James S. McCarthy, Richard N. Price
Antimalarial Drugs
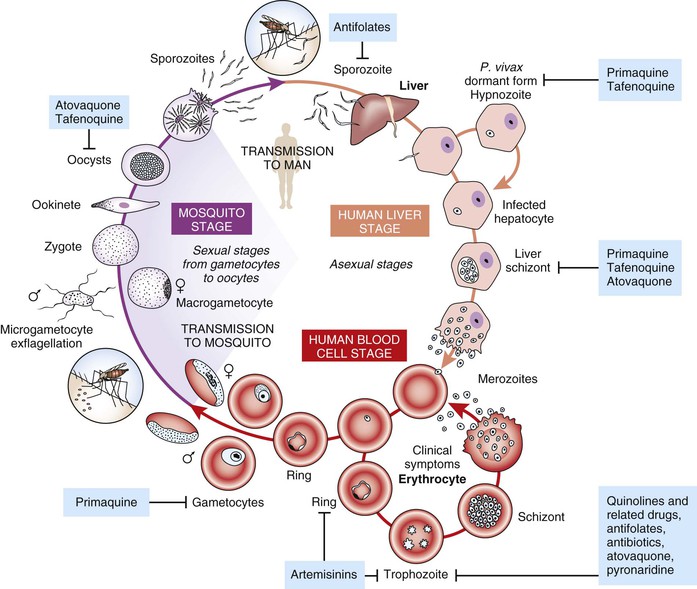
Antimalarial drugs can be classified in the following ways: chemical class, mechanism of action, or life cycle stage of the parasite against which they are active. In clinical medicine, the focus of antimalarial chemotherapy is predominantly on the life cycle stage that causes illness, namely blood stage infection, and indeed most of the available antimalarials are active against this life cycle stage. However, a number of drugs are also active against the pre-erythrocytic life cycle stage in the liver, and such activity is important for a number of drugs used in chemoprophylaxis, as well as in circumstances where the objective is to eliminate latent infection with hypnozoites of Plasmodium vivax and Plasmodium ovale. A small number of drugs are also active against the sexual stage of the parasite, the gametocyte that resides in the blood and is picked up when a mosquito feeds. Killing parasites in this stage of the life cycle is important if the intention is to interrupt transmission (e.g., where gametocytemic individuals are living in an environment where Anopheles vectors are present). The life cycle stages that available antimalarial drugs target are shown in Figure 40-1.
Pharmacokinetic properties of major antimalarials are presented in Table 40-1 and discussed with each drug.
Artemisinin Derivatives
Artemisinin is the active principle extract of Artemisia annua (Qinghao), a plant used for centuries in traditional Chinese herbal remedies for the treatment of febrile illness. In the 1960s during the Cultural Revolution, Chinese scientists began an extensive search for new antimicrobial compounds from their traditional pharmacopoeia, a search that brought to light the novel antimalarial properties of artemisinin, which were announced to the Western literature in 1979.1 The artemisinin drugs are unusual in that they consist of a peroxide bridge within a 1,2,4-trioxane configuration, which contains a sesquiterpene lactone ring with an endoperoxide bridge that is essential for their antimalarial activity.2 Subsequent clinical studies have shown that artemisinin and its derivatives are highly potent antimalarials with a broad-stage specificity of action, resulting in a faster clinical and parasitologic response than any other antimalarial agents in clinical use.
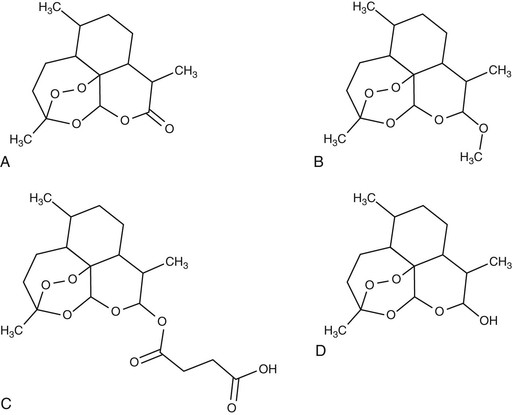
The use of the parent compound artemisinin has been superseded in most countries by the development of four main derivatives: the water-soluble hemisuccinate artesunate, dihydroartemisinin (DHA), and the lipophilic methyl ethers artemether and artemotil (formerly known as arteether). Artesunate, the most widely used of the derivatives, is available in oral, intravenous, intramuscular, or rectal formulations. It is the only formulation that can be administered intravenously. Artemether, a methyl ether derivative, can be administered by intramuscular injection suspended in peanut oil, or as capsules for oral administration. Artesunate, artemether, and artemotil are synthesized from the active metabolite DHA. Novel synthetic and semisynthetic derivatives are also available, with potent antimalarial activity in vitro.3 Artemisone and artemiside (semisynthetic 10-alkylaminoartemisinin derivatives) demonstrate even greater potency than the artemisinin derivatives but suffer from chemical instability.4 Artelinic acid was designed and synthesized as a more stable alternative to artesunate for intravenous use5 and demonstrates potent in vitro activity,6 but it has yet to be tested in vivo. Two synthetic peroxides (OZ277 and OZ439) are undergoing early clinical trials.7
Structures of Artemisinin Compounds
The artemisinin derivatives demonstrate excellent efficacy against all human malaria parasites, including multidrug-resistant strains of Plasmodium falciparum8 and P. vivax9 (Fig. 40-2). These drugs have also been shown to have in vitro activity against other parasites, including Schistosoma, Fasciola, Opisthorchis, Clonorchis, and Leishmania, although susceptibility is more modest, and they have not received the same degree of preclinical and clinical development for these indications.10–12 Although the activity of the artemisinins is known to be dependent on the endoperoxide dioxygen bridge, the exact mechanism of their antimalarial activity remains unclear. The active endoperoxides accumulate in various parasite compartments, including the cytosol, digestive vacuole, and membranes. The interaction between the drug and intraparasitic heme-derived iron appears to be a crucial step both in vitro and in vivo,13,14 in which the endoperoxide bridge is cleaved to hydroperoxide, the resultant hydro-peroxide-metal complex acting as a powerful oxidizing agent, releasing carbon-centered free radicals and other reactive metabolites.15,16 These reactive metabolites bind with hemin, parasite proteins, and red cell membranes, which are thought to cause critical damage to parasite organelles.17 Other studies have implicated interference with calcium homeostasis, a translationally controlled tumor protein homologue (TCTP), the mitochondrial electron transport chain, and inhibition of angiogenesis.
