Fig. 13.1
Functional classification of cell death modalities as described by the Nomenclature Committee on Cell Death (NCCD) [19]
13.2 Mechanisms of Apoptosis
The term “apoptosis” was introduced by Kerr and co-workers in 1972, derived from a Greek term meaning “dropping off” of leaves or petals from trees or flowers [9]. Earlier methods to define cell death rely much on morphological criteria and the use of microscopes [7]. The earliest recognized morphological changes in apoptosis involve compaction and segregation of nuclear chromatin and condensation of the cytoplasm [9, 20]. The process is followed by the convolution of the plasma membrane and cell blebbing in a florid manner, producing fragments of cells known as apoptotic bodies. These fragments are membrane-bounded and contain nuclear components [20, 21]. Apoptotic bodies are quickly taken up by nearby cells and degraded within their lysosomes, usually with no associated inflammation [9, 20].
It is important to note that despite the various types of apoptosis characterized by their biochemical features and signaling pathways, they share similar morphological features. Biochemically, apoptosis is universally characterized by the double-stranded cleavage at the linker regions between nucleosomes, resulting in the formation of multiple DNA fragments [21] and phosphatidylserine externalization [22], and is accompanied by a series of gene and protein expressions. Figure 13.2 illustrates the morphological characteristics of apoptosis and how it is compared with necroptosis and autophagic cell death.
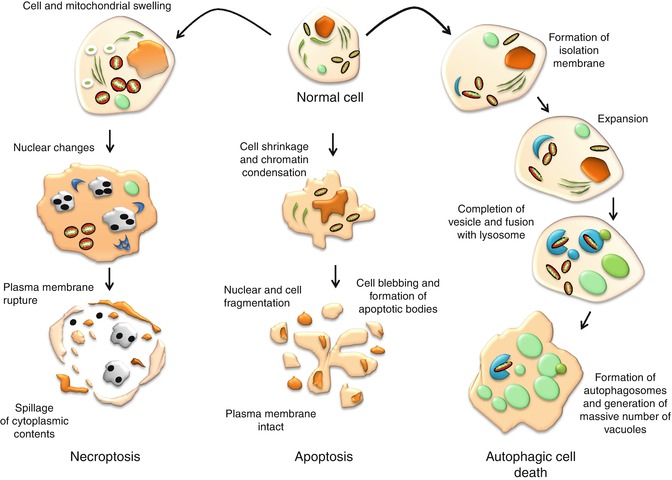
Fig. 13.2
Morphological characteristics of cells undergoing apoptosis, autophagic cell death, and necroptosis
The NCCD has formulated several rounds of recommendations to propose guidelines and unify criteria on the use of cell death terminologies [19, 23]. According to the latest NCCD publication, apoptosis is functionally classified into intrinsic or extrinsic apoptosis. Intrinsic apoptosis is either caspase-dependent or caspase-independent, whereas extrinsic apoptosis is categorized depending on source of trigger, as mediated either by death receptors or by dependence receptors.
13.2.1 Extrinsic Apoptosis Pathway
Extrinsic apoptosis is essentially caspase-dependent and is induced by extracellular stress signals which are mediated by specific transmembrane receptors. In the extrinsic apoptosis induced by death receptors, the signaling pathway is mediated by receptors belonging to the tumor necrosis factor (TNF) receptor superfamily and is characterized by extracellular cysteine-rich domains (CRDs) and intracellular death domain (DD). Ligands such as TNF ligand, TNF ligand superfamily member 10 (TNFSF10), FAS ligand, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) interact with their respective death receptors [FAS/CD95, TNF-α receptor 1 (TNFR1), or TRAIL receptor (TRAIL-R1 or TRAIL-R2)], recruit Fas-associating protein with a death domain (FADD), and form the death-inducing signaling complex (DISC) [24, 25]. This complex recruits pro-caspase-8 and pro-caspase-10, leading to the activation of the executioner caspase-3, caspase-6, and caspase-7 [26, 27].
On the other hand, extrinsic apoptotic signals can be alternatively mediated by dependence receptors such as UNC-5 homolog family receptors (UNC-5A, UNC-5B, UNC-5C, and UNC-5D) and deleted in colorectal cancer (DCC) family receptors. These receptors are activated by netrins, a family of extracellular proteins that direct cell and axon migration during embryogenesis [28]. Netrins are members of the laminin superfamily and contribute to the regulation of cell-cell adhesion and tissue organization [29]. Netrin-1 has been recently identified to be an anti-apoptotic survival factor in tumorigenesis [30]. DCC and UNC-5 homologs mediate cell death in the absence of netrin-1 and the binding of the ligand to these receptors switches between a pro-apoptotic signal and the promotion of survival and motility [30]. UNC-5B (also known as UNC-5H2) complex responds to the withdrawal of netrin-1 by recruiting a signaling complex consisting of protein phosphatase 2A (PP2A) and death-associated protein kinase 1 (DAPK1) [31]. In the presence of netrin-1, the PP2A complex is repressed by the recruitment of cancerous inhibitor of PP2A (CIP2A) into the UNC-5B/DAPK1 complex, of which DAPK1 is autophosphorylated and remained inactive. Conversely, netrin-1 withdrawal is associated with a conformational change in UNC-5B, resulting in the exposure of the death domain, releasing of CIP2A, and the recruitment of PP2A to the UNC-5B-DAPK1 complex. PP2A-mediated dephosphorylation of DAPK1 results in the activation of downstream apoptotic pathway. PP2A-like activity has been linked to the formation of DISC, and is known to inhibit B-cell lymphoma 2 (Bcl-2) phosphorylation, leading to apoptotic cell death [32, 33]. In certain cell types, where the extrinsic apoptotic pathway is triggered but lower levels of DISC followed by lower levels of active caspase-8 are formed, amplification of the death signal is possible through the cleavage of Bid by caspase-8, which directly mediates Bak/Bax oligomerization, and triggers the release of cytochrome (Cyt) c [34, 35].
Another signaling pathway mediated by dependence receptors are the DCC and the Patched dependence receptor (Ptc). DCC encodes an approximately 200 kDa type I membrane protein, which displays homology with cell adhesion molecules in its extracellular domain, suggesting that DCC may play a role in cell-cell or cell-matrix interactions [36, 37]. DCC appears to drive apoptosis independent of both mitochondrial-dependent and death receptor/caspase-8 pathways. DCC interacts and drives the activation of caspase-3 through caspase-9 without requiring Cyt c or Apaf-1 [38]. Ptc, identified as a tumor suppressor, induces apoptosis, but is suppressed by its ligand, sonic hedgehog (Shh) [39, 40]. Ptc interacts with the adapter protein DRAL/FHL2 in the absence of Shh and recruits a protein complex that includes DRAL/FHL2, the CARD-containing domain protein TUCAN, and apical caspase-9. Ptc triggers caspase-9 activation and enhances cell death via a caspase-9-dependent mechanism [41, 42].
