Antigen–Antibody Interactions, Immune Assays, and Experimental Systems
Introduction
In previous chapters, we have, by necessity, touched upon several techniques and assays that have been used to help us understand some fundamental aspects of innate and adaptive immunity. In this chapter we discuss, in greater detail, in vitro techniques, assays, and experimental systems that are used in research and diagnostic laboratories. Some of these are strictly antibody-based (e.g., serological methods), whereas others employ molecular biological methods, genetic engineering, cell culture techniques, and in vivo animal models that have greatly contributed to our understanding of the physiology and pathophysiology of the immune system. Since the sequencing of the human genome in 2000, and with aggressive efforts to sequence microbial genomes, approaches that use bioinformatics and computational biology (so-called in silico analyses) have emerged as promising methods for the study of our immune system. Using information derived from genomic and proteomic databases, powerful software tools and algorithms, these technologies hold great promise for the field of immunology. This is particularly true with regard to important efforts to identify immunogenic epitopes expressed by pathogens that can be studied further as candidate vaccines. Although this topic is beyond the scope of this chapter, it is important to keep in mind that future progress in the field of immunology will come from a combination of in vitro, in vivo, and in silico approaches.
We begin this chapter with a discussion of physical dynamics of antigen–antibody interactions.
Antigen–Antibody Interactions
The reaction between antigen and antibodies serves as the basis for many immune assays. Because of the exquisite specificity of the immune response, the interaction between antigen and antibody in vitro is widely used for diagnostic purposes, for the detection and identification of either antigen or antibody. An example of the use of serological methods for the identification and classification of antigens is the serotyping of various microorganisms by the use of specific antisera.
The interaction of antigen with antibodies may result in a variety of outcomes, including precipitation (if the antigen is soluble), agglutination (if the antigen is particulate), and activation of complement. All of these outcomes are caused by the interactions between multivalent antigens and antibodies that have at least two combining sites per molecule. The consequences of antigen–antibody interaction listed above do not represent the primary interaction between antibodies and a given antigen but rather depend on secondary phenomena, which result from the interactions between multivalent antigens and antibodies. Such phenomena as the formation of precipitate, agglutination, and complement activation would not occur if the antibody with two or more combining sites reacted with a hapten (i.e., a unideterminant, univalent antigen), nor would they occur as a result of the interaction between a univalent fragment of antibody, such as Fab, and an antigen, even if the antigen is multivalent. The reasons for these differences are depicted in Figure 6.1. Cross-linking of various antigen molecules by antibody is required for precipitation, agglutination, or complement activation, and it is possible only if the antigen is multivalent and the antibody is divalent [either intact, or F(ab′)2]. Figure 6.1 illustrates several different types of antigens and their interactions with antibody molecules or fragments thereof [Fab versus F(ab′)2]. Figure 6.1E illustrates a typical multivarient antigen, one that expresses multiple antigenic determinants or epitopes (in this case, epitopes A, B, and C) to which antibody responses have been generated (anti-A, anti-B, and anti-C, respectively). Pathogenic microorganisms are good examples of multivariant antigens since they express an array of epitopes to which B and T cells respond during an immune response to these pathogens. The assembly of immune complexes resulting from cross-linking facilitates several goals of the immune system that are collectively aimed at destroying, neutralizing, and eliminating the pathogenic organism. As we have discussed in earlier chapters, immune complexes activate the classical complement pathway leading to the assembly of a membrane attack complex and lysis of the organism. In addition, the immune complex brings together a high density of Fc regions of the antibodies interacting with antigen, which increases the chance that phagocytic cells expressing Fc receptors will bind to these Fc regions to initiate phagocytosis of the antigen. Finally, since complement activation also generates C3b components that also bind to immune complexes, red blood cells expressing CR1 receptors on their surface bind to the C3b-decorated immune complexes and transport them to phagocytes in liver and spleen for removal.
