Biology and Genetics of Adult Male Germ Cell Tumors
Darren R. Feldman
Jane Houldsworth
George J. Bosl
R.S.K. Chaganti
Germ cell tumors (GCTs) arise by transformation of a germ cell that during its normal lifespan undergoes a series of proliferation and differentiation events that culminate in the formation of gametes (1). They are the most common malignancy in young adult males of ages 15 to 40, presenting predominantly in the testis (2). Uniquely, transformed germ cells exhibit the potential to initiate molecular pathways resembling in part those occurring during normal human development, as evidenced by the array of histologies observed within tumor specimens (3). These histologies range from seminomas that resemble undifferentiated germ cells, to nonseminomas that resemble early zygotic differentiation (embryonal carcinoma), embryonic somatic differentiation (teratoma), and extraembryonic differentiation (yolk sac tumor, choriocarcinoma). Occasionally, within differentiated components of teratoma, further malignant transformation can occur leading to the appearance of a non-germ cell malignancy (3). Clinically, approximately 95% of newly diagnosed patients are cured, including 70% to 80% of advanced cases following treatment with cisplatin-based chemotherapy (4). Thus, GCTs represent an ideal system in which the molecular mechanisms underlying this exquisite sensitivity can be studied. In this chapter, a review is undertaken of the current state of knowledge of the biology and genetics of germ cell transformation, differentiation, and response to cisplatin.
LIFESPAN OF THE NORMAL HUMAN MALE GERM CELL
During gastrulation of the developing embryo, primordial germ cells (PGC)—the embryonic precursors of gametes—are first recognized in the mouse as a cluster of alkaline phosphatase-positive cells that arise from the embryonic ectoderm (5). Upon proliferation, the PGCs migrate to the mesoderm of the primitive streak, to the endoderm via the base of the allantois, through the hindgut endoderm, and finally to the genital ridge. PGCs isolated at this stage are the source of embryonic germ cells that display pluripotentiality in vitro (6). During the initial formation of the genital ridge, the male and female gonads appear identical. However, shortly thereafter, stromal expression of the SRY gene on chromosome Y leads to testicular development; in the absence of SRY expression, development defaults to female gonadogenesis. In the male genital ridge, the PGCs, along with Sertoli cells, are incorporated into the seminiferous cords and, along with other somatic cells such as Leydig cells, form the developing gonad by the seventh or eighth week of human fetal life (7). Once enclosed within the seminiferous tubules, the PGCs cease to proliferate and undergo differentiation into gonocytes. In the neonatal testis, gonocytes are easily recognized as large round cells centrally located within tubules (7). After birth, the gonocytes reinitiate proliferation, migrate to the tubular wall, and mature into undifferentiated Type A spermatogonia usually within 2 to 3 months of age. In the prepubertal testis and culminating at the onset of puberty induced by gonadotrophins, spermatogonial proliferation and maturation into Type B spermatogonia are evident with the onset of meiosis leading to the appearance of fully differentiated spermatozoa in the tubules (8,9). In the pubertal and young adult testis, Type A dark type (d) and Type A pale type (p) spermatogonia usually exist as single, pairs, and chains of cells; by analogy with nonprimates, the single spermatogonia represent stem cells. Type Ad spermatogonia rarely divide and are thought to represent a reserve of stem cells in the case of germ cell loss. Type Ap spermatogonia actively divide, possibly representing renewing stem cells, though the true self-renewing stem cell in humans is still the subject of debate (8,9). Type B spermatogonia undergo only one mitotic division in humans that leads to primary spermatocytes. Premeiotic DNA replication follows with entrance into meiosis comprising a protracted prophase (leptotene, zygotene, pachytene, diplotene, and diakinesis). Following two sequential meioses (I and II), four haploid gametes are left. These round spermatids then undergo spermiogenesis, maturing into spermatozoa. In humans, the entire cycle of spermatogenesis takes approximately 74 days, with waves of meiosis taking 26 days (7,9). Spontaneous apoptosis is normal in the adult male testis, occurring mostly in primary spermatocytes, spermatids, and few spermatogonia (9). In addition, apoptosis is thought to be involved in the control of the stem cell pool size, with some evidence for a mechanistic role of BCL2 family members (8).