The death receptor and dependence receptor pathways converge at the activation of caspase-3, followed by cleavage and activation of downstream caspases. Caspases or cysteine aspartic acid-specific proteases are synthesized as inactive zymogens (or proenzymes) and are usually cleaved to form active enzymes or undergo autoproteolysis in a cascade manner. Initiator caspases such as caspase-8, caspase-9, and caspase-10 couple cell death stimuli to the downstream effector caspases such as caspase-3, caspase-6, and caspase-7. The major proteolysis activity that takes place during apoptosis is carried out by effector caspases. Caspase-3 appears to be the major executioner caspase during the demolition phase of apoptosis [43, 44]. Caspase-3 cleaves a number of structural proteins such as fodrin, gelsolin, rabaptin, nuclear lamin B, and vimentin [44–46]. On the other hand, caspase-6 appears to merely cleave the nuclear lamin A during apoptosis [44]. Caspase-3 also cleaves diverse regulatory proteins and enzymes, including focal adhesion kinase (FAK), protein kinase C delta, retinoblastoma protein (Rb) (a protein involved in cell survival), p21-activated kinase (PAK), U1 small nuclear ribonucleoprotein (U1snRNP), DNA fragmentation factor 45 (DFF45)/inhibitor of caspase-activated DNase (ICAD), receptor interacting protein (RIP), X-linked inhibitor of apoptosis protein (X-IAP), signal transducer and activator of transcription-1 (STAT1), and topoisomerase I [44, 45, 47]. Initially, poly (ADP-ribose) polymerase (PARP) is reported to be an exclusive substrate for caspase-7 [44], but a later study proved that it is cleaved by both caspase-3 and caspase-7 [48].
Caspase-mediated cleavage of structural proteins is essential for the apoptosis-associated morphological changes. For example, cleavage of gelsolin in multiple cell types causes cells to round up, detach from the plate, and undergo nuclear fragmentation [49]. Inactivation of rabaptin-5 causes fragmentation of endosomes during the execution phase of apoptosis [50]. Fodrin is a major component of the cortical cytoskeleton of most eukaryotic cells; it has binding sites for actin, calmodulin, and microtubules [51]. Its proteolysis contributes to structural rearrangements including blebbing during apoptosis [52, 53].
FAK is a tyrosine kinase of which its phosphorylation state and activity are linked to cell adhesion to the extracellular matrix through integrin receptors. It has a direct influence on the cytoskeleton, structures of cell adhesion sites, and membrane protrusions, leading to regulation of cell movement [54, 55]. Caspase-mediated cleavage of FAK is known to contribute to the morphological changes in apoptosis. On the other hand, PAK, a serine-threonine kinase, regulates morphological and cytoskeletal changes in a variety of cell types [56, 57]. Blocking PAK function during Fas-induced apoptosis inhibits the morphological changes, but accelerates the phosphatidylserine externalization in the membrane. Stable Jurkat cell lines that expressed a dominant-negative PAK mutant are resistant to Fas-induced formation of apoptotic bodies and cleavage of PAK [58].
PARP cleavage is believed to attenuate the cell’s ability to carry out DNA repair [45, 59]. Caspase-8 is also found to cleave PARP-2, a member of the PARP family involved in DNA repair, suggesting that caspase-8 is both an initiator and effector caspase [60]. Active caspase-3 or caspase-7 proteolytically cleaves DFF45, which subsequently releases active DFF40, the inhibitor’s associated endonuclease. It is responsible for the degradation of chromosomes into nucleosomal fragments, considered as the characteristic hallmark of apoptosis [61, 62]. Cleavage of both structural and regulatory proteins is essential for the apoptotic-associated chromatin condensation, DNA fragmentation, nuclear collapse, and morphological changes such as cell shrinkage and detachment, membrane blebbing, and formation of apoptotic bodies. Figure 13.3 illustrates the extrinsic apoptosis signaling pathway.
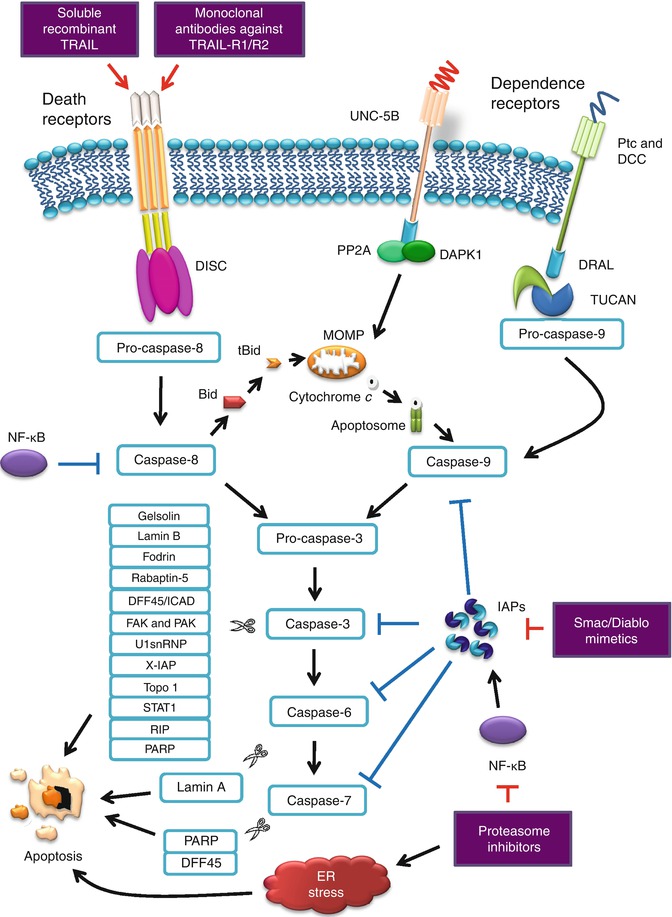
Fig. 13.3
Extrinsic apoptosis signaling pathway and antitumor therapeutic targets
13.2.2 Intrinsic Apoptosis Pathway
Intrinsic apoptosis is known as either caspase-dependent or caspase-independent, and both signaling pathways are centrally mediated by the mitochondria. Intrinsic apoptosis can be triggered by DNA damage, γ-irradiation, oxidative stress, cytosolic Ca2+ overload, serum deprivation, and many other intracellular stress conditions. Upon stimulation, various molecules are released into the cytoplasm including Cyt c [27, 63], second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (Smac/Diablo) [64, 65], apoptosis-inducing factor (AIF; promotes chromatin condensation) [66], endonuclease G (EndoG; facilitates chromatin degradation) [67, 68], and high-temperature requirement protein A2 (HtrA2/Omi) [69]. Cyt c binds to and activates Apaf-1 protein in the cytoplasm, inducing the formation of apoptosome which subsequently recruits the initiator pro-caspase-9, yielding activated caspase-9 and finally mediating the activation of caspase-3 and caspase-7 [35]. Loss of Cyt c from the mitochondria also results in the inhibition of the respiratory chain. The condition elicits and aggravates reactive oxygen species (ROS) overproduction and is thought to activate a feedforward circuit for the amplification of the apoptotic signal [70]. The function of Cyt c and its role in apoptosis are widely reviewed and discussed elsewhere [71–73].