Pharmacokinetics and Dynamics.
The artemisinins are absorbed rapidly after oral administration, the maximum plasma concentration occurring at 2 to 3 hours for artemisinin and artemether, and at less than 1 hour for artesunate.18,19 When administered rectally, peak concentrations are achieved between 4 and 7 hours for artemisinin, but within 1 hour for artesunate. Absorption of artemether when administered by intramuscular is more variable, with maximum concentrations varying between 2 and 10 hours after injection.20
Artesunate is metabolized almost immediately to dihydroartemisinin, such that it is often considered a prodrug. Pharmacokinetic studies therefore tend to present the profile of dihydroartemisinin rather than artesunate. Maximum concentrations of dihydroartemisinin occur at 10 minutes after IV injection of artesunate, 30 to 40 minutes after IM injection, and 1 to 2 hours after oral and rectal administration.21,22 Bioavailability of these compounds varies with the route of administration and duration of therapy. For instance, the bioavailability of oral artesunate is approximately 60%22,23 but falls to 16% when administered rectally,21 whereas the bioavailability of dihydroartemisinin after artesunate is 86% to 88% when it is given parenterally.24 The bioavailability of artemisinin after oral administration is significantly less (32%) than that for oral artesunate.25 The bioavailability of oral artemether is far more variable, ranging from 43% to 200%.26,27
Host factors can affect the bioavailability of some artemisinin derivatives. The bioavailability of dihydroartemisinin is increased twofold in patients with malaria, compared with healthy volunteers,28 and is twofold higher during the acute phase of infection compared with convalescence.23 However, in pregnant women with malaria the bioavailability of artesunate is reduced fourfold compared with nonpregnant malaria patients.29 The bioavailability of oral artemisinin, but not artesunate, falls by more than 80% after 5 days of treatment, a process thought to relate to autoinduction of metabolism.30
Once artemether, artesunate, and arteether are absorbed, there is extensive first-pass metabolism and rapid biotransformation to dihydroartemisinin. The elimination half-life of dihydroartemisinin after all routes of administration of artesunate is less than 1 hour.21,24 The elimination half-life of artemether and dihydroartemisinin after artemether, given either orally or intramuscularly, is longer, varying from 2 to 12 hours. Metabolic pathways for the biotransformation of artesunate, artemether, arteether, and dihydroartemisinin differ from that observed for the parent compound artemisinin. The metabolism of artemisinin primarily involves CYP2B6,31 with the inactive metabolites excreted via bile. Artemether and arteether are metabolized predominantly by the hepatic cytochrome CYP3A4 to DHA. Dihydroartemisinin is metabolized by hepatic cytochrome P-450, involving biotransformation to biologically inert glucuronides that are eliminated in bile.32
Available pharmacokinetic data suggest that the plasma concentrations of the artemisinin derivatives and DHA are similar in children and adults, although no data exist in infants younger than 1 year of age. Conversely, the kinetics of DHA are significantly modified in late pregnancy, with plasma concentrations lower than in nonpregnant adults,29 suggesting dose-optimization studies in pregnant women may be necessary.
Clinical Use.
Comparative clinical studies have shown the artemisinin compounds to act faster than any other antimalarial, with typical fever clearance times being approximately 20 hours and parasite clearance achieved within 48 hours.33 Although this class of drugs is extremely potent, treatment failure appears to depend on the duration of artemisinin therapy, rather than on the magnitude or frequency of the dose given. Because these compounds are rapidly eliminated, a prolonged course (minimum 7 days) of monotherapy is required to effect cure,34 particularly in patients presenting with high initial parasitemia.35 For this reason the World Health Organization recommends that artemisinin derivatives always be administered with a longer half-life partner drug to improve cure rates,36 so-called artemisinin combination therapy (ACT).
Combination regimens with an artemisinin derivative offer several inherent benefits. The rapid clinical response can improve the tolerability and absorption of the combination partner drug, which can often be compromised in an acutely febrile patient.37,38 The rapid reduction of the pathogen biomass achieved with an artemisinin derivative also reduces the number of asexual parasites exposed to the second drug, thereby reducing the chances of a resistant mutant emerging during treatment.39 Although difficult to prove, the sustained clinical efficacy of several ACTs suggests that this strategy has indeed slowed the rise of antimalarial resistance in areas where use has been widespread.40 A combination treatment regimen also serves to protect the artemisinin compounds because neither drug will be exposed to the parasites alone.40a–40d Artemisinin derivatives have the additional benefit of dramatically reducing the production of gametocytes, the sexual stages of the parasite.41 Because recrudescent infections are associated with an increase in gametocyte carriage, the reduction in their carriage after ACT therapy may help to slow the emergence of resistance and reduce malaria transmission in settings of low endemicity.41a
More than 80 malaria endemic countries have now endorsed the use of ACT as the first treatment of uncomplicated malaria, with huge clinical experience supporting their safety and efficacy.36 To date more than 100 million treatments of artemether-lumefantrine have been dispensed in malaria-endemic countries. A fixed-dose combination has been registered by regulatory authorities in Europe, the United States, and Australia (Coartem), where it is prescribed for the treatment of returning travelers and migrants with malaria. More recently two new ACTs have been licensed through the European Drug agency: dihydroartemisinin-piperaquine (Eurartesim) and artesunate-pyronaridine (Pyramax), although their use in nonendemic countries is so far limited.
