or viral inhibitor of apoptosis (vIAP). K7/vIAP is a membrane-associated protein that contains a putative mitochondrial-targeting signal and localizes to mitochondria, ER, and possibly other membranes as well (Feng et al. 2002; Wang et al. 2002). It has been reported that K7/vIAP binds to cellular Bcl-2 via its C-terminal BH2-like domain and to activated (proteolytically cleaved) caspase 3, bridging the two proteins and functionally inhibiting caspase 3 proteolytic activity (Wang et al. 2002). Interaction with and inhibition of terminal caspase 3 in the apoptotic cascade is analogous to the activities of cellular IAPs, which include survivin, XIAP, and cIAPs 1 and 2. However, the interaction between K7/vIAP and Bcl-2 is a property not reported for its cellular counterparts. The functional and biological significance of this interaction remains to be determined. Nonetheless, K7/vIAP is able to inhibit pro-apoptotic signaling in transfected cells treated with agents such as Fas antibody and TNF-α, indicating its potential to act as a promoter of lytic replication via its pro-survival activity during lytic cycle-induced stress (Wang et al. 2002). K7/vIAP also interacts with calcium-modulating cyclophilin ligand (CAML), which regulates intracellular calcium ion concentrations (Bram and Crabtree 1994; Feng et al. 2002). The functional significance of this interaction is evident from the ability of wild-type but not CAML binding-refractory K7/vIAP to inhibit chemically-induced mitochondrial depolarization (i.e., apoptotic triggering) in transfected cells (Feng et al. 2002). Thus, in addition to its inhibitory binding to caspase 3, K7/vIAP appears to mediate apoptotic inhibition via CAML interaction. K7/vIAP also interacts with the cellular protein PLIC1 (protein-linking integrin-associated protein and cytoskeleton-1, also called ubiquilin), which associates with ubiquitin-conjugated proteins to inhibit their proteasomal degradation (Feng et al. 2004; Kleijnen et al. 2000; Wu et al. 1999). K7/vIAP appears to antagonize PLIC1 activity, thereby destabilizing ubiquitinated proteins, as demonstrated for p53 and NF-κB-inhibitory IκB (Feng et al. 2004). Together, the inhibitory interactions between K7/vIAP and cellular proteins PLIC1, caspase 3/Bcl-2, and CAML may promote cell survival during lytic replication and consequently enhance the efficiency of virus production.
4 Terminal Membrane Proteins
4.1 K1/Variable ITAM-Containing Protein (VIP)
The K1 ORF of HHV-8 is located at the left end of the genome and is collinear with other gammaherpesvirus genes encoding signaling membrane proteins. These include saimiri transformation-associated protein (STP) of herpesvirus saimiri (HVS), latency membrane protein-1 (LMP-1) of EBV, and the K1-homologous R1 receptor of rhesus rhadinovirus (Albrecht et al. 1992; Damania et al. 1999; Kaye et al. 1993; Lagunoff and Ganem 1997; Murthy et al. 1989). The K1 protein is a type I transmembrane signaling protein containing a functional immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic C-tail (Lagunoff et al. 2001; Lee et al. 1998a). Sequencing of K1 in different HHV-8 isolates identified an unusual degree of amino acid sequence variability in the extracellular regions of the encoded proteins (Nicholas et al. 1998; Zong et al. 1999), hence the naming of the K1 protein as variable ITAM-containing protein (VIP). While the functional significance of this variability has not been established, the K1 locus has served as a basis of epidemiological studies of HHV-8 strain distribution and infectivity (Hayward and Zong 2007; Mbulaiteye et al. 2006; Whitby et al. 2004). Based initially on the genomic position of K1 and subsequently on the constitutive signaling and transforming properties of K1/VIP, the protein was implicated as a potential contributor to HHV-8 pathogenesis. K1/VIP, like RRV R1, was able to substitute functionally for the positionally equivalent ORF1/STP of HVS in in vivo tumorigenesis assays and to promote cell growth and transformation in isolation (Lee et al. 1998b; Prakash et al. 2002). K1/VIP activation of the AKT pathway and consequent activation of mTOR (associated with cell growth) and the inactivation of pro-apoptotic GSK3, Bad, and forkhead transcription factors have been implicated in these activities (Tomlinson and Damania 2004; Wang et al. 2006). However, as K1 appears to be expressed primarily or exclusively during lytic replication (Jenner et al. 2001; Lagunoff and Ganem 1997; Nakamura et al. 2003; Paulose-Murphy et al. 2001), its potential role in KS, PEL, and MCD may be restricted to paracrine effects of K1/VIP-induced cellular cytokines (see below) rather than direct effects suggested by initial functional analyses. While immunodetection of K1/VIP in KS and MCD tissues has been reported, this has not been associated with latently infected cells (Lee et al. 2003a; Wang et al. 2006). It should be noted, however, that in situ detection of K1 transcripts in some KS cells lacking detectable lytic marker (major capsid protein) mRNA expression suggests the possibility that K1/VIP may be expressed in at least some latently infected KS cells (Wang et al. 2006).