Bcl-2 family of proteins plays an important role in the regulation of mitochondrial-linked apoptosis [74]. Bcl-2 subfamilies such as Bax, Bak, and Bcl-2 homolog (BH)3-only subfamily proteins (e.g., Bid) play a pro-apoptotic role, while Bcl-2 and Bcl-XL are functionally anti-apoptotic. Activated Bax and Bak form homo-oligomer which creates pores on the mitochondrial membrane and releases toxic proteins from the mitochondria. Bcl-2 and Bcl-XL inhibit the action by blocking the activation of Bax and Bak and preventing the release of pro-apoptotic proteins [75]. Nevertheless, the activation of Bax and Bak can be restored with the presence of pro-apoptotic BH3-only proteins. BH3-only proteins function as antagonists of specific subsets of their pro-survival relatives [76, 77]. The pore-forming activities of Bax and Bak trigger a condition known as mitochondrial outer membrane permeabilization (MOMP). MOMP can also be triggered by the opening of a multiprotein complex known as permeability transition pore complex (PTPC) [78, 79]. MOMP causes generalized and irreversible inner mitochondrial transmembrane potential (ΔΨm) dissipation. In the inner mitochondrial membrane (IM) of a healthy cell, the frontier between the intermembrane/intercristal space and the matrix is nearly impermeable to all ions, including protons which help create the proton gradient required for oxidative phosphorylation [70]. The charge imbalance that results from the generation of an electrochemical gradient across the IM forms the basis of the ΔΨm [70]. A loss of the ΔΨm or long-lasting or permanent ΔΨm dissipation can lead to cell death [80]. MOMP causes the release of toxic proteins from the mitochondria to the cytosol as mentioned above. Pro-apoptotic Bcl-2 proteins appear to cause the release of Cyt c, Smac/Diablo, and HtrA2/Omi but not EndoG and AIF [81]. On the other hand, BH3-only protein Bid cleavage by caspase-8 serves to engage a mitochondrial amplification loop during extrinsic apoptosis. Caspase-8 cleaves Bid, generating a truncated fragment known as truncated Bid (tBid) that can permeabilize the mitochondrion, resulting in MOMP [82].
Inhibitors of apoptosis proteins (IAPs) play an important role in the regulation of apoptosis. Eight human IAPs have been identified consisting of X-IAP, IAP-like protein-2 (ILP-2), cIAP-1, cIAP-2, melanoma inhibitor of apoptosis protein (ML-IAP), neuronal apoptosis inhibitory protein (NAIP), survivin, and apollon [83]. Human IAP family members such as X-IAP, cIAP-1, and cIAP-2 are potent caspase inhibitors [84, 85]. X-IAP, cIAP-1, and cIAP-2 block Cyt c-induced activation of caspase-9, thus preventing the activation of caspase-3, caspase-6, and caspase-7. Furthermore, these IAPs bind to and inhibit the enzymatic activity of caspase-3 following its activation by caspase-8, thereby arresting the proteolytic cascade initiated by the initiator caspase [86]. X-IAP primarily inhibits caspase by disrupting the conformation of the active caspase and masking the substrate-binding active site [83].
Smac/Diablo and HtrA2/Omi inhibit the anti-apoptotic function of several members of the IAP family [87, 88]. Smac/Diablo and HtrA2/Omi are two nuclear-encoded mitochondrial proteins functioning as IAP antagonists, identified in mammals [69, 89–92]. After their release into the cytosol stimulated by apoptotic triggers, Smac/Diablo and HtrA2/Omi competitively bind to the BIR domains of IAPs via the IAP-binding motif, so that the BIR-bound caspases are released and reactivated [93–95]. Smac/Diablo and HtrA2/Omi manifest distinct physical characteristics and biochemical activities, of which the active Smac/Diablo is a homodimer, whereas HtrA2/Omi is a homotrimer [87, 96]. Despite Smac/Diablo, HtrA2/Omi is a mitochondrial serine protease [97, 98]. HtrA2/Omi has diverse roles, including maintenance of mitochondrial homeostasis and regulation of cellular apoptosis [99]. A comprehensive proteome-wide analysis of Jurkat cell lysates leads to the identification of potential HtrA2/Omi substrates, for example, the cytoskeleton-associated proteins such as actin, α- and β-tubulin, and vimentin, further suggest its role in the caspase-independent pathway [100].
AIF and EndoG function in a caspase-independent manner, by relocating to the nucleus, where they mediate large-scale DNA fragmentation, independent of caspases [101, 102]. Mammalian EndoG is a nuclear-encoded protein targeted to mitochondria and compartmentalized in the intermembrane space (IMS) and is known to possess DNase/RNase activity [103]. It is implicated in the mitochondrial DNA replication and is shown to be involved in apoptotic DNA degradation [102]. In isolated non-apoptotic nuclei, EndoG first generates large fragments of DNA (>50 kb) and then cleaves at inter- and intra-nucleosomal sites [104]. Although EndoG apoptotic activity appears to occur in the absence of caspase activation, the pathway leading to EndoG-dependent DNA damage remains controversial [105, 106].
AIF was originally discovered as an IMS component capable of inducing chromatin condensation and DNA loss in the nuclei isolated from healthy cells [104, 107]. AIF is a flavoprotein which was first proposed to act as a protease or protease activator [108]; notably, its apoptogenic activity is not affected by z-VAD-fmk [109]. Contribution of AIF to apoptosis depends on the cell types and death triggers [104]. Both endogenous and recombinant AIF are found to trigger peripheral chromatin condensation and large-scale DNA fragmentation in a caspase-independent manner [110, 111]. AIF is not known to possess nuclease activity; therefore, AIF is postulated to directly interact with DNA and disrupt/collapse chromatin structure by displacing chromatin-associated proteins and/or by recruiting proteases and nucleases to form DNA-degrading complexes or degradosomes [104, 112].
Another important signaling pathway affecting the regulation of apoptosis worth mention is the nuclear factor-kappa B (NF-κB). NF-κB is a sequence-specific transcription factor known to be involved in the inflammatory and innate immune responses. Under normal conditions, NF-κB becomes activated only upon stimulation and subsequently upregulates the transcription of its target genes. NF-κB is activated by many divergent stimuli, including proinflammatory cytokines such as TNF-α, TRAIL, interleukin-1β (IL-1β), epidermal growth factor (EGF), T- and B-cell mitogens, bacteria and lipopolysaccharides (LPS), viral proteins, double-stranded RNA, drugs, and a variety of physical and chemical stresses [113]. However, in tumor cells, molecular alterations result in impaired regulation of NF-κB and become constitutively activated in such cases, leading to deregulated expression of NF-κB-controlled genes [114]. Some genes targeted by NF-κB include cytokines/chemokines and their modulators, immunoreceptors, transcription factors, and regulators of apoptosis such as Bcl-XL, Fas, FasL, and IAPs [113].