Severe Malaria.
The rapid action and broad-stage specificity of the artemisinin derivatives are critically important for their role in the treatment of severe malaria and for prevention of the development of severe disease. Patients with high parasitemia (>4%) are at far greater risk of mortality compared with those with lower parasite biomass. In this context, oral artesunate results in a far faster parasite reduction compared with intravenous quinine, with potential to arrest the progression to severe disease.42 More recently the clinical benefits of accelerated parasite clearance have been highlighted in patients with severe malaria in multicenter, randomized clinical trials comparing intravenous quinine to artesunate. The first, conducted at four sites in Southeast Asia in 1461 patients, highlighted a 35% reduction in case fatality with intravenous artesunate (from 22% to 15%).43 The second, conducted in 5425 patients enrolled at 11 African sites, involved a greater proportion of children and showed a 22% reduction in fatality (from 10.9% to 8.5%).44 A meta-analysis of these results, together with other smaller clinical trials, highlights overwhelming evidence for the clinical efficacy of intravenous artesunate in reducing the mortality from severe malaria in an endemic setting (odds ratio [OR] 0.69 [95% confidence interval [CI: 0.57 to 0.87]).44
Artesunate can be given by slow IV or IM injection, at an initial dose of 2.4 mg/kg (≈140 mg in adults), followed by 2.4 mg/kg at 12, 24, and 48 hours, with further daily doses if required. Once oral treatment can be tolerated, an appropriate artemisinin combination partner can be administered to complete eradication of remaining infecting parasites. In addition to parenteral formulations, the rapid absorption of rectal artesunate and high plasma concentrations gives it significant advantages over the other artemisinin derivatives in prehospital treatment of malaria.45 In settings without ready access to parenteral therapy, an artesunate suppository at a dose of 10 mg/kg may be administered before reaching a health care facility where more intensive treatment can be given.46
Resistance.
The artemisinin derivatives have been in clinical use in China and Cambodia for almost 40 years, while deployment in the rest of the world has increased substantially over the past 15 years. Artemisinin resistance has been induced in laboratory strains, the speed of acquisition varying between different plasmodium species.47–50 Evidence of declining artemisinin susceptibility in P. falciparum has been observed from in vitro studies, suggesting reduced drug susceptibility in field isolates in areas with high levels of multidrug resistance.8,51,52
Recent clinical studies from Cambodia have raised concerns over declining efficacy to artemether-lumefantrine, with the risk of recurrence rising to 15% to 30% within 1 month of treatment.53 Subsequent studies have documented concerning trends of increasingly delayed parasite clearance on the western provinces of Cambodia, historically an epicenter of drug-resistant malaria.54,55 These reports have been followed by others from the border regions between Thailand and Myanmar and greater Mekong region showing a similar trend, albeit at less prominent levels as that reported from Cambodia.56,57 There is ongoing controversy regarding the phenotypic characterization of such artemisinin resistance. However, patients who are slow to clear their initial parasitemia are at greater risk of producing gametocytes and having recrudescent infection after combination therapy. These parasites thus have the potential of spreading to other malaria endemic regions, undermining the current treatment strategies, which are heavily dependent on the ACTs.44
Modeling of parasite clearance data suggests that the artemisinin resistance phenotype is associated with reduced inhibition of the immature ring stages of parasite development,58 and there is some in vitro evidence to support this.59,59a This parasite trait appears to be heritable,60 with genetic studies identifying several key areas of the parasite genome under selective pressure.61,61a Work is ongoing to identify the genetic loci responsible for artemisinin resistance.
The public health consequences of declining treatment efficacy, particularly in areas of high transmission, are alarming, a threat that is heightened by the fact that no other antimalarial medicines are currently available that offer the same level of efficacy and tolerability as ACTs. This has prompted the World Health Organization to produce the Global Plan for Artemisinin Resistance Containment (GPARC),62 which sets out a proposed strategy for better coordination and resources to contain and prevent artemisinin resistance at both global and local levels.
Toxicity.
The artemisinin drugs have an excellent toxicity profile, the most commonly reported adverse effects being nausea, vomiting, and diarrhea, all of which are frequently reported during an acute episode of malaria.63,64 No differences in the incidence of possible clinical adverse reactions have been noted between derivatives. There have been a few cases of acute urticaria and anaphylaxis after oral artesunate and artemether alone65 or in combination63; readministration to these individuals should be avoided.
High doses of the liposoluble compounds (artemether and arteether) have been associated with neurologic toxicity in animal studies involving rodents, dogs, and monkeys.66–69 The pattern of neuronal damage is unusual, preferentially affecting the brainstem, in particular the reticular formation, vestibular system nuclei, and trapezoid nucleus. In animals, manifestation includes gait disturbance, loss of spinal, brainstem, and pain responses and cardiorespiratory depression. However, prolonged exposure at high doses (>15 mg/kg/day for more than 15 days) through parenteral administration is required to elicit these phenomena, the risk being far greater after parenteral rather than oral administration.
