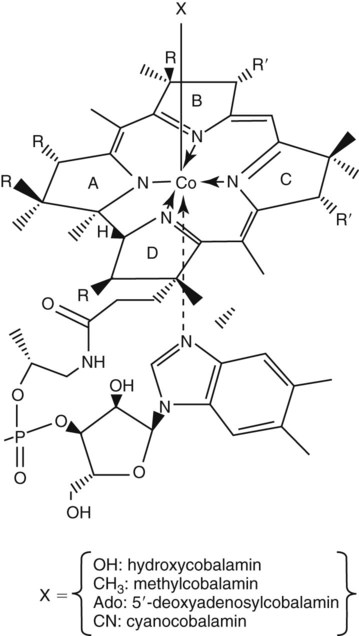
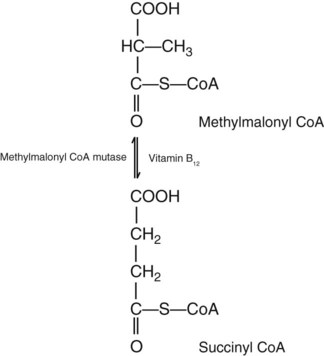
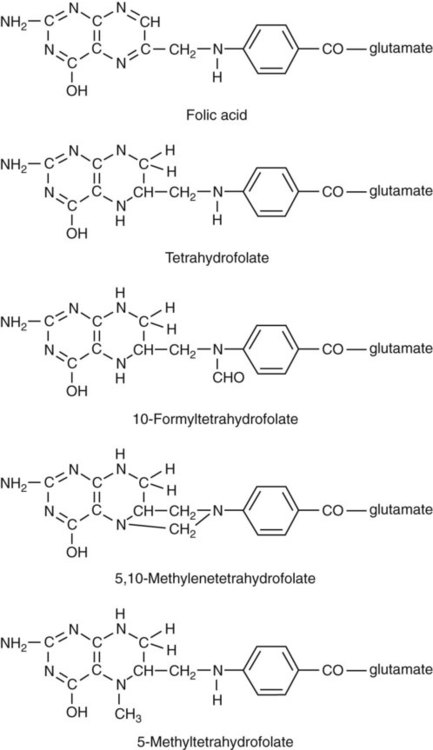
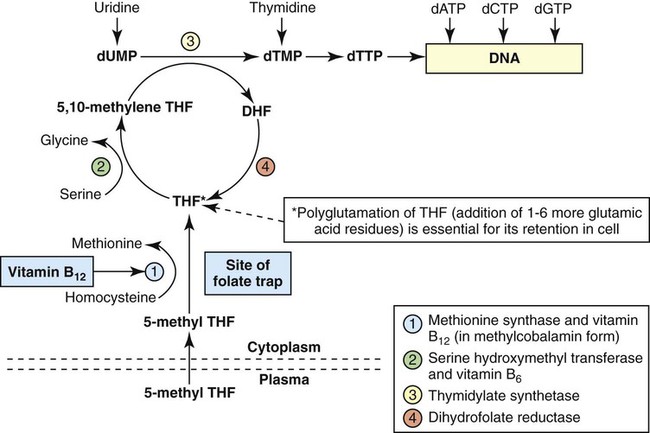
After completion of this chapter