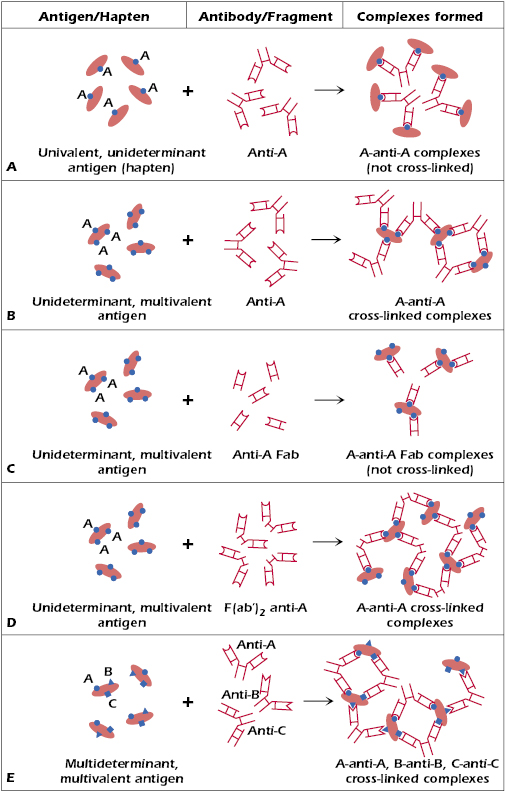
In contrast with multivalent antigens, Figure 6.1A shows molecules expressing a single low molecular weight hapten (A). If an antibody response is generated to this hapten (recall that this would require immunization using the molecule expressing the hapten together with a carrier protein in order to make the hapten immunogenic), the interactions between the anti-A antibodies and the antigen expressing the single hapten would result in complexes that are not cross-linked. Similarly, if one were to combine Fab fragments of an antibody specific for a unideterminant, multivalent antigen shown in Figure 6.1C, cross-linking of antibody–antigen complexes would not occur. Finally, in the case of unideterminant (epitope A), multivalent antigens shown in Figure 6.1B–D, cross-linking will occur when this antigen interacts with intact anti-A antibodies (Figure 6.1B) or divalent F(ab′)2 anti-A fragments (Figure 6.1C) but not with monovalent anti-A Fab fragments (Figure 6.1D).
Primary Interactions Between Antibody and Antigen
No covalent bonds are involved in the interaction between antibody and an antigen. Consequently, the binding forces are relatively weak. They consist mainly of van der Waals forces, electrostatic forces, and hydrophobic forces, all of which require a very close proximity among the interacting moieties. Thus the interaction requires a very close fit between an epitope and the antibody, a fit that is often compared to that between a lock and a key. Because of the low levels of energy involved in the interaction between antigen and antibody, antigen–antibody complexes can be readily dissociated by low or high pH, by high salt concentrations, or by chaotropic ions, such as cyanates, which efficiently interfere with the hydrogen bonding of water molecules.
Association Constant
The reaction between an antibody and an epitope of an antigen is exemplified by the reaction between antibody and a univalent hapten. Because an antibody molecule is symmetric, with two identical Fab antigen-combining sites, one antibody molecule binds with two identical monovalent hapten molecules, each Fab binding in an independent fashion with one hapten molecule. The binding of a monovalent antigen (Ag) with each site can be represented by the equation:
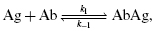
where k1 represents the forward (association) rate constant and k−1 represents the reverse (dissociation) rate constant. The ratio of k1/k−1 is the association constant K, a measure of affinity. It can be calculated by determining the ratio of bound AbAg complex to the concentration of unbound antigen and antibody. Thus,
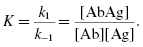
The association constant (K) is really a measure of the affinity of the antibody for the epitope (see below). When all the antibody molecules that bind a given hapten or epitope are identical (as in the case of monoclonal antibodies), then K represents the intrinsic association constant. However, because serum antibodies, even those binding to a single epitope, are heterogeneous, an average association constant of all the antibodies to the epitope is referred to as K0. The interaction between antibodies and each epitope of a multivalent antigen follows the same kinetics and energetics as those involved in the interaction between antibodies and haptens because each epitope of the antigen reacts with its corresponding antibody in the same manner as that described above.