The clinical relevance of such neurotoxicity in humans exposed to treatment doses of artemisinin derivatives (generally no more than a total dose of 12 mg/kg) is unclear. There have been several reports of neurologic abnormalities after artesunate treatment of patients with malaria, some of which attribute these to artemisinin neurotoxicity.70–72 However, the doses of drug administered suggest that toxicity would be unlikely. Furthermore, cerebellar dysfunction, although rare, is a well-recognized complication of malaria73; hence, these reports may reflect the presentation of patients with postmalarial neurologic syndrome, rather than drug toxicity.74
The artemisinin derivatives have been used in several million patients over the past 20 years, with more than 10,000 enrolled in clinical trials and more than 300 patients undergoing formal neurophysiologic testing. Despite this, there have been no convincing clinical reports of neurotoxicity reliably associated with these drugs.63,75 These studies have been supported further by the analysis of patients undergoing audiometry and auditory-evoked responses, which have also failed to reveal significant neurologic pathology.76,77 However, although reassuring, ongoing surveillance of cumulative dosing is warranted, in view of the animal data.
Cardiotoxicity does not appear to be a significant issue with the artemisinins; electrocardiographic changes are confined to minor QT prolongation, sinus bradycardia, and a few cases of transient first-degree heart block.78 Hematologic abnormalities have been noted, particularly mild transient neutropenia (1.3%), when doses of artesunate in excess of 18 mg/kg total dose have been administered. A dose 30% higher than the 12 mg/kg total is usually recommended.79–81
Embryotoxicity and fetal resorption, but not teratogenicity, have been reported in animal studies at relatively low doses of artemether (7.5 to 15 mg/kg/day).82 However, clinical studies from the Thai-Myanmar border and The Gambia have indicated that artesunate and artemether are well tolerated in pregnancy, with no evidence of an increased risk of abortion, stillbirth, congenital abnormality, or premature delivery.83–86 In one study, where infants were monitored in the first year of life, normal developmental milestones were attained.87
Available data are reassuring for administration of these drugs in later pregnancy, although data are less comprehensive in the first 3 months.88 In view of the serious adverse effects of malaria on fetal well-being and the lack of evidence of toxicity, the 2006 World Health Organization guidelines recommended using ACT to treat pregnant women in the second and third trimesters but endorsed quinine plus clindamycin in the first trimester.36
4-Aminoquinolines
Chloroquine
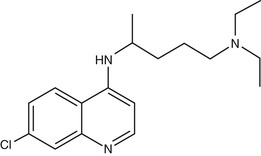
Chloroquine (7-chloro-4-[4-diethylamino-1-methylbutylamino] quinoline) (Fig. 40-3) was first synthesized in Germany in 1934 but not developed until after World War II.89 Hydroxychloroquine (Plaquenil), which differs from chloroquine only by hydroxylation at the end of the side chain, has identical antimalarial properties to chloroquine and is increasingly used in parts of the world where the availability of chloroquine is becoming limited. Hydroxychloroquine has anti-inflammatory and antimalarial properties and is less prone to cause ocular toxicity than chloroquine in those receiving prolonged treatment.90 Chloroquine and other 4-quinoline antimalarials are only active against the blood stages of the parasite life cycle during which hemoglobin is degraded.
Chloroquine, a weak base, concentrates in the food vacuoles of intraerythrocytic parasites due to a pH gradient between the parasite cytoplasm and the acidic food vacuole of the parasite. Once it enters the food vacuole it is rapidly converted to a membrane-impermeable protonated form, where it is trapped. It binds to heme released after the parasite’s digestion of host hemoglobin, preventing its conversion to the inert crystal haemozoin. It is the resulting accumulation of toxic heme monomers or the heme-chloroquine complex, or both, that is believed to kill the parasite.91
Pharmacokinetics.
Chloroquine is well absorbed whether given by oral, subcutaneous, or intramuscular routes. Rectal bioavailability exceeds 90% but falls to 22% to 24% of the oral dose92 unless a noncoated tablet is used, in which case plasma concentrations are only slightly lower than those found with oral administration.93 Chloroquine is 46% to 74% protein bound; it has an extremely large apparent volume of distribution and very long terminal half-life. As a consequence, a loading dose is required to yield effective plasma concentrations and its pharmacokinetic profile is largely determined by its distribution rather than the elimination phase. Therapeutic drug levels in plasma are reached 2 to 3 hours after oral administration, with a mean initial half-life of 4 days. As plasma levels decline, the rate of excretion decreases, thus enabling once-weekly administration for prophylaxis. The estimated terminal elimination half-life is 45 to 55 days for chloroquine and 59 to 67 days for its active metabolite desethylchloroquine. Chloroquine is detectable in the urine for up to a year after drug administration.94 Urinary excretion is the main route of elimination for both chloroquine and desethylchloroquine. The elimination half-life of chloroquine is significantly prolonged in chronic renal disease, with an associated increase in the area under the curve (AUC); hence, a dose reduction is warranted in patients with renal insufficiency taking chloroquine for prophylaxis, although not required for acute malaria.95
Clinical Use.
Chloroquine remains the drug of choice for the treatment and prophylaxis of P. falciparum malaria in the few regions of the world where this species remains sensitive to this drug. Against sensitive parasites, chloroquine induces rapid parasite clearance only slightly slower than artemisinin drugs. Chloroquine remains the treatment of choice for both prophylaxis and treatment of the nonfalciparum malarias, although this is under threat for P. vivax due to an increasing prevalence of chloroquine resistance in this species.96 It lacks activity against sporozoites and mature gametocytes. Although it is not active against liver stage infection, it may potentiate the activity of primaquine as a causal prophylactic agent.97
Resistance.