K1/VIP recruits and activates Src-family kinases, PI3K, and PLCγ to mediate signal transduction via several pathways by ligand-independent, constitutive signaling (Lagunoff et al. 2001; Lee et al. 2005, 1998a; Samaniego et al. 2001; Tomlinson and Damania 2004). It has been suggested that K1/VIP may contribute to HHV-8-associated disease, especially KS, by induction of cellular cytokines. K1/VIP-induced cytokines include pro-inflammatory IL-1β, IL-6, and GM-CSF and angiogenic factors VEGF, CXCL8, and bFGF (Lee et al. 2005; Prakash et al. 2002; Samaniego et al. 2001; Wang et al. 2006). Contributions of the HHV-8 receptor to pathogenesis via cellular cytokine induction could theoretically occur during lytic replication or during latency. In KS, PEL, and MCD, small proportions of cells support lytic reactivation, enabling lytically expressed proteins, like K1, to exert paracrine influence on surrounding latently infected and uninfected cells (Aoki et al. 2003; Aoki and Tosato 2003; Ensoli et al. 2001).
4.2 K15-Encoded Membrane Protein
The K15-encoded protein in its full-length form is a twelve-transmembrane domain-containing signaling receptor. Like K1, K15 may play a role in pathogenesis via cytokine dysregulation, and it could conceivably contribute to malignant disease through pro-survival signaling during latency. Transcripts from the K15 locus are expressed predominantly during the lytic cycle, but some K15 products have been detected in resting (latent) PEL cultures (Choi et al. 2000; Glenn et al. 1999; Sharp et al. 2002). The issue is complex because the K15 primary transcript contains eight exons and can be differentially spliced; the resulting mRNAs and encoded proteins may be expressed differently based on cell type and whether the virus is in the latent or productive phase. All forms of K15 contain C-terminal protein sequences with functional signaling motifs (see below), but the protein isoforms differ in their complement of transmembrane domains. K15 transcripts and a 23-kDa protein isoform have been detected during latency in PEL cells, but K15 mRNA levels are induced considerably upon lytic reactivation (Choi et al. 2000; Glenn et al. 1999; Sharp et al. 2002; Tsai et al. 2009). Full-length K15 protein has been detected in HHV-8 bacmid-containing HEK293 cells, though only after lytic induction with butyrate treatment (Brinkmann et al. 2007). However, the full-length protein has not been observed in cells naturally infected with HHV-8. The ability of immediate early, lytic trigger protein RTA to activate transcription from the K15 promoter is consistent with predominant lytic expression of K15 (Wong and Damania 2006). Nonetheless, uncertainty remains regarding the expression characteristics of K15 transcripts and proteins and whether K15 receptor signaling could contribute to latency and HHV-8 neoplasia in an autocrine manner.
Signaling motifs in the cytoplasmic tail of all K15 isoforms and in both M and P allelic protein types (Poole et al. 1999) include two SH2- and single SH3- and TRAF-binding sites (Brinkmann et al. 2003; Choi et al. 2000; Glenn et al. 1999). SH2 binding-mediated interactions with Src-family kinases occur via the Y481EEV motif, which is the primary site of K15 phosphorylation (Brinkmann et al. 2003; Choi et al. 2000). This, together with the SH3-binding sequence (PPLP), leads to inhibition of B-cell receptor (BCR) signaling. PPLP-motif interactions with intersectin 2 (endocytic adaptor protein) and with Src kinases (such as Lyn and Hck) are important for this activity (Lim et al. 2007; Pietrek et al. 2010). BCR inhibition by the K15 receptor parallels that of the collinearly encoded LMP-2 of EBV, and each is likely to promote latency by inhibiting lytic cycle reactivation promoted by BCR signaling. The Y481EEV motif has been implicated in the activation of NF-κB and mitogen-activated protein kinases (MAPKs) ERK and JNK, which occurs after Y481 phosphorylation (Brinkmann and Schulz 2006; Choi et al. 2000; Pietrek et al. 2010). Interaction of the K15 receptor with TRAFs 1, 2, and 3 is likely to contribute to NF-κB and MAPK signaling (Brinkmann and Schulz 2006; Glenn et al. 1999). The second tyrosine-containing motif, Y432ASI, is not detectably phosphorylated, and its significance is uncertain. However, its interaction with apoptotic regulatory protein HAX-1 and ER and mitochondrial colocalization of HAX-1 with K15 suggest that the viral receptor may function to promote cell survival via this motif (Sharp et al. 2002).