The association constant K can be determined using the method of equilibrium dialysis. In this procedure, a dialysis chamber is used in which two compartments are separated by a semipermeable membrane allowing the free passage of appropriately sized molecules from one side to the other. Antibody is placed on one side of the semipermeable membrane and cannot pass through due to its size. On the antigen side of the membrane, a known amount of small, permeable, radiolabeled hapten molecules, oligosaccharides, or oligopeptides comprising the epitope of the complex carbohydrate or protein is added. At time zero, the hapten or antigenic epitope used (referred to as the ligand hereafter) will then diffuse across the membrane, and at equilibrium, the concentration of free ligand will be the same on both sides. However, the total amount of ligand will be greater on the side containing Ab because some of the ligand will be bound to the antibody molecules. The difference in the ligand concentration in the two compartments represents the concentration of the ligand bound to antibody (i.e., the [AgAb] complex). The higher the affinity of the antibody, the more ligand is bound.
Since the concentration of antibody added to the equilibrium dialysis chamber can be predetermined and kept constant, varying concentrations of ligand can also be used in this analysis. This approach facilitates the so-called Scatchard analysis of the antibody. This is useful in determining whether a given antibody preparation is homogeneous (e.g., monoclonal antibody) or heterogeneous (e.g., polyclonal antiserum) and in measuring the average affinity constant (K0).
Affinity and Avidity
As noted above, the intrinsic association constant that characterizes the binding of an antibody with an epitope or a hapten is termed affinity. When the antigen consists of many repeating identical epitopes or when antigens are multivalent, the association between the entire antigen molecule and antibodies depends not only on the affinity between each epitope and its corresponding antibody but also on the sum of the affinities of all the epitopes involved. For example, the affinity of binding of anti-A with multivalent A (shown in Figure 6.1) may be four or five orders of magnitude higher than between the same antibody (i.e., anti-A) and univalent A (Figure 6.1). This is because the pairing of anti-A with A (where A is multivalent) is influenced by the increased number of sites on A with which anti-A can react.
While the term affinity denotes the intrinsic association constant between antibody and a univalent ligand such as a hapten, the term avidity is used to denote the overall binding energy between antibodies and a multivalent antigen. Thus, in general, IgM antibodies are of higher avidity than IgG antibodies, although the binding of each Fab in the IgM antibody with ligand may be of the same affinity as that of the Fab from IgG.
Secondary Interactions between Antibody and Antigen
Agglutination Reactions
Referring again to the representations given in Figure 6.1, the reactions of antibody with a multivalent antigen that is particulate (i.e., an insoluble particle) results in the cross-linking of the various antigen particles by the antibodies. This cross-linking eventually results in the clumping or agglutination of the antigen particles by the antibodies.
Titer. The agglutination of an antigen as a result of cross-linking by antibodies is dependent on the correct proportion of antigen to antibody. A method sometimes used to measure the level of serum antibody specific for a particulate antigen is the agglutination assay. More sensitive, quantitative assays (e.g., enzyme-linked immunosorbent assay [ELISA], discussed later in this chapter) have largely replaced this approach for measuring antibody levels in serum. Indeed, the agglutinating titer of a certain serum is only a semiquantitative expression of the antibodies present in the serum; it is not a quantitative measure of the concentration of antibody (weight/volume). The assay is performed by mixing twofold serial dilutions of serum with a fixed concentration of antigen. High dilutions of serum usually do not cause antigen agglutination because at such dilutions there are not enough antibodies to cause appreciable, visible agglutination. The highest dilution of serum that still causes agglutination, but beyond which no agglutination occurs, is termed the titer. It is a common observation that agglutination may not occur at high concentrations of antibody, even though it does take place at higher dilutions of serum. The tubes with high concentrations of serum, where agglutination does not occur, represent a prozone. In the prozone, antibodies are present in excess. Agglutination may not occur at high ratio of antibody to antigen because every epitope on one particle may bind only to a single antibody molecule, preventing cross-linking between different particles.
Because of the prozone phenomenon, in testing for the presence of agglutinating antibodies to a certain antigen, it is imperative that the antiserum be tested at several dilutions. Testing serum at only one concentration may give misleading conclusions if no agglutination occurs, because the absence of agglutination might reflect either a prozone or a lack of antibody.