Chloroquine accumulates at concentrations 4 to 10 times lower in drug-resistant parasites compared with chloroquine-sensitive (CQ-S) parasites; it is this marked decrease in chloroquine accumulation that underlies the phenomenon of chloroquine resistance. CQ-R parasites can be partially resensitized to chloroquine in vitro by a range of weak bases, including the antihistamine chlorpheniramine and the calcium channel blocker verapamil.98 This ”resistance reversal” effect is characterized by both an increase in chloroquine accumulation and an increase in the chloroquine sensitivity of CQ-R parasites. However, the concentration of these reversal agents required to reverse chloroquine resistance is generally higher than that tolerated in vivo. The key molecular determinants of chloroquine resistance involve a number of mutations in the so-called “chloroquine resistance transporter” gene, or pfcrt. Of these, the predominant resistance-conferring mutation (K76T)99 results in the loss of a positive charge from the putative substrate-binding site on the vacuolar side of the protein.100 Chloroquine-resistant P. falciparum is now found throughout the malaria-endemic world, with the exception of Central America and the Panama Canal. High-grade drug resistance of P. vivax has been documented from the Island of Papua (both Indonesian and Papua New Guinea) and across the Indonesia archipelago, Thailand, Myanmar, Horn of Africa, and Brazil.96,101 The mechanism of resistance and genetic basis of chloroquine resistance in P. vivax appears to be different from that underlying resistance to P. falciparum.102
Toxicity.
Chloroquine is generally well tolerated.64 The most commonly reported adverse effect is pruritus, which is more common in dark-skinned populations.103 It predominantly affects the palms, soles, and scalp and starts within 24 hours of exposure and can last for several days. As systemic equilibration of chloroquine is relatively slow, this combined with the cardiac and central nervous system (CNS) toxicity of high plasma levels renders parenteral administration or overdose hazardous, with seizures and death from cardiovascular collapse resulting from high plasma levels.104 Although chloroquine crosses the placenta,105 doses used for chemoprophylaxis do not cause a significant increase in fetal abnormalities. Chloroquine is safe in nursing mothers. When used for prolonged periods (>5 years prophylaxis), the cumulative accumulation can cause retinal damage,106 and regular ophthalmologic examination is appropriate if long-term therapy is undertaken.
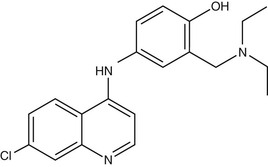
Amodiaquine
Amodiaquine (AQ) (Fig. 40-4) is a 4-aminoquinoline that was widely used for prevention of malaria until the mid-1980s, when reports of fatal adverse drug reactions described in travelers using AQ as prophylaxis107 resulted in a dramatic decrease in its use for these indications. However, reevaluation of the drug for treatment rather than prophylaxis108 resulted in increased recognition that the drug is not significantly more toxic than chloroquine when used for treatment. Its activity against chloroquine-resistant parasites gives it an important role in the antimalarial pharmacopoeia. However, this activity against CQ-R parasites shows significant geographic variation, activity being greater in isolates from Africa,109–111 whereas those from Southeast Asia, South America, and Papua New Guinea display high levels of AQ resistance.112 The apparent geographic specificity of AQ resistance may be due to differences among the PfCRT haplotypes found in these locations.113 The mechanism of action of AQ is believed to be similar to that of chloroquine.
Pharmacokinetics.
In contrast to chloroquine, AQ has a relatively short half-life (5 hours), being rapidly cleared by the liver by the action of CYP2C8 to an active metabolite, desethylamodiaquine AQ. The half-life of this metabolite has been variously estimated to be 1 to 3 weeks in adults and 3 to 12 days in children. Polymorphism of this enzyme may be a factor in drug interactions.
Clinical Use.
The principal use of the drug is as a component of fixed-dose co-formulation with artesunate (4 mg/kg daily for 3 days). Recently, AQ has been used and co-formulated with artemisinin drugs, particularly artesunate, as an ACT. AQ retains efficacy against chloroquine-resistant P. vivax,9 although recurrence rates after AQ monotherapy and AQ plus artesunate are high.114,115
Toxicity.
The most important adverse effects of AQ are hepatotoxicity (1/15,000 exposures) and bone marrow toxicity (1/2200 exposures; fatality rate, 1 in 31,000). Of note, none of these toxic effects have been reported when AQ has been used for malaria treatment but only in more prolonged use as a prophylactic agent. However, the actual incidence of such serious side effects in the setting of treatment is unknown. An important recently reported side effect is the significant risk of severe neutropenia in HIV-infected children treated for malaria with artesunate-AQ.116
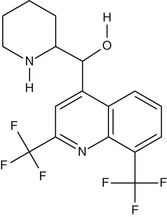
Mefloquine
Mefloquine (Fig. 40-5) is a chiral quinoline methanol. It is active against the asexual erythrocytic stages of malarial parasites but not against sporozoites, liver-stage parasites, or gametocytes. Mefloquine forms a complex with heme that may be toxic to the parasite,117 although its mechanism of action is believed to be distinct from that of chloroquine. Of note, chloroquine-resistant parasites are more sensitive to mefloquine and vice versa.118
Pharmacokinetics.
Mefloquine is only available as an oral tablet formulation. Absorption is adversely affected by vomiting and diarrhea but significantly enhanced when administered with or after food.119 The mean absorption half-life of mefloquine after a single oral dose is 2.1 hours in healthy volunteers and 5.4 hours in patients with uncomplicated falciparum malaria.120 About 98% of the drug binds to protein.121 The terminal elimination half-life is 14 to 28 days. The apparent volume of distribution is compatible with plasma protein binding of more than 98%.121 Pharmacokinetic parameters are similar in single- and multiple-dose studies of healthy volunteers, suggesting that there are no significant changes in clearance when mefloquine is taken for prophylaxis. Mefloquine at a dose of 250 mg taken weekly reaches steady-state concentrations in 10 weeks without any indication of accumulation.122 Mefloquine pharmacokinetics are highly stereoselective, with the (−) enantiomer demonstrating a significantly greater Cmax, AUC, and longer elimination half-life than the (+) enantiomer.123
No dose adjustment is necessary in renal impairment or hemodialysis. Excretion of the drug and its metabolites is largely in the bile and feces. No studies have examined mefloquine disposition in patients with significant hepatic dysfunction, ascites, or cardiac failure. Because mefloquine is largely metabolized by the liver, the use of usual prophylactic doses over time may cause significant accumulation and consequent increased toxicity in patients with liver disease.