Understanding of the genetic control of germ cell development and differentiation is limited in humans, based primarily on genetic disorders that lead to infertility (10). Microdeletions or translocations of the Y chromosome, in particular involving the SRY and AZF loci, imbalance of X and Y chromosome number, mutations of genes associated with gonadotrophin production, follicle-stimulating hormone and luteinizing hormone function, and androgen synthesis and function can lead to oligospermia or azoospermia (9,10). It has been reported that multiple genes are expressed in specific stages of the germ cell lifespan and that their roles have often been indicated or confirmed in mutant or transgenic mice. With the advent of microarray-based expression analyses, the expression profiles of isolated populations of germ cells at different times during their lifespan have been performed, though the role of these genes in human germ cell development remains to be confirmed.
During their lifespan, germ cells undergo genetic and epigenetic modifications. Recombination between paired chromosomes during prophase of meiosis I is a specialized process for which the underlying enzymatic and structural machinery and regulatory checkpoints are being defined predominantly in lower eukaryotes, though often with homologs in mammals
(11). Germ cells are also responsible for the inheritance of the paternal-specific epigenetic imprint of specific subsets of genes. Analysis of the methylation of CpG dinucleotides during human spermatogenesis has revealed that the methylation pattern inherited from the parents is initially erased in fetal spermatogonia prior to entry of meiosis (12). The parentspecific imprint is reestablished in adult spermatogonia and then maintained in spermatocytes and spermatozoa. Errors can occur in both forms of genomic modification, leading to miscarriage and birth defects.
(11). Germ cells are also responsible for the inheritance of the paternal-specific epigenetic imprint of specific subsets of genes. Analysis of the methylation of CpG dinucleotides during human spermatogenesis has revealed that the methylation pattern inherited from the parents is initially erased in fetal spermatogonia prior to entry of meiosis (12). The parentspecific imprint is reestablished in adult spermatogonia and then maintained in spermatocytes and spermatozoa. Errors can occur in both forms of genomic modification, leading to miscarriage and birth defects.
PATHOBIOLOGY OF MALE GERM CELL TUMORS
Transformation of male germ cells gives rise to tumors that display an array of histopathologies in the adult (Fig. 30.1). GCTs are broadly classified into two groups: seminomas and nonseminomas (3). The first recognizable lesion in the testis is intratubular germ cell neoplasia (ITGCN), where transformed germ cells are evident within the seminiferous tubules. ITGCN is generally accepted as the precursor of all invasive GCTs, although morphologically it more closely resembles seminomas than nonseminomas (3). Seminomatous GCTs retain the morphology of undifferentiated spermatogonial germ cells that frequently express protein markers of germ cells early in development, such as placental alkaline phosphatase, KIT, RET (GDNF receptor), and POU5F1 (OCT3/OCT4) (13,14,15). They exhibit low mitotic and apoptotic indices, as well as low metastatic potential, and are generally cured by orchiectomy alone or a combination of orchiectomy and radiation therapy (2,3). Nonseminomas display a variety of histologies that resemble different stages and patterns of embryonic and extraembryonic differentiation that normally occur during human development. Embryonal carcinoma resembles zygotic cells early in embryonic development, consistent with the pluripotentiality displayed by embryonal carcinoma-derived cell lines in vitro (16). This histology displays the highest mitotic and apoptotic indices of all GCT components (3,17). Teratomas display multiple patterns of somatic differentiation normally seen in the three developing germ layers of the embryo. Components resembling incomplete differentiation (immature teratoma) or well-differentiated tissues (mature teratoma) are observed that tend to have both low mitotic and apoptotic indices (3,17). Mature teratomas on occasion will undergo malignant transformation of one of the histologic components, requiring subsequent tailoring of treatment to the malignant tissue counterpart (18). Yolk-sac tumors and choriocarcinomas express alpha-fetoprotein and human chorionic gonadotrophin, respectively, and exhibit morphologies resembling extraembryonically differentiated tissues (3). Nonseminomas are cured in the majority of cases by combining surgery and cisplatin-based chemotherapy, although the relative resistance of teratomatous components to chemotherapy is well documented (2). With the exception of seminomas, GCTs of pure histology are rare, with the majority being a mixture of nonseminomatous histologic components. A combination of seminomatous and nonseminomatous components is also frequent.