Zeta Potential. The surfaces of certain particulate antigens may possess an electrical charge, as, for example, the net negative charge on the surface of red blood cells caused by the presence of sialic acid. When such charged particles are suspended in saline solution, an electrical potential termed the zeta potential is created between particles, preventing them from getting very close to each other. This introduces a difficulty in agglutinating charged particles by antibodies, in particular red blood cells by IgG antibodies. The distance between the Fab arms of the IgG molecule, even in its most extended form, is too short to allow effective bridging between two red blood cells across the zeta potential. Thus, although IgG antibodies may be directed against antigens on the charged erythrocyte, agglutination may not occur because of the repulsion by the zeta potential. On the other hand, some of the Fab areas of IgM pentamers are far enough apart and can bridge red blood cells separated by the zeta potential. This property of IgM antibodies, together with their pentavalence, is a major reason for their effectiveness as agglutinating antibodies.
Through the years attempts were made to improve agglutination reactions by decreasing the zeta potential in various ways, none of which was universally applicable or effective. However, an ingenious method was devised in the 1950s by Coombs to overcome this problem. This method, described below, facilitates the agglutination of erythrocytes by IgG antibodies specific for erythrocyte antigens. It is also useful for the detection of nonagglutinating antibodies that are present on the surface of erythrocytes.
Coombs Test. The Coombs test employs antibodies to immunoglobulins (hence it is also called the anti-immunoglobulin test). It is based on two important facts: (1) that immunoglobulins of one species (e.g., human) are immunogenic when injected into another species (e.g., rabbit) and lead to the production of antibodies against the immunoglobulins, and (2) that many of the anti-immunoglobulins (e.g., rabbit anti-human Ig) bind with antigenic determinants present on the Fc portion of the antibody, and leave the Fab portions free to react with antigen. Thus, for example, if human IgG antibodies are attached to their respective epitopes on erythrocytes, then the addition of rabbit antibodies to human IgG will result in their binding with the Fc portions of the human antibodies bound to the erythrocytes by their Fab portion (see Figure 6.2). These rabbit antibodies not only bind with the human antibodies that are bound to the erythrocyte but also, by so doing, they cross-link (form bridges) between human IgG on relatively distant erythrocytes, across the separation caused by the zeta potential, and cause agglutination. The addition of anti-immunoglobulin brings about agglutination, even if the antibodies directed against the erythrocytes are present at sufficiently high concentrations to cause the prozone phenomenon.
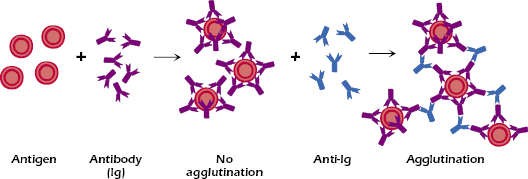
There are two versions of the Coombs test: the direct Coombs test and the indirect Coombs test. The two versions differ somewhat in the mechanics of the test but both are based on the same principle: using heterologous anti-immunoglobulins to detect a reaction between immunoglobulins and antigen. In the direct Coombs test, anti-immunoglobulins are added to the particles (e.g., red blood cells) that are suspected of having antibodies bound to antigens on their surfaces. For example, a newborn baby is suspected of having hemolytic disease of the newborn caused by maternal anti-Rh IgG antibodies that are bound to the baby’s erythrocytes. If that suspicion proved to be correct, the direct Coombs test would have the following results: the addition of anti-immunoglobulin to a suspension of the baby’s erythrocytes would result in the binding of the anti-immunoglobulin to the maternal IgG on the surface of the erythrocytes and would cause agglutination.
The indirect Coombs test is used to detect the presence, in the serum, of antibodies specific to antigens on a particle. The serum antibodies, when added to the particles, may fail to cause agglutination because of the zeta potential. The subsequent addition of anti-Ig will cause agglutination. A common application of the indirect Coombs test is in the detection of anti-Rh IgG antibodies in the blood of an Rh− woman (see Chapter 16). This consists, first, of the reaction of the woman’s serum with Rh+ erythrocytes, and then the addition of the anti-immunoglobulin reagents (as in the direct Coombs test). Thus, the direct Coombs test, it measures Ab already bound to the red blood cells (RBCs) in the patient whereas the indirect test measures antibody in the serum of the patient.