Clinical Use.
Mefloquine is indicated for the treatment and prophylaxis of malaria, including chloroquine-resistant strains. Treatment regimens initially recommended a single dose of 15 mg/kg, although a higher dose (25 mg/kg) has been recommended to overcome declining efficacy. A combination with artesunate (4 mg/kg/day) as a 3-day regimen has been used in parts of Southeast Asia for almost 20 years and appears to retain efficacy despite underlying high-grade mefloquine resistance.40 A fixed-dose combination of artesunate-mefloquine is now available.124
Resistance.
High-grade mefloquine-resistant strains of P. falciparum have been documented in Southeast Asia, particularly the western border of Thailand, since the early 1990s. Cross-resistance of mefloquine with halofantrine and with quinine has been documented.125 P. falciparum isolates resistant to mefloquine have both increased copy number of the pfmdr1 gene and increased expression of the gene product.126
Toxicity.
The adverse effects of mefloquine have been the subject of much study. More than 20 million doses have been prescribed since 1984. Consistent with the frequency of side effects during other chemoprophylactic regimens, weekly mefloquine is associated with symptoms such as dizziness, nausea, and diarrhea in 20% of travelers.127 These symptoms are usually mild and self-limiting. Dose-related drug-induced vomiting can be problematic, particularly in young children, in whom rates can reach 30% in infants younger than 2 years of age. When given as part of an ACT, vomiting can be reduced by delaying the administration of the mefloquine component of the ACT until day 1 or 2.128 Less serious side effects include rash, pruritus, and oral ulcers.129 More serious dermatologic reactions have been reported but are exceptionally rare.
The most serious adverse effect of mefloquine is neuropsychiatric toxicity. This can develop at any time during prophylaxis or treatment, and symptoms range from mild to life threatening. The incidence of neuropsychiatric effects appears higher in Caucasians and Africans than in Asians and higher in women than men.127 Although difficult to assess in the context of an infection that can itself cause profound neurologic disturbances, psychosis, depression, encephalopathy, convulsions, and other severe neuropsychiatric effects occur after the use of mefloquine for treatment of falciparum malaria, with a frequency of 1 : 200 to 1800 courses.130 Symptoms typically resolve within 3 to 4 weeks.127 Serious reactions similar to those seen after treatment courses can occur during prophylaxis, with rates of 1 : 10,000 to 20,000 courses reported in large trials.129,130 Sleep abnormalities (e.g., insomnia, abnormal dreams) have occasionally been reported. Mefloquine should not be prescribed to patients with neuropsychiatric conditions, including depression, anxiety, psychosis, schizophrenia, and seizure disorder. If acute anxiety, depression, restlessness, or confusion develops during prophylaxis, the drug should be discontinued. Mefloquine-induced neurotoxicity can be difficult to distinguish from the postmalarial neurologic syndrome (PMNS), which typically lasts 1 to 10 days and consists of acute confusion, psychosis, and tremor, with or without convulsions. PMNS has been reported to occur at a rate of 1 : 1000 patients131 but appears to be more common among patients with severe malaria, with frequencies of PMNS being reported in 1 in 20. This late complication is almost 10 times more likely to occur after treatment with mefloquine than quinine.131 Of note, re-treatment with mefloquine within 1 month of a course of therapy markedly increases the risk of adverse effects sevenfold, rising from 1/1217 to 1/173.132 Of, importance, the incidence of neurotoxicity is significantly higher following severe malaria (1/20), and thus the drug should not be used in this setting.
Mefloquine should not be used concurrently with quinine, quinidine, or drugs producing β-adrenergic blockade owing to the risk of electrocardiographic abnormalities or cardiac arrest. Administration of mefloquine with quinine or chloroquine may increase the risk of convulsions. Mefloquine may lower plasma levels of anticonvulsants.133 Minor or inconsistent interactions have been observed between mefloquine and sulfadoxine-pyrimethamine, primaquine, metoclopramide, and oral contraceptives. Mefloquine has been shown to exert variable effects on ritonavir pharmacokinetics.
Although mefloquine prophylaxis is not recommended during pregnancy, the drug is probably safe and effective and discovery of pregnancy during mefloquine prophylaxis is not an indication for pregnancy termination.134
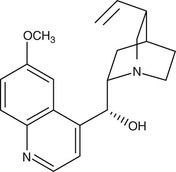
Quinine and Quinidine
Derived from the bark of the cinchona tree, quinine remained the sole effective antimalarial until the development of synthetic antimalarials during and after World War I. As the availability of the racemic mixture of quinine is reducing in the developed world, its D-enantiomer, quinidine, which is also used as a cardiac antiarrhythmic, can be used as a replacement135 (Fig. 40-6). Intravenous quinine is the second-line alternative to intravenous artesunate for treatment of severe malaria.135,136 In recent large-scale. multicentered randomized trials undertaken in both children and adults, parenteral artesunate was shown to be more efficacious in the SEQUAMAT and AQUAMAT studies,43,44 reducing mortality by 22% to 35%. As is the case for chloroquine, the only life cycle stage of P. falciparum that quinine is active against is the asexual erythrocytic form. It has little effect on sporozoites or liver-stage parasites. It is gametocidal against P. vivax and P. malariae but not against P. falciparum. Quinine is active against the zoonotic primate malaria species P. knowlesi.137 Although the mechanism of action of quinine against Plasmodium spp. is not known, it appears to inhibit the digestion of hemoglobin. It has also been shown to inhibit the ATPase of P. falciparum food vacuoles138; this mechanism appears to be independent of its alkalinization of the food vacuole.139
Pharmacokinetics.