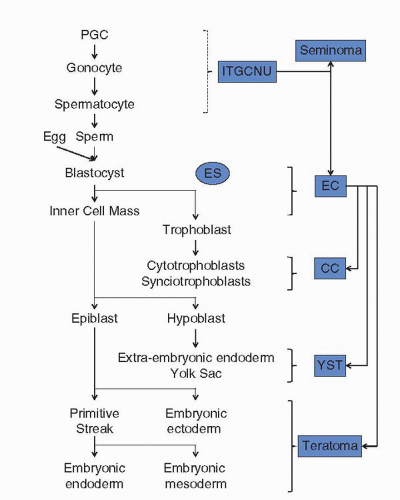 FIGURE 30.1. Model of normal germ cell and embryological development with notation of the stage that most closely correlates biologically with particular germ cell tumor histologies. |
GENETIC BASIS OF ADULT MALE GERM CELL TRANSFORMATION
In 1982, Atkin and Baker (19) first reported the detection of an isochromosome of the short arm of chromosome 12 [i(12p)] in GCTs using conventional karyotypic analysis of metaphase chromosomes. This chromosomal abnormality has subsequently been recognized in 85% of GCTs (20). Using a 12p painting probe, fluorescence in situ hybridization (FISH) of GCTs that do not exhibit this marker chromosome revealed the presence of tandem duplications of 12p either in situ or in other chromosomes (21). Thus, the presence of extra copies of the short arm of chromosome 12 is diagnostic of GCTs, and FISH analysis for this marker in interphase and metaphase tumor cells is used routinely for diagnostic purposes. This assay has been useful in the identification of some mediastinal tumors of uncertain histogenesis as GCTs, permitting appropriate treatment choices (22). Several studies have indicated that i(12p) is evident as early as in ITGCN, yet others have indicated that the appearance of this marker is associated with tumor invasion out of the tubules (23,24,25,26).
Gain of the entire 12p chromosome arm containing more than 400 genes has made identification of a single responsible gene quite challenging. While several candidate genes have been reported, it is now commonly believed that a group of genes acting in concert, as well as additional chromosomal abnormalities, may be necessary for invasive GCT to develop. CCND2 was initially identified as a possible 12p lineagesurvival oncogene in GCTs (27). This D-type cyclin, along with the cyclin-dependent kinases 4 and 6, regulates transition of the G1-S phase of the cell cycle (28). Perturbations in cell cycling due to overexpression of these cyclins have been noted in multiple tumor systems and the oncogenic capacity confirmed in transgenic murine models. For GCTs, cyclin D2 is aberrantly expressed in the transformed germ cells of ITGCN, with elevated expression also detected in seminoma and embryonal carcinoma (27). With somatic differentiation, the levels of cyclin D2 paralleled those detected in the normal tissue counterpart. Cyclin D2 expression was only rarely noted in spermatogonial cells of the normal adult testis, but
examination of murine neonate testis showed that mitotically dividing spermatogonial cells express cyclin D2 (1). Thus, analogous to cyclin D1 in breast cancer (29), increased 12p copy number may lead to aberrant expression of cyclin D2 in a germ cell that has previously expressed this D-type cyclin during its development but now with oncogenic potential.
examination of murine neonate testis showed that mitotically dividing spermatogonial cells express cyclin D2 (1). Thus, analogous to cyclin D1 in breast cancer (29), increased 12p copy number may lead to aberrant expression of cyclin D2 in a germ cell that has previously expressed this D-type cyclin during its development but now with oncogenic potential.