NF-κB is also known to play a pro-apoptotic role, in addition to its more common anti-apoptotic role. Examples of its pro-apoptotic effects in cells include those found in B cells [115], T cells [116, 117], and neuronal cells [118, 119]. On the other hand, the anti-apoptotic effects of NF-κB appeared to be cell-type specific and/or dependent on the inducing signal. Normally, NF-κB is transcriptionally inactive in the cytoplasm of most cells as it is bound to its cytoplasmic inhibitor IκBα. Upon stimulation with proinflammatory cytokines, such as TNF-α or IL-1, IκBα protein is phosphorylated, ubiquitinated, and subsequently degraded by the proteasome (the role of proteasome is further discussed under proteasome inhibitors). This process exposes the previously masked nuclear localization signal of NF-κB, allowing it to translocate into the nucleus upon IκBα proteolysis and subsequently activate the expression of important target genes involved in cell growth, survival, and adhesion [120, 121]. Activated NF-κB leads to the activation of A1/Bfl-1, a member of the Bcl-2 family, which suppresses Cyt c release from the mitochondria [122]. NF-κB activation blocks caspase-8 cleavage and Cyt c release, indicating that NF-κB suppresses the earliest signaling components of the caspase cascade. The IAP family genes (cIAP–1 and cIAP–2) and TRAF family genes (TRAF1 and TRAF2) are positively regulated by NF-κB with rapid kinetics following TNF addition [123, 124]. Another member of the IAP family, X-IAP, has been shown to be activated by NF-κB in endothelial cells [125, 126]. Thus, NF-κB activation functions to suppress apoptosis at multiple levels.
The Nomenclature Committee on Cell Death (NCCD) suggests to define “intrinsic apoptosis” as cell death mediated by MOMP and associated with generalized and irreversible ΔΨm dissipation, release of IMS proteins, and respiratory chain inhibition [19]. On the other hand, differentiation between caspase-dependent and caspase-independent intrinsic apoptosis pathways is based on the extent of cytoprotection as conferred by inhibition of caspases. The caspase-independent mechanisms mediated by AIF, EndoG, or ATP depletion tend to prevail over caspase inhibition and kill cells in conditions that would have been rapidly executed by the caspase cascade [19]. However, the caspase-independent signaling pathway is still vague, and the exact mechanisms remain to be investigated. Figure 13.4 illustrates the caspase-dependent and caspase-independent intrinsic apoptosis pathway.
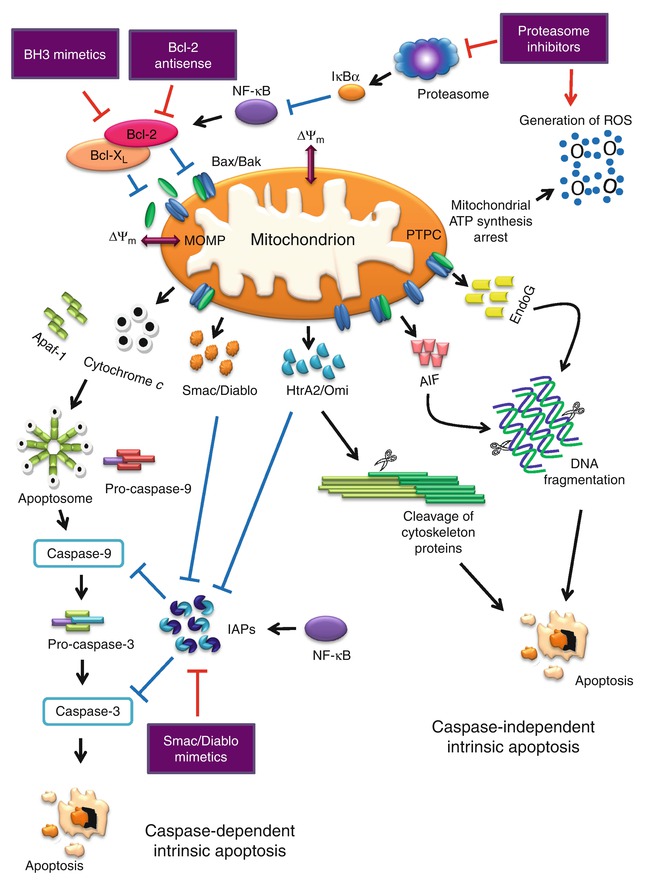
Fig. 13.4
Caspase-dependent and caspase-independent intrinsic apoptosis signaling pathway and antitumor targets
13.3 Apoptosis and Cancer
Apoptosis is an essential developmental process to maintain tissue homeostasis. Therefore, defect in apoptosis regulation plays an important role in cancer development. Deregulation in the apoptosis pathway is one of the reasons why neoplastic cells gain extended lifespan, develop genetic mutations capable of growth under stress conditions, and undergo angiogenesis [12]. Several key pathways controlling apoptosis are commonly altered in cancer [127]. Tumor resistance to apoptotic cell death is often a hallmark of cancer and contributes to chemoresistance [12]. Alteration of many proteins involved in both intrinsic and extrinsic signaling pathways has been described, and many more to be discovered in near future. For example, overexpression of certain anti-apoptotic proteins, such as Bcl-2, Bcl-XL, Akt, NF-κB, and IAP protein family, is found in various human tumors [128].
The apoptotic pathway of Fas, one of the TNF receptor family members, is frequently blocked by several mechanisms in cancer, one of which is Fas gene mutation [129–131]. Fas mutations have been detected in several types of human cancers with frequent allelic losses of chromosome 10q24 where the gene resides [130–132]. Both TRAIL–R1 and TRAIL–R2 genes are mapped on chromosome 8p21-22 [133, 134]. Allelic losses of the chromosome 8p21-22 have been reported as a frequent event in several cancers, including non-Hodgkin lymphoma (NHL), lung cancer, breast cancer, colon cancer, prostate cancer, hepatocellular carcinoma, and head and neck cancer [135–141]. Mutations of TRAIL–R2 gene have been reported in head and neck cancer [142] and non-small cell lung cancer (NSCLC) [143]. In addition, somatic mutations of TRAIL–R1 and TRAIL–R2 genes are found in NHL [144] and breast cancer [145]. The number of pancreatic tumor tissues with positive membrane staining for TRAIL-R1 and TRAIL-R2 is lower than non-tumor tissues [146]. Loss of TRAIL-R2 expression is associated with poorer prognosis in patients [146]. A significant association is also observed between lower expression of TNF gene and poor prognosis in childhood adrenocortical tumors [147].
On the other hand, PP2A inactivation in cancer occurs frequently through the upregulation of CIP2A, a PR65 interactor and PP2A inhibitor [148]. PR65β, a scaffold protein which interacts with the catalytic subunit of PP2A, appears to play a key regulatory role in cancer. This scaffold protein is decreased or mutated in a large fraction of human cancers and has been recently linked to cancer development [149]. On the other hand, Ptc is a tumor suppressor and mutations of Ptc are associated with neoplasia, especially in basal cell carcinoma and medulloblastoma [39, 40]. DCC expression is shown to be markedly reduced in more than 50 % of colorectal tumors. The loss of DCC is not restricted to colon carcinoma, but has been observed in other tumor types, including carcinoma of the stomach, pancreas, esophagus, prostate, bladder, and breast, male germ tumors, neuroblastomas, gliomas, and some leukemias [36, 150, 151].