The pharmacokinetic properties of quinine have been extensively reviewed.140 Two clinically important pharmacokinetic issues are the risk of underdosing in severe malaria necessitating a loading dose (see later)141 and the variation in pharmacokinetic parameters in patients with severe malaria.140 Although quinine levels are higher in severe malaria, toxicity is uncommon compared with equivalent doses in healthy subjects. This is due to increased plasma binding to alpha-1 acid glycoprotein, an acute-phase reactant elevated in malaria,142 which reduces free quinine levels by 25% to 30% in uncomplicated malaria and by 40% in severe malaria.143–145 With increasing severity of malaria, there is also a reduction in the volume of distribution and clearance (particularly hepatic clearance) because of the changes in free quinine levels.140 Both quinine and quinidine are metabolized by the hepatic microsomal enzyme cytochrome P-450 3A4; quinidine is also metabolized by CYP 2D6.140
It is important to avoid confusion arising from calculation of the dose of quinine depending on its various salts. The following doses are equivalent: quinine base 100 mg; quinine sulphate 121 mg; quinine bisulphate 169 mg; and quinine dihydrochloride 122 mg. The dose of quinine for treatment of malaria is 10 mg/kg, given three times a day for 7 days.
Pharmacokinetic parameters of quinine in patients with acute renal failure appear to be similar to patients with severe malaria without renal failure.146 A dose reduction (of 30% to 50%) is suggested only after 2 days of standard quinine therapy (including standard loading doses) for patients with acute renal failure or who remain severely unwell.140
Clinical Use.
Quinine is less well tolerated than other antimalarials (see later) and requires prolonged course of treatment (at least 7 days) to effect a cure. Hence, completion of a therapeutic course is problematic. It remains an option as an oral treatment of all species of malaria, particularly in pregnant women, infants, and patients with suspected drug resistant infections recurring following a compromised treatment regimen. To reduce the risk of recrudescence, quinine (10 mg salt/kg three times daily for 7 days) is often combined with clindamycin (10 mg/kg twice a day for 7 days). In children older than 8 years, clindamycin can be substituted for either doxycycline (3 mg/kg orally daily) or tetracycline (4 mg/kg orally four times per day).147,148 Further, because of the poor tolerability of quinine, especially after 3 days, many recommended oral regimens for mild malaria entail 3 days of quinine, with a full 7 days of the partner drug. When used for treatment of severe malaria, it is imperative that therapeutic drug concentrations are reached as quickly as possible and hence an initial loading dose should be prescribed141 followed by a maintenance dose or infusion. Two loading-dose regimens can be used:
• The standard regimen is quinine dihydrochloride 20 mg of salt/kg by IV infusion over 4 hours OR
• An alternative regimen has been suggested with quinine dihydrochloride 7 mg of salt/kg by IV infusion over 30 minutes followed by 10 mg/kg infused over 4 hours.149
A common clinical dilemma is whether to administer a loading dose if there is a history of self-medication with antimalarials. The risk of inadequate treatment due to underdosing must be weighed against the risk of serious cardiac toxicity due to overdose. Authorities suggest omitting the loading dose if more than 2 g of quinine has been administered within the past 48 hours.150,151
If an intravenous infusion cannot be safely administered, quinine can also be administered intramuscularly. Although pharmacokinetic studies of intramuscular quinine are lacking in adult patients with severe malaria, a loading dose appears to be well absorbed in patients with uncomplicated malaria152 and efficacious in severe malaria.153 Quinine should be diluted with water to reach a concentration for administration of less than 150 mg/mL to reduce local pain and improve absorption, and be administered in divided doses into each thigh.140,154
Quinine Resistance.
Quinine resistance in P. falciparum was first reported in Brazil in 1908.155 Although quinine resistance of P. falciparum varies globally, published data on its prevalence since the increased use of artemisinin antimalarials are sparse. Resistance is more common and severe in Southeast Asia and has paralleled the rise of resistance to mefloquine,156 suggesting cross-resistance between these two drugs may be playing a role. As is the case for mefloquine, amplification of the drug transporter pfmdr1 is associated with increased quinine IC50. However, there is a paucity of data associating pfmdr1 amplification and clinical outcome.157
Quinine for Treatment of Babesiosis.
Quinine in combination with clindamycin is the treatment of choice for severe babesiosis at doses similar to those used for treatment of malaria; quinine 8 mg salt/kg (up to 650 mg in adults) every 8 hours, with clindamycin 7 to 10 mg/kg (up to 600 mg in adults) every 6 to 8 hours.158 Azithromycin with atovaquone is a better-tolerated regimen and equally effective in patients with mild-moderate babesiosis.159
Toxicity.