Examination of the downstream effects of K15 signaling has provided insight into possible functions of the receptor in HHV-8 biology and its potential contributions to viral pathogenesis. In addition to suspected antiapoptotic activity via interaction with HAX-1, K15 can induce the expression of several antiapoptotic genes, including A20, Bcl–2A1, Birc2, and Birc3 (Brinkmann et al. 2007). Induction of these genes may help promote cell survival during the lytic cycle, further enhancing productive replication. If K15 is expressed during latency, its pro-survival signaling could contribute to prolonged latent cell viability and maintenance of latency pools in vivo and to viral pathogenesis. On the other hand, the observed induction of cellular cytokines in K15-transduced cells suggests a mechanism by which K15 can affect surrounding cells (latently infected and uninfected) by paracrine signaling from lytically infected cells. K15-expressing latently infected cells could exert similar effects on the microenvironment. Cytokines induced by K15 receptor signaling include IL-6, CCL2, CXCL3, and CXCL8; each of these possesses angiogenic activity and has been implicated in KS pathogenesis (Brinkmann et al. 2007; Caselli et al. 2007; Ensoli and Sturzl 1998). It is intriguing that K15 also induces expression of genes representing downstream targets of angiogenic VEGF signaling. This would clearly implicate K15 as an autocrine contributor to pathogenesis should the receptor be expressed during latency, but such activity of K15 could also contribute to productive replication. Angiogenic targets of K15 include Dscr–1 and Cox–2 (Brinkmann et al. 2007). It is notable that Cox-2 has been reported to be induced during de novo and subsequent latent infection of endothelial cells by HHV-8, and Cox-2 is important for the production of several inflammatory and angiogenic factors (Sharma-Walia et al. 2010). Thus, pro-survival and paracrine-mediated pro-angiogenic roles of K15 in HHV-8 lytic replication and pathogenesis seem likely, and there is potential for autocrine activity via pro-survival signaling contributing to latency and neoplasia if the receptor is latently expressed.
5 Summary
The discovery and study of HHV-8 has provided the opportunity to identify unique virus-specified activities encoded by proteins either not previously known among viruses or those not previously investigated or characterized in depth in other viral systems. HHV-8 has also provided a model for the identification and characterization of viral miRNAs, a new area of research that has provided unique and important insights into viral manipulation of host cell processes as part of normal virus biology and potentially in viral pathogenesis. The properties of the characterized protein and miRNA players in these processes have been described in detail, and several key points emerge. First, the notion that only autocrine, latent viral activities are relevant to virus-associated neoplasia needs revision, certainly in the case of KS and possibly for PEL and MCD. Paracrine factors (viral and/or cellular) produced during lytic replication can contribute to pro-proliferative, pro-survival, and other functions of pathogenic relevance. The latent and lytic viral proteins implicated in HHV-8 pathogenesis and their likely autocrine and paracrine contributions to disease are summarized in Fig. 3. Secondly, “lytic” and “latent” classifications for viral products are not as distinct as once thought. For example, vIL-6, vIRF-1, vIRF-3, K1, and K15 are clearly expressed maximally during productive replication, but there is evidence for their expression during latency as well in some cell types. Furthermore, it is notable that latently expressed vIL-6 and vIRF-3 are of demonstrable importance for PEL cell growth and viability. A third key point is that the virally encoded chemokines vCCL-1 and vCCL-2, while secreted during productive replication and thought to function to promote virus production via paracrine effects on the microenvironment (most notably to evade host immune responses), can also act directly on the cells in which they are produced to enhance virus production via antiapoptotic signaling. Such direct pro-replication activity has also been demonstrated for vGPCR. For the v-chemokines and vGPCR, it is possible that induced cellular cytokines may serve similar and/or additional activities to promote virus replication in an autocrine fashion as well as having broader effects on the host microenvironment. Finally, several of the HHV-8 proteins have multiple interactions with a broad range of host factors, a point summarized in Table 2 and exemplified by LANA and vIRF-1. Thus, viral proteins can have extraordinarily multifaceted activities via numerous protein interactions, and detailed characterization of these interactions and their functional effects is important for understanding their individual and combined contributions to virus biology and pathogenesis. Such characterization can potentially provide the basis for the development of new antiviral and therapeutic drugs designed to interfere with specific virus–host interactions of critical importance for viral replication or pathogenesis. This chapter has attempted to overview the various novel HHV-8–host interactions and related activities that contribute to these processes and that could perhaps be targeted in this way. The interactions described also illustrate the breadth and complexity of virus–host interactions and suggest that similar activities and mechanisms may be operative in other viral systems.