Passive Agglutination. The agglutination reaction can be used with particulate antigens (e.g., erythrocytes or bacteria) and also with soluble antigens, provided that the soluble antigen can be firmly attached to insoluble particles. For example, the soluble antigen thyroglobulin can be attached to latex particles, so that the addition of antibodies to the thyroglobulin antigen will cause agglutination of the latex particles coated with thyroglobulin. Of course, the addition of soluble antigen to the antibodies before the introduction of the thyroglobulin-coated latex particles will inhibit the agglutination because the antibodies will first combine with the soluble antigen, and if the soluble antigen is present in excess, the antibodies will not be able to bind with the particulate antigen. This latter example is referred to as agglutination inhibition. It should be distinguished from agglutination inhibition in which antibodies to certain viruses inhibit the agglutination of red blood cells by the virus. In these cases, the antibodies are directed to the area or areas on the virus that bind with the appropriate virus receptors on the red blood cells.
When the antigen is a natural constituent of a particle, the agglutination reaction is referred to as direct agglutination. When the agglutination reaction takes place between antibodies and soluble antigen that had been attached to an insoluble particle, the reaction is referred to as passive agglutination.
The agglutination reaction (direct or passive, either employing or not employing the Coombs test) is widely used clinically. In addition to the examples already given, major applications include erythrocyte typing in blood banks, diagnosis of various immunologically mediated hemolytic diseases such as drug-induced autoimmune hemolytic anemia, tests for rheumatoid factor (human IgM anti-human IgG), confirmatory test for syphilis, and the latex test for pregnancy, which involves the detection of human chorionic gonadotropin (HCG) in the urine of pregnant women.
Precipitation Reactions
Reaction in Solutions. In contrast to the agglutination reaction, which takes place between antibodies and particulate antigen, the precipitation reaction takes place when antibodies and soluble antigen are mixed. As in the case of agglutination, precipitation of antigen–antibody complexes occurs because the divalent antibody molecules cross-link multivalent antigen molecules to form a lattice. When it reaches a certain size, this antigen–antibody complex loses its solubility and precipitates out of solution. The phenomenon of precipitation is termed the precipitin reaction.
Figure 6.3 depicts a quantitative precipitin reaction. When increasing concentrations of antigen are added to a series of tubes that contain a constant concentration of antibodies, variable amounts of precipitate form. The weight of the precipitate in each tube may be determined by a variety of methods. If the amount of the precipitate is plotted against the amount of antigen added, a precipitin curve like the one shown in Figure 6.3 is obtained.
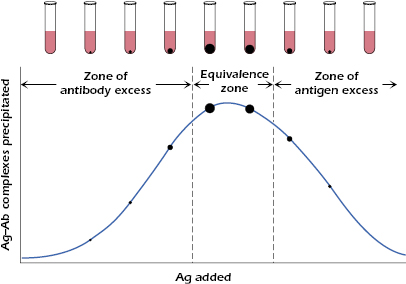
There are three important areas under the curve shown in Figure 6.3: (1) the zone of antibody excess, (2) the equivalence zone, and (3) the zone of antigen excess. In the equivalence zone, the proportion of antigen to antibody is optimal for maximal precipitation; in the zones of antibody excess or antigen excess, the proportions of the reactants do not lead to efficient cross-linking and formation of precipitate.
It should be emphasized that the zones of the precipitin curve are based on the amount of antigen–antibody complexes precipitated. However, the zones of antigen or antibody excess may contain soluble antigen–antibody complexes, particularly the zone of antigen excess where a minimal amount of precipitate is formed, but large amounts of antigen–antibody complexes are present in the supernatant. Thus, the amount of precipitate formed is dependent on the proportions of the reactant antigens and antibodies: the correct proportion of the reactions result in maximal formation of precipitate; excess of antigen (or antibody) results in soluble complexes.
In clinical laboratories, nephelometers are used to measure the amount of light scatter caused by antigen–antibody complexes in solution. They are also used to determine the levels of several blood plasma proteins, including complement. The technique is referred to as nephelometry and has largely replaced radial immunodiffusion (discussed later in this chapter). While both nephelometry and radial immunodiffusion measure immune complex formation, the former measures soluble immune complexes whereas the latter measures precipitated complexes. Therefore, these two approaches target different points on the precipitation curve.