The syndrome of cinchonism (tinnitus, high-tone deafness, visual disturbances, headache, dysphoria, vomiting, and postural hypotension) commonly occurs at therapeutic plasma levels, with plasma levels correlating with severity.160 Reversible deafness, which is predominantly high-tone, also correlates with plasma concentrations.161–164 The most common cardiovascular adverse events associated with quinine and quinidine are postural hypotension and syncope.78 Although cardiotoxicity due to quinine is unusual at plasma concentrations in the therapeutic range,164,165 quinidine causes QT interval prolongation in approximately 3% of patients.166 Fatal ventricular arrhythmias have only been described in a patient with a preexisting prolonged QT interval (490 ms).167 Acute toxicity caused by inadvertent rapid infusion may cause life-threatening cardiac arrhythmias, including ventricular tachycardia and ventricular fibrillation.168 Patients being treated with quinidine should have regular clinical assessment for hypotension, a baseline electrocardiogram (ECG) for assessment of corrected QT interval, and cardiac monitoring. Patients being treated with quinine do not generally require cardiac monitoring unless there is preexisting heart disease or the potential of interactions with other medications that are known to affect cardiac electrophysiology or in children younger than 2 years receiving intravenous treatment. Quinine has been associated with transient retinal toxicity, which typically occurs in drug overdose; the majority of patients report complete blindness, but alterations in color vision, blurring of vision, or visual field restriction are described.169
An important adverse effect of quinine treatment in severe malaria is hypoglycemia.170,171 Pregnant women are particularly susceptible to this complication.172 All patients receiving quinine or quinidine should be monitored for hypoglycemia, and any deterioration in clinical status (including seizures or altered mental status) should prompt assessment of plasma glucose. Patients receiving intravenous therapy should also receive an intravenous infusion of dextrose.
Blackwater fever is the syndrome of massive hemolysis (with visible hemoglobinuria) in the setting of severe malaria, glucose-6-phosphate dehydrogenase deficiency (G6PD), and quinine treatment. Because malaria is also associated with hemolysis, the relative roles of quinine and G6PD are unclear.153,173,174
Allergic reactions, such as drug-induced thrombocytopenia,175 hepatitis, urticaria, and other more severe dermatologic phenomena (erythema multiforme, fixed drug eruptions, and toxic epidermal necrolysis),168,176,177 are all much less common following malaria chemotherapy than when the drug has been used long term.
The risks of using quinine in pregnancy must be weighed against the known deleterious effects of malaria on pregnancy. Some, but not all, animal studies have demonstrated abnormalities of the inner ear, auditory nerve, and CNS.134,178–180 At standard doses, quinine does not increase the rate of spontaneous abortion, and its use in the first trimester does not appear to be associated with birth defects.181 Controlled trials have not found evidence of an increased risk of birth defects.182 As noted earlier, in pregnant women with severe malaria, hypoglycemia appears to be a common complication of quinine treatment.183
Piperaquine
Piperaquine is a bisquinoline 4-aminoquinoline antimalarial structurally related to chloroquine. It was synthesized independently in France and China in the 1960s184 and widely used for malaria control activities in China in the 1970s and 1980s.185 In the 1990s, piperaquine was reconsidered as a partner drug in artemisinin-based combination therapy and led to the development of a novel combination formulation of dihydroartemisinin plus piperaquine, each tablet containing 40 mg dihydroartemisinin and 320 mg piperaquine phosphate (DHP). The mechanism of action and resistance of piperaquine have not been well studied but are likely to be similar to those of drugs of the same class.185
Pharmacokinetics.
The pharmacokinetic properties of piperaquine are similar to those of chloroquine. It has a large volume of distribution, ranging from 103 to 716 L/kg, values that are significantly larger even than comparable drugs such as chloroquine.186–188 It has a long terminal elimination half-life of 22 days and 20 days in adults and children, respectively.189 The prolonged half-life results in a beneficial post-treatment prophylactic period, estimated to be about 20 days, and protects against both P. vivax and P. falciparum. Although early recurrent infections are reduced, infections treated with DHP are more likely to produce gametocytes than artemether-lumefantrine, an observation hypothesized to reflect the lower dosing of artemisinin derivative in DHP (total ≈7.5 mg/kg of DHA compared with ≈11.5 mg/kg of artemether in artemether-lumefantrine). Furthermore, the drug has a smaller volume of distribution and shorter half-life in children, resulting in a higher risk of recrudescence and earlier reinfection. Thus, an increase of the weight-adjusted dosage in young children may be required.189a
Piperaquine is highly lipophilic, and its oral bioavailability is approximately doubled by administration with a high-fat meal.190,191 However, data on the food effect of bioavailability of piperaquine are conflicting.187,192,193 In a study carried out in Papua New Guinea, a surprisingly low efficacy of DHP was reported (88% at day 42), significantly lower than that for artemether-lumefantrine. However, the difference had wide CIs and was apparent at day 28 but not day 42. This reduced efficacy is in contrast to other studies carried out in Africa194–196 and Asia,197,198 where DHP had similar or higher efficacy to other ACTs.
Clinical Use.
Eurartesim is available in tablets of two strengths, either 20/160 or 40/320 mg of dihydroartemisinin/piperaquine, respectively. It should be administered over three consecutive days for a total of three doses taken at the same time each day. The recommended dose is 2/16 mg/kg, once daily for 3 days (equivalent to 3 to 4 40/320 tablets for adults). Some manufacturers recommend four doses (at 0, 6 to 8, 24, and 32 hours), although there is no evidence that this regimen has any better efficacy than the simpler once-daily dosing. Development of a pediatric formulation is under way.
Resistance.
Although cross-resistance between piperaquine and chloroquine has been reported,9,184 piperaquine has potent activity against highly chloroquine-resistant P. falciparum189 and P. vivax.9 More recently, declining efficacy of DHP has been noted in the eastern provinces of Cambodia in the epicenter of artemisinin resistance, with the risk of true recrudescence rising to 25%.199 However, CIs were wide, pharmacokinetic measures were not taken, and it is unclear whether the failures were due to the DHA or piperaquine component of the drug combination. Further studies are ongoing.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