Members of the Bcl-2 family of proteins as prominent regulators of apoptosis signaling are often deregulated in many cancers, including lung carcinoma, lymphoma, and glioblastoma [152–156]. Aberrant expression of Bcl-2 is common in chronic lymphocytic leukemia (CLL) and is associated with poor response to chemotherapy and decreased overall survival [157]. Bcl–2 gene amplification is reported in diffuse large B-cell lymphomas (DLBCL) and overexpression of Bcl-2 protein has been associated with poor prognosis in some forms of NHL [158–160]. Myc/Bcl-2 co-expression in DLBCL is associated with aggressiveness, is more common in the unfavorable activated B-cell-like subtypes, and contributes to the overall inferior prognosis of patients with activated B cell-DLBCL [161]. Single-nucleotide polymorphisms in Bcl-2 are found to have an association with survival in advanced-stage NSCLC patients who received chemotherapy [162]. Furthermore, mutations that inactivate the pro-apoptotic Bax gene have been observed in solid tumors and hematologic malignancies [163, 164]. Higher Bcl-2 to Bax ratios has been associated with progression of CLL, shorter remission duration, and shorter survival [165, 166]. Therefore, cancer therapeutics that specifically inhibit the anti-apoptotic proteins or activate the pro-apoptotic members of the Bcl-2 family proteins are an attractive strategy to reverse the intrinsic or acquired resistance of cancer cells to apoptosis [167].
Studies have reported that polymorphic variants of the caspase–8 gene are associated with the risk of multiple cancers [168–172]. For example, a six-nucleotide insertion-deletion variant polymorphism (6 N ins/del) of caspase–8 promoter is linked to a significant decreased risk of bladder and lung cancer in Chinese populations [171, 172]. Since cancer cells are highly dependent on these genetic changes in the apoptotic pathways for survival, designing novel anticancer drugs that selectively kill cancer cells while sparing normal cells seem appealing [173]. Survivin, a member of the IAP family, is undetectable in terminally differentiated adult tissues, but abundantly expressed in human cancers such as lung, colon, pancreas, prostate, and breast [167]. Increased survivin mRNA is associated with decreased overall survival in colon cancer patients [174]. Furthermore, increased levels of cIAPs in malignant cells are associated with a shorter relapse-free survival in patients with prostate cancer [175]. Livin or ML-IAP, another member of the IAP family of proteins, is found to be expressed in tumor cells [176, 177]. Thus, the possibility of IAP inactivation through therapeutic intervention is rather attractive and has gained much interest over the years.
Another important pathway linked to the apoptotic cell death is the p53 pathway, which is often inactivated and deregulated in human cancers [178, 179]. The p53 protein is a transcription factor with tumor suppressor activities. Its role in tumor suppression relies partly on its ability to regulate the transcription of genes important in cell cycle arrest and in apoptosis. The p53 protein upregulates the expression of a number of genes in response to genotoxic stress, including the pro-apoptotic Bax [180]. It is also found to inhibit the expression of the Bcl–2 gene [181]. Studies have also shown that Bid is a p53-responsive chemosensitivity gene which may enhance the cell death response to chemotherapy [182]. The fact that a majority of human cancers harbor mutations in the p53 gene suggests that such mutations would have contributed to the apoptosis-resistant environment. However, the p53 network and the mechanism by which p53 determines the fate of cells remain to be explored.
13.4 Apoptosis Signaling Pathways and Therapeutic Targets in Cancer
13.4.1 TRAIL (TRAIL Ligands, Monoclonal Antibodies Against TRAIL-R1 and TRAIL-R2)
TRAIL (Apo2 ligand) induces cell death via the extrinsic pathway by recruiting and activating caspase-8 and caspase-10 to its R1 and R2 receptors [183]. It activates the intrinsic pathway via the TRAIL-caspase-8-tBid-Bax cascade, through the cleavage of Bid, which promotes Bax and Bak oligomerization, leading to Cyt c release and activation of caspase-9 [184]. These processes collectively amplify the activities of the related executioner caspases. TRAIL is a promising cancer therapeutic agent, known to induce apoptosis in a wide variety of tumor cells while sparing normal cells [185, 186]. TRAIL activity is also known to be independent of the p53 status, making it potentially effective against chemotherapy-resistant tumors [187]. Early clinical trials have been initiated in cancer patients, using soluble recombinant TRAIL (rhApo2L, codeveloped by Genentech and Amgen) [188, 189] and monoclonal antibodies (mAbs) (agonists) targeting TRAIL-R1, such as mapatumumab [HGS-ERT1 is developed by Human Genome Sciences (HGS)], and anti-TRAIL-R2 agents, such as lexatumumab (HGS-ETR2 is developed by HGS), conatumumab (developed by Amgen), and apomab (developed by Genentech) [190].
In an early phase I safety and pharmacokinetic trial of rhApo2L used as a single agent in patients with advanced solid tumors and NHL, of 32 patients with post-baseline tumor assessment, 17 (53 %) had stable disease and 13 (41 %) proceeded with disease progression. Only a single patient was reported to have a partial response to the drug [188]. Phase I/Ib trials of rhApo2L in advanced cancer [191], advanced NSCLC [192], and NHL [193] reported that this drug was well tolerated by patients and no anti-rhApo2L Abs were detected. Promising outcome in phase Ib trial of rhApo2L in combination with cytotoxic chemotherapy (paclitaxel and carboplatin) and targeted anti-angiogenesis agent (bevacizumab) in advanced NSCLC has led to a randomized phase II study [192]. Despite the encouraging phase Ib results, the addition of rhApo2L to paclitaxel/carboplatin or paclitaxel/carboplatin/bevacizumab combination did not improve the outcome and produced a higher incidence of treatment-related adverse effects [194]. Similarly, the addition of rhApo2L to rituximab did not improve the objective outcome in phase II NHL study despite its promising activity in phase Ib study [193, 195]. Adverse effects commonly associated with rhApo2L include neutropenia and serum lipase elevation [194, 195]. Phase I trials of rhApo2L in colorectal cancer are ongoing (Table 13.1).