Microarray-based expression studies identified a cluster of stem cell-associated genes, including STELLAR, NANOG, and GDF3 that map to a 200 kb region at 12p13.31 and exhibit elevated expression exclusively in seminomas and embryonal carcinomas (30). It is upregulation of these genes as a result of gain of 12p that may underlie the stem cell-like or undifferentiated germ cell-like phenotype of seminomas and embryonal carcinomas. Consistent with reduced levels of expression of this cluster of genes in somatic and extraembryonic histologic components is their downregulation following induction of differentiation of embryonal carcinoma cell lines with resultant loss of pluripotency (30,31,32). Gains in 12p13.31 have also been observed in late passage ES cell lines, suggesting that gain of this region may be critical to acquisition and maintenance of pluripotency (33). Together with other candidate genes overexpressed in GCTs and mapped to 12p such as GLUT3, a gene involved in glucose transport, it would seem that gain of 12p has a multifunctional role in germ cell tumorigenesis (30).
Chromosomal comparative genomic hybridization (CGH) analyses of GCTs have indicated that some GCTs (predominantly seminomas) exhibit a high-level amplification of the 12p11-12.2 region, in addition to gain of the entire short arm (34,35). Recently, molecular cytogenetic and global genomic screening methods have been employed to delineate the amplicon and identify potential candidates for the target gene(s) with inconclusive results (36,37,38). It is unclear whether this region contains a responsible protooncogene that drives the gain of 12p observed in all GCTs or represents a subregional amplification involved predominantly in the progression of seminomas.
Extensive cytogenetic and molecular genetic studies have described other frequent chromosomal abnormalities in adult GCTs (24,25,35,39,40). Chromosomal CGH performed on microdissected ITGCN revealed gain of genetic material derived from chromosomes 1, 5, 7, 8, 12q, and X and loss of chromosome 18 material in these preinvasive lesions (24,25). Adjacent invasive tumors exhibited additional sites of gain including chromosomes 2, 3, 4, 6, 13q, 14q, 17q, 18q, 20, and 21 and additional losses including chromosomes 1p, 4, 6q, 9, 11, 13q, and 19. For 17q, microarray-based expression studies have provided evidence for GRB7 and JUP as the target genes of the regional gain (41). Conventional karyotype analysis revealed that, overall, GCTs are hypertriploid in chromosomal content and display recurrent chromosomal abnormalities, some of which are associated with histological subtype (39,40). The most frequently observed nonrandom chromosomal aberrations were deletion/rearrangement of 12q, 1p32-36, and 7q, along with deletion at 6q13-25. Breakpoints involving 1p32-36 and 7q11.2 were associated significantly with a teratomatous histology, and 1p22 with yolk sac tumors.
Comprehensive loss of heterozygosity analyses of the chromosomal complement of GCTs have indicated a number of sites that may harbor tumor suppressor genes (42). These include regions containing known tumor suppressor genes (RB1, DCC, and NME), regions previously identified as containing putative tumor suppressor genes (1p, 3p, 5q, 9p, 10q, 11p, 11q, and 17p), and several novel sites (1q, 2q, 3q, 5p, 9q, 12q, 18p, and 20p). Molecular genetic analysis of the long arm of chromosome 12 has provided evidence for two putative tumor suppressor genes mapped at 12q13 and 12q22, and it narrowed the minimally deleted regions down to <1Mbp (43,44). For 5q, three common sites of loss have now been indicated (5q14, 5q21, and 5q34-qter), although the corresponding putative tumor suppressor genes remain to be identified (45). Silencing of expression of tumor suppressor genes can also be achieved by hypermethylation of the promoter of the respective gene. Examination of the methylation status and expression of candidate tumor suppressor genes in GCTs has been carried out by several investigators. Overall, seminomas displayed a much lower level of promoter methylation than nonseminomas, reflecting an epigenetic modification associated with differentiation rather than tumorigenesis (46,47,48,49). In addition, methylation of a gene did not always correlate with expression. However, it was apparent in two independent studies that expression of MGMT at 10q was regulated by methylation, implicating it as the candidate tumor suppressor gene at this locus (47,49). For the candidate on 3p, RASSF1A, the methylation status and associated expression were inconsistent between studies (46,47).
Mutations of KRAS2 (50), KIT (51,52), and SMAD4 (53) have been found in GCTs that may be involved in germ cell transformation. For KIT, activating somatic mutations have been found, which have been reported to be predictive of bilateral GCT (51). This was refuted in two more recently published studies, where no association was found between KIT mutation and bilateral GCT (54,55). Thus, the precise role of this mutation in germ cell transformation and progression remains to be clarified.