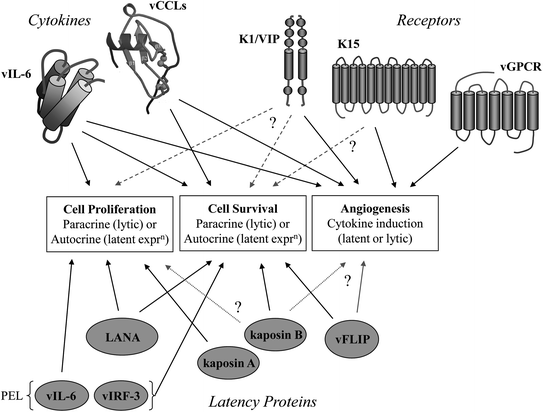
Fig. 3
Overview of potential contributions of HHV-8 proteins to virus-associated neoplasia. The general activities of relevance to HHV-8 malignant pathogenesis are indicated. Both latent and lytic proteins have the potential to contribute to disease, via autocrine and paracrine activities, respectively. Latent expression of signaling receptors encoded by ORFs K1 and K15 has so far not been demonstrated conclusively. Therefore, whether the autocrine activities detected experimentally (dotted lines) reflect direct roles of the receptors in HHV-8 pathogenesis remains uncertain (?). However, contributions of K1 and K15 receptors could contribute to neoplasia via induction of mitogenic, pro-survival, and angiogenic secreted cellular factors. This is also true of the viral chemokine receptor, vGPCR. The viral cytokines (lytic) are secreted and can act by both autocrine and paracrine mechanisms to influence cell growth and survival. These activities can promote virus replication (by autocrine signaling) in addition to contributing to viral pathogenesis (via paracrine effects). Latent expression of vIL-6 in PEL is likely to contribute to pathogenesis, mainly via intracrine signaling. vIRF-3 is also expressed during latency in PEL and like vIL-6 promotes PEL cell viability. The latency proteins have the potential to contribute to HHV-8-associated malignancies by direct autocrine effects on cell proliferation and survival by mechanisms typical of oncogenes and tumor suppressors (see the text). Kaposin B and vFLIP have the potential to function as promoters of cell proliferation and/or angiogenesis, but these functions have not been demonstrated directly (?)
Table 2
Virus–host protein interactions and their activities
Protein | Class | Target(s) | Activity/Function | References |
|---|---|---|---|---|
LANA | Latent, replication | p53 | Pro-survival | Friborg et al. (1999) |
Angiogenin/annexin A2 | Pro-survival | Paudel et al. (2012) | ||
pRb | Pro-mitogenic | Radkov et al. (2000) | ||
GSK3β | Pro-mitogenic | Fujimuro et al. (2003) | ||
c-Myc | Pro-mitogenic | Liu et al. (2007b) | ||
Histones H2A/B | Viral genome-chromosome tethering | Barbera et al. (2006) | ||
DNMT3a | Transcriptional repression | Shamay et al. (2006) | ||
SUV39H1 | Transcriptional repression | Sakakibara et al. (2004) | ||
mSin3/SAP30/CIR | Transcriptional repression | Krithivas et al. (2000) | ||
K13/vFLIP | Latent, signaling | IKKα/β | NF-κB activation/survival | Matta et al. (2007) |
TRAF2/IKKγ? | NF-κB/Jnk activation | Guasparri et al. (2006) | ||
Procaspase-8 | Inhibition of caspase activation/survival | Belanger et al. (2001) | ||
Kaposin A | Latent, signaling | Cytohesin-1, Septin 4 variant | GTPase-mediated signal transduction, activation of several signaling kinases; roles in survival/proliferation? | |
Kaposin B | Latent, mRNA reg. | MK2 kinase | MK2 activation, stabilization of ARE-containing mRNAs (e.g. IL-6, PROX1); potential pro-survival and/or mitogenic activity, endothelial reprogramming | McCormick and Ganem (2005) |
vIL-6 | Ligand, cytokine | gp130/gp80 | STAT/MAPK activation; proliferation/survival, pro-inflammatory/angiogenic | |
VKORC1v2 | Proliferation/survival (PEL), unknown mechanism | Chen et al. (2012) | ||
vCCL-1 | Ligand, chemokine | CCR8 | Agonist, Th2 polarization; pro-survival, promotes virus replication; pro-angiogenic | Dairaghi et al. (1999),
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|