Table 13.1
Current therapeutic targets in the apoptosis signaling pathway and clinical trial stages
Therapeutic targets | Current drugs | Clinical trial stages (published reports)/type of cancer | Combined with | References |
|---|---|---|---|---|
TRAIL-R1 and TRAIL-R2 | rhApo2L (dulanermin) | Phase I: advanced cancer | – | [191] |
Phase Ib: NHL | Rituximab | [193] | ||
Phase Ib: advanced NSCLC | Paclitaxel, carboplatin, and bevacizumab | [192] | ||
Phase II: NHL | Rituximab | [195] | ||
Phase II: advanced NSCLC | Paclitaxel, carboplatin, and bevacizumab | [194] | ||
Ongoing phase I: colorectal cancer | Cetuximab and irinotecan or FOLFIRI regimen and bevacizumab | |||
Ongoing phase I: colorectal cancer | FOLFOX regimen and bevacizumab | |||
TRAIL-R1 | Mapatumumab | Phase I: advanced solid tumor | Gemcitabine and cisplatin | [196] |
Phase I: advanced solid tumor | Paclitaxel and carboplatin | [197] | ||
Phase Ib: advanced hepatocellular carcinoma | Sorafenib | [198] | ||
Phase Ib/II: NHL | – | [199] | ||
Phase II: colorectal cancer | – | [200] | ||
Phase II: advanced NSCLC | Paclitaxel and carboplatin | [201] | ||
Ongoing phase I/II: advanced cervical cancer | Cisplatin and radiotherapy | |||
Ongoing phase II: advanced hepatocellular carcinoma | Sorafenib | |||
TRAIL-R2 | Lexatumumab | Phase I: advanced solid tumor | – | [201] [203] |
Apomab (drozitumab; PRO95780) | Phase I: solid tumor Phase I: advanced cancer | – – | [204] [205] [206] | |
Conatumumab (AMG 655) | Phase II: NSCLC | Paclitaxel, carboplatin, and bevacizumab | [207] | |
Phase II: NHL | Rituximab | [208] | ||
Phase I: advanced solid tumor | – | [209] | ||
Phase I: advanced solid tumor | – | [210] | ||
Phase Ib: pancreatic cancer | Gemcitabine | [211] | ||
Phase Ib: advanced NSCLC | Paclitaxel and carboplatin | [212] | ||
Phase Ib: colorectal cancer | mFOLFOX6 and bevacizumab | [213] | ||
Phase I/II: soft tissue sarcoma | Doxorubicin | [214] | ||
Phase Ib/II: colorectal cancer | Panitumumab | [215] | ||
Phase II: pancreatic cancer | Gemcitabine | [216] | ||
Phase II: colorectal cancer | FOLFIRI | |||
Phase II: advanced NSCLC | Paclitaxel and carboplatin | |||
Ongoing phase I/II: colorectal cancer | mFOLFOX6 and bevacizumab | |||
Ongoing phase II: advanced solid tumors | AMG 479 | |||
Anti-apoptotic Bcl-2 family members | AT-101 | Phase I: CLL | – | [217] |
Phase I/II: SCLC | Topotecan | [218] | ||
Phase II: SCLC | – | [219] | ||
Phase II: NSCLC | Docetaxel | [220] | ||
Ongoing phase II: advanced laryngeal cancer | Docetaxel and cisplatin/carboplatin | |||
Ongoing phase II: SCCHN | Docetaxel | |||
Obatoclax mesylate (GX15-070) | Phase I: leukemia and myelodysplasia | – | [221] | |
Phase I: advanced CLL | – | [222] | ||
Phase I: solid tumor | Topotecan | [223] | ||
Phase I: advanced solid tumor or lymphoma | – | [224] | ||
Phase II: SCLC | Topotecan | [225] | ||
Phase II: Hodgkin lymphoma | – | [226] | ||
Ongoing phase I: CLL | Fludarabine and rituximab | |||
Ongoing phase I: solid tumor, lymphoma, or leukemia | Vincristine sulfate, doxorubicin, and dexrazoxane hydrochloride | |||
Ongoing phase I/II: SCLC or advanced solid tumor | Topotecan | |||
ABT-263 (navitoclax) | Phase I: lymphoid tumor | – | [227] | |
Phase II: SCLC | – | [228] | ||
Ongoing phase I: solid tumor | Erlotinib or irinotecan | |||
Ongoing phase I/IIa: CLL | – | |||
Ongoing phase II: lymphoid cancer | – | |||
Bcl-2 mRNA Proteasome | Oblimersen sodium | Phase I: advanced melanoma | Temozolomide and albumin-bound paclitaxel | [229] |
Phase II: CML | Imatinib | [230] | ||
Phase II: SCLC | Carboplatin and etoposide | [231] | ||
Phase III: advanced melanoma | Dacarbazine | [232] | ||
Phase III: CLL | Fludarabine and cyclophosphamide | [233] | ||
Phase III: CLL | Fludarabine and cyclophosphamide | [234] | ||
Phase III: MM | Dexamethasone | [235] | ||
Ongoing phase I: AML | Cytarabine and daunorubicin hydrochloride | |||
Ongoing phase I: advanced melanoma | Abraxane and Temodar | |||
Bortezomib | Approved by FDA for multiple myeloma (2005) | – | [236] | |
Approved by FDA for mantle cell lymphoma (2006) | – | [237] | ||
Phase I: acute leukemia | – | [238] | ||
Phase I: advanced solid tumor | Paclitaxel | [239] | ||
Phase I/II: MM | Cyclophosphamide and prednisone | [240] | ||
Phase II: MM | Dexamethasone | [241] | ||
Phase II: mantle cell lymphoma | – | [242] | ||
Phase II: Hodgkin lymphoma | Gemcitabine | [243] | ||
Phase II: esophageal, gastric, and gastroesophageal cancer | Paclitaxel and carboplatin | [244] | ||
Phase II: mantle cell lymphoma | – | [245] | ||
Phase II: MM | – | [246] | ||
Phase III: MM | Dexamethasone | [247] | ||
Ongoing phase I: prostate cancer | Mitoxantrone | |||
Ongoing phase I: neuroblastoma | Irinotecan | |||
Ongoing phase I: T-cell lymphoma | Azacitidine | |||
Ongoing phase I: acute leukemia or myelodysplastic syndrome | Belinostat | |||
Ongoing phase I/II: mantle cell lymphoma | Rituximab and lenalidomide | |||
Ongoing phase I/II: Waldenstrom’s macroglobulinemia | Everolimus and rituximab | |||
Ongoing phase II: plasma cell leukemia | Liposome doxorubicin and dexamethasone | |||
Ongoing phase III: MM | Panobinostat and dexamethasone | |||
Ongoing phase III: MM | Vorinostat | |||
Carfilzomib (PR-171) | Phase I: hematologic malignancies | – | [248] | |
Ongoing phase I: AML or ALL | – | |||
Ongoing phase I/II: MM | Immunomodulatory drugs | |||
Ongoing phase II: MM | – | |||
Ongoing phase III: MM | Dexamethasone | |||
X-IAP mRNA | AEG35156 | Phase I: advanced cancer | – | [249] |
Phase I/II: AML | Idarubicin and cytarabine | [250] | ||
Phase II: AML | Idarubicin and cytarabine | [251] | ||
Pan-IAP | LCL161 | Phase I: advanced cancer | – | [252] |
Phase I: advanced solid tumor | Paclitaxel | [253] | ||
Ongoing phase II: breast cancer | Paclitaxel | |||
HGS1029 | Phase I: advanced solid tumor | – | [254] | |
TL32711 | Phase I: advanced solid tumor and lymphoma | – | [255] | |
Ongoing phase I: advanced solid tumor | Gemcitabine | |||
Ongoing phase I/II: myelodysplastic syndrome | Azacitidine | |||
Ongoing phase I/II: AML, ALL, and myelodysplastic syndrome | – | |||
Ongoing phase I/II: solid tumor | – | |||
Ongoing phase II: ovarian, fallopian tube, and peritoneal cancer | – | |||
Survivin | YM155 | Phase I: advanced solid tumor or lymphoma | – | [256] |
Phase I: advanced solid tumor | – | [257] | ||
Phase II: NSCLC | – | [258] | ||
Phase II: melanoma | – | [259] | ||
Ongoing phase II: breast cancer | Docetaxel | |||
Ongoing phase II: NHL | Rituximab |
Mapatumumab, a fully human agonistic mAb targeting TRAIL-R1, either used alone or in combination with other chemotherapy drugs in phase I or phase II trials, has yet to produce impressive trial outcomes, as in most cases, few patients ended with partial response or stable disease [260–263]. Despite its favorable safety profile, mapatumumab demonstrated limited or no clinical activity in phase I and II trials in advanced solid malignancies [196, 197], NHL [199], NSCLC [201], refractory colorectal cancer [200], and advanced hepatocellular carcinoma [198]. Additional trials of mapatumumab in advanced hepatocellular carcinoma and advanced cervical cancer may provide additional data on the usefulness of this drug (Table 13.1).