Precipitation Reactions in Gels. Precipitation reactions between soluble antigens and antibodies can take place not only in solution but also in semisolid media such as agar gels. When soluble antigen and antibodies are placed in wells cut in the gel (Figure 6.4A), the reactants diffuse in the gel and form gradients of concentration, with the highest concentrations closest to the wells. Somewhere between the two wells, the reacting antigen and antibodies will be present at proportions that are optimal for formation of a precipitate.
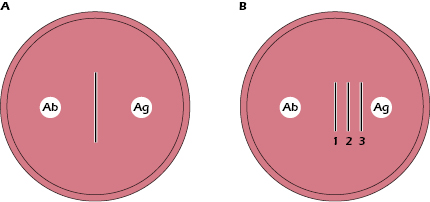
If the antibody well contains antibodies 1, 2, and 3 specific for antigens 1, 2, and 3, respectively, and if antigens 1, 2, and 3, placed in the antigen well diffuse at different rates (with diffusion rates of 1 > 2 > 3), then three distinct precipitin lines will form. These three precipitin lines form because anti-1, anti-2, and anti-3, which diffuse at the same rate, react independently with antigens 1, 2, and 3, respectively, to form three equivalence zones and thus three separate lines of precipitate (Figure 6.4B). Different rates of diffusion of both antibody, and antibody and antigen, result from differences in concentration, molecular size, or shape.
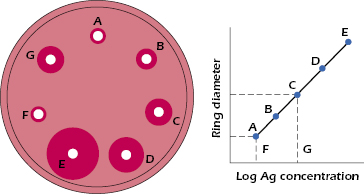
Radial Immunodiffusion. The radial immunodiffusion test, depicted in Figure 6.5, represents a variation of the double-diffusion test although it is not commonly used due to the availability of other more quantitative test options. The wells contain antigen at different concentrations, while the antibodies are distributed uniformly in the agar gel. Thus, the precipitin line is replaced by a precipitin ring around the well. The distance the precipitin ring migrates from the center of the antigen well is directly proportional to the concentration of antigen in the well. The relationship between concentration of antigen in a well and the diameter of the precipitin ring can be plotted as shown in Figure 6.5. If wells, such as F and G, contain unknown amounts of the same antigen, the concentration of that antigen in these wells can be determined by comparing the diameter of the precipitin ring with the diameter of the ring formed by a known concentration of the antigen.
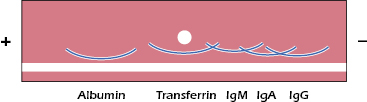
Immunoelectrophoresis. Immunoelectrophoresis involves separating a mixture of proteins in an electrical field (electrophoresis) followed by their detection with antibodies diffusing into the gel. It is very useful for the analysis of a mixture of antigens by antiserum that contains antibodies to the antigens in the mixture. This is an extension of the protein electrophoresis described in Chapter 5 (Figure 5.1) in which proteins are separated and their presumed composition is determined by their position on the gel (e.g., albumin, gamma globulin regions). In immunoelectrophoresis or immunofixation electrophoresis, a specific protein is identified by the addition of a specific antibody following the separation. For example, in the clinical characterization of human serum proteins, a small drop of human serum is placed in a well cut in the center of a slide that is coated with agar gel. The serum is then subjected to electrophoresis, which separates the various components according to their mobilities in the electrical field. After electrophoresis, a trough is cut along the side of the slides, and antibodies to human serum proteins are placed in the trough. The antibodies diffuse in the agar, as do the separated serum proteins. At an optimal antigen-to-antibody ratio for each antigen and its corresponding antibodies, they form precipitin lines. The result is a pattern similar to that depicted in Figure 6.6. Comparison of the pattern and intensity of lines of normal human serum with the patterns and intensity of lines obtained with sera of patients may reveal an absence, overabundance, or other abnormality of one or more serum proteins. In fact, it was through the use of the immunoelectrophoresis assay that the first antibody-deficiency syndrome was identified in 1952 (Bruton’s agammaglobulinemia) (see Chapter 18).
Western Blots (Immunoblots)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