More recently, KIT signaling has been implicated in susceptibility to testicular GCT. KIT and its ligand (KITLG/SCF) have well documented roles in PGC development and recent genome-wide association studies have now demonstrated an increased likelihood of polymorphisms within the KITLG region on chromosome 12q22 among patients with testicular cancer as compared to controls (56,57). Patients homozygous for the major allele at either of two reference sequences had a 4.5-fold increased risk of having GCT as compared to those homozygous for the minor allele (56). Polymorphisms within regions on chromosome 5q31.3 near the SPRY4 gene (encoding a downstream effector of KIT) were also associated with an increased risk of testicular GCT, as were sequences on chromosome 6 (56). Thus, male infertility or subfertility associated with an increased risk of testicular GCT could be founded in abnormal KIT signaling.
GERM CELL OF ORIGIN OF ADULT MALE GERM CELL TUMORS
Expression and genetic studies have lead to the formulation of two models for the stage in the lifespan of a germ cell at which transformation occurs. For pediatric GCTs presenting prior to puberty, there is common acceptance that these tumors arise by transformation of a germ cell in utero. The genetics and biology of these prepubertal GCTs are distinct from those found in adult GCTs and have recently been reviewed (58). One model for the genesis of adult GCTs proposes that gonocytes in the developing embryo escape normal development into spermatogonia and undergo abnormal cell proliferation mediated through a paracrine loop involving KIT and its ligand (KITLG/SCF), leading to ITGCN (59,60). Such gonocytes derailed in their normal development are then suggested to be susceptible to invasive growth mediated by postnatal and pubertal gonadotrophin stimulation. Data in support of this model include the shared expression of many genes between ITGCN aberrant germ cells and PGCs or gonocytes such as PRDM1/BLIMP1 and PRMT5, the types of abnormal germ cells seen in developmental disorders that predispose to GCTs, and the epidemiology of GCT incidence (56,57,59,60,61). The second model takes into account various genetic and expression properties of GCTs (1,23). A zygotene/pachytene spermatocyte
with a duplicated chromosomal complement and undergoing homologous recombination is postulated to be the germ cell of origin. During this phase of meiosis initiated at puberty, an aberrant recombinational event could lead to a chromosome abnormality with several exclusive outcomes: (a) act to trigger apoptosis leading to demise of the germ cell, (b) be transmitted to the zygote with or without obvious consequences to the embryo, or (c) in the specific case of increased 12p, lead to aberrant expression of cyclin D2, reinitiation of mitosis, reestablishment of a pluripotent state, and genomic instability portending tumor formation. Similarities between GCTs and zygotene/pachytene spermatocytes support this model, including the triploid-tetraploid chromosome complement of GCTs, and the abundant expression of wild-type p53 (39,40,62). With the current capacity to examine the global expression patterns of GCTs in relation to PGCs, gonocytes, and isolated populations of spermatogonia and spermatocytes in the adult testis, further insights may be gained into the germ cell of origin of adult GCTs.
with a duplicated chromosomal complement and undergoing homologous recombination is postulated to be the germ cell of origin. During this phase of meiosis initiated at puberty, an aberrant recombinational event could lead to a chromosome abnormality with several exclusive outcomes: (a) act to trigger apoptosis leading to demise of the germ cell, (b) be transmitted to the zygote with or without obvious consequences to the embryo, or (c) in the specific case of increased 12p, lead to aberrant expression of cyclin D2, reinitiation of mitosis, reestablishment of a pluripotent state, and genomic instability portending tumor formation. Similarities between GCTs and zygotene/pachytene spermatocytes support this model, including the triploid-tetraploid chromosome complement of GCTs, and the abundant expression of wild-type p53 (39,40,62). With the current capacity to examine the global expression patterns of GCTs in relation to PGCs, gonocytes, and isolated populations of spermatogonia and spermatocytes in the adult testis, further insights may be gained into the germ cell of origin of adult GCTs.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