Lexatumumab, apomab, and conatumumab are agonistic human mAbs against TRAIL-R2. Generally, the percentage of patients who developed partial response or stable disease in several early phase I trials involving these novel drugs is low, despite being well tolerated by patients. For example, no objective activity of apomab was demonstrated in a phase II study among patients with NHL [206], despite some evidence of activity in phase I study in patients with advanced malignancies [204]. The effects of apomab in phase II NSCLC trial coincide with rhApo2L, where addition of apomab to paclitaxel/carboplatin/bevacizumab combination did not improve the efficacy, while increasing the rate of some adverse effects [194, 205].
As for conatumumab, a phase I study in advanced solid tumors showed that this drug is generally well tolerated [207, 208]. Conatumumab in combination with gemcitabine shows evidence of an improved 6-month survival rate and tolerable toxicity in phase Ib and II metastatic pancreatic cancer trials [209, 214]. In metastatic colorectal cancer, conatumumab improves progression-free survival (PFS) when combined with FOLFIRI [215], but limited activity when combined with modified FOLFOX6 and bevacizumab [211], and no activity when combined with panitumumab [213]. The effect of conatumumab in NSCLC is similar as compared with rhApo2L [192, 194], of which combination of this drug with paclitaxel and carboplatin did not produce promising results [210, 216]. Combination of conatumumab with other chemotherapy drugs also produces no evidence of activity in soft tissue sarcomas [212]. The common adverse effects with this drug are neutropenia and thrombocytopenia [214–216]. Generally, these early trials lacked data on the correlation between patient’s TRAIL status and response to therapy. Preferential TRAIL sensitivity and presence of TRAIL-R1 and TRAIL-R2 expression in certain cancers are considered factors in patient’s response. Therefore, rhApo2L and agonistic anti-TRAIL-R therapies may be limited to patients with TRAIL-sensitive tumors. The efficacy of TRAIL targeting therapies may be improved if diagnostic methods determining TRAIL sensitivity of clinically detectable human cancers are available [190]. Trials are still ongoing, especially those involving the combination of these agents with current chemotherapy drugs.
13.4.2 Bcl-2 Family Proteins (BH3 Mimetics and Bcl-2 Antisense)
Bcl-2 family proteins can regulate apoptosis both positively and negatively. The Bcl-2 family members consist of anti-apoptotic (Bcl-2, Bcl-XL, Bcl-W, Bag-1, Mcl-1, and A1/Bfl-1) as well as pro-apoptotic (Bad, Bax, Bak, Bcl-xs, Bid, Bik, and Hrk) molecules [264, 265]. The balance and interaction between Bcl-2 gene family members and posttranslational modifications of Bcl-2-related proteins have been demonstrated to play important roles in regulating cell survival and death.
The Bcl-2 family is characterized by specific regions of homology termed Bcl-2 homology (BH1, BH2, BH3, and BH4) domains. Anti-apoptotic proteins have BH1–BH4 domains (e.g., Bcl-2 and Bcl-XL). Pro-apoptotic proteins have either BH1–BH3 domains (e.g., Bax and Bak) or BH3-only domains (e.g., Bid, Bim, Puma, Bad, Noxa, Hrk, Bik) [77, 266, 267]. These domains are critical to the function of these proteins, especially their impact on cell survival and cell death and their ability to interact with other family members and regulatory proteins. The molecular surface of the multidomain anti-apoptotic Bcl-2 protein contains a BH3-binding groove, which accommodates BH3 domain from pro-apoptotic Bcl-2 protein family members. The BH3-only proteins are known to function as antagonists of anti-apoptotic Bcl-2 family proteins and act as tumor suppressors [77]. This forms the basis or platform for subsequent drug discovery strategies based on mimicking BH3 peptides with chemical compounds that bind in the same groove [268].
The earlier observation that apoptosis deregulation in cancer cells primarily affects the upstream of the signaling pathways of Bax/Bak and mitochondria, leaving the downstream core of the apoptotic machinery mostly intact, has led to a therapeutic strategy of which manipulation of the equilibrium between the pro- and anti-apoptotic Bcl-2 family members could possibly restore apoptosis [128, 173]. Since pro-apoptotic BH3 domains directly bind to the hydrophobic grooves of pro-survival proteins with high affinity, and are necessary and sufficient for initiation of apoptosis, agents mimicking the BH3 domains may provide some degree of selectivity against cancer cells. This is mainly because cancer cells are postulated to be more sensitive to inhibition of pro-survival proteins compared with their normal counterparts [12]. Cancer cells often express high levels of Bcl-2-like anti-apoptotic proteins to evade the apoptotic fate imposed by aberrant cell proliferation, activation of oncogenes, or DNA damage [269]. Therefore, it is possible to design BH3 mimetics to target specific anti-apoptotic proteins that are overexpressed in a particular type of cancer for improved specificity [173]. Several chemicals mimicking BH3 peptides exclusively targeting the Bcl-2 anti-apoptotic proteins have since been described [268, 270, 271]. Another antitumor strategy is direct inhibition of Bcl-2 mRNA, in the form of antisense.
One of the earliest small-molecule BH3 mimetics or more accurately Bcl-2 and Bcl-XL inhibitor that went through several phase I/II clinical trials is gossypol, an orally available compound derived from cottonseed extracts [272]. It binds to the BH3-binding grooves of Bcl-2, Bcl-XL, and Mcl-1 [273]. However, several past clinical trials have not indicated this compound as an effective anticancer agent. Either used alone or in combination, patients treated with gossypol failed to show evidence of tumor regression or any therapeutic responses in several clinical trials [274–276]. A derivative of R-(-)-gossypol (AT-101) is found to be well tolerated in a phase I trial involving CLL patients [217]. However, later studies showed that either AT-101 is not active in patients or the response rates are too low that it did not meet the criteria for additional enrolment in further trials for small cell lung cancer (SCLC) [218, 219]. In NSCLC, patients did not meet the primary endpoint of improved PFS when given a combination of AT-101 and docetaxel [220]. Current trials to evaluate the potency of this drug in other forms of cancer are still ongoing, for example, as a combination therapy in squamous cell carcinoma of the head and neck (SCCHN) and advanced laryngeal cancer. A semisynthetic analog of gossypol with improved pharmacologic properties, such as apogossypolone (ApoG2), was found to inhibit the growth of diffuse large cell lymphoma cells in vitro and in vivo [277]. However, this compound has yet to proceed to clinical trials.
GX15-070 (obatoclax mesylate) is an indole derivative and a broad-spectrum inhibitor of pro-survival Bcl-2 family proteins; it has been extensively evaluated in clinical trials. A phase I clinical trial of obatoclax mesylate in 44 patients with refractory leukemia and myelodysplasia has demonstrated that the drug is well tolerated up to the highest dose. However, only a single patient with acute myeloid leukemia (AML) with mixed lineage leukemia t(9;11) rearrangement achieved complete remission, which lasted 8 months, and of 14 patients with myelodysplasia, only three showed hematologic improvement [221]. In another phase I trial, where obatoclax was administered to patients with advanced CLL, activation of Bax and Bak was demonstrated in peripheral blood mononuclear cells, and induction of apoptosis was related to overall obatoclax exposure, as monitored by the plasma concentration of oligonucleosomal DNA/histone complexes. Obatoclax is noted to have some biological activity and modest single-agent activity in heavily pretreated patients with advanced CLL [222]. In advanced solid tumor and lymphoma, of 35 patients given obatoclax infusions, only one patient with relapsed NHL achieved partial response of 2 months duration, and one patient had stable disease for 18 months [224]. In a phase II study in patients with relapsed SCLC, obatoclax added to topotecan produced no difference in response rates as compared to topotecan alone, even though the drug was generally well tolerated [223, 225]. Obatoclax has also showed limited clinical activity in heavily pretreated patients with classic Hodgkin lymphoma (HL) [226]. Neurological symptoms are reported as the most common adverse effects in patients. Obatoclax appears to have limited efficacy as a single agent or even in combination with some of the more common anticancer drugs. Clinical trials using obatoclax in combination with other chemotherapy drugs in solid tumor, leukemia, and SCLC are currently ongoing (Table 13.1).
Another BH3 mimetic, ABT-737, possesses greater binding affinity to BH3-only proteins, enhances the effect of death signal, and is synergistic with cytotoxic agents and radiation [268]. To overcome the low solubility and oral bioavailability of ABT-737, the ABT-263 analog (navitoclax) was developed for clinical investigation. Preclinical studies confirmed that navitoclax has high affinity for the anti-apoptotic Bcl-2 family proteins and kills cancer cells in a Bax/Bak-dependent manner [227]. In a phase II clinical study, navitoclax exhibits limited single-agent activity against advanced and recurrent SCLC [228]. Clinical trials of navitoclax as a single agent or as combination therapy in a variety of cancers such as lymphoid, leukemia, and other solid tumors are ongoing.
A nuclease-resistant phosphorothioate antisense oligonucleotide targeting Bcl-2 mRNA (oblimersen sodium) has shown promising activity for CLL and malignant melanoma in randomized phase III trials [232–234]. It is an 18-mer phosphorothioate antisense oligonucleotide designed to bind to the first six codons of the human Bcl-2 mRNA [278]. The use of oblimersen in combination with chemotherapy in a variety of cancers has shown diverse response rates with good tolerability. In the Oblimersen Melanoma Study Group, the addition of oblimersen to dacarbazine improved the multiple clinical outcomes in patients with advanced melanoma and increased overall patient’s survival [232]. In another phase III trial, the addition of oblimersen to fludarabine and cyclophosphamide significantly increased the complete response/nodular partial response rate in patients with relapsed or refractory CLL [233]. In the same study, a significant 5-year survival benefit was observed with oblimersen in combination with fludarabine and cyclophosphamide. Among patients with fludarabine-sensitive disease who had previously demonstrated maximum benefit with the same treatment, a 50 % reduction in the risk of death was observed [234].
However, not all combination therapies produce desirable outcomes. In the Cancer and Leukemia Group B Study 10107 (CALGB), although the combination of oblimersen and imatinib was safe and feasible, no clinical benefits were observed in imatinib-resistant chronic myeloid leukemia (CML) patients [230]. In a randomized phase II study of carboplatin and etoposide with or without oblimersen for extensive-stage SCLC (CALGB 30103), the addition of oblimersen to a standard regimen did not improve any clinical outcome measure [231]. A randomized study of dexamethasone with or without oblimersen sodium in patients with advanced multiple myeloma (MM) demonstrated no significant differences in time to tumor progression or objective response rate [235]. Interestingly, in a recent phase I study, the combination of oblimersen, temozolomide, and albumin-bound paclitaxel was well tolerated and demonstrated encouraging activity in patients with advanced melanoma, with objective response rate and disease control rate at 40.6 % and 75 %, respectively [229]. Some of the common adverse effects associated with oblimersen sodium administration include fatigue, transaminase elevation [279, 280], and hematologic disorders [231–233]. There are a number of trials listed in the NIH ClinicalTrials.gov website; some trials are terminated, and some are completed, while the outcome of trials involving some other tumor types is unknown at this point of time.
13.4.3 Proteasome Inhibitors
The proteasome is a multicatalytic enzyme complex that degrades intracellular proteins by a targeted and controlled mechanism. The 26S proteasome, a large protein complex, composes approximately 50 subunits that function as a highly specific molecular shredder by hydrolyzing ubiquitinated proteins into small peptides [281]. The 26S proteasome can be further divided into two sub-complexes, a central 20S proteolytic core particle (CP) that is capped at either end by one or two 19S regulatory particles (19S RP). The 20S CP is the degradation unit and contains the active sites required to hydrolyze proteins into peptides [281]. On the other hand, the 19S RP controls the degradation of ubiquitin-tagged substrates by acting as a receptor for poly-ubiquitinated proteins and facilitating their ATP-dependent translocation into the catalytic chamber of the 20S CP [281].
Related posts:
 Regulatory T Cells and Th17 Cells in Cancer Microenvironment
Epigenetics and microRNAs in Cancer
The Role of Exhaustion in Tumor-Induced T Cell Dysfunction in Cancer
Primary Immunodeficiencies and Cancers
B Cells in Cancer Immunology: For or Against Cancer Growth?
Cancer Immunoediting: Immunosurveillance, Immune Equilibrium, and Immune Escape
Regulatory T Cells and Th17 Cells in Cancer Microenvironment
Epigenetics and microRNAs in Cancer
The Role of Exhaustion in Tumor-Induced T Cell Dysfunction in Cancer
Primary Immunodeficiencies and Cancers
B Cells in Cancer Immunology: For or Against Cancer Growth?
Cancer Immunoediting: Immunosurveillance, Immune Equilibrium, and Immune Escape
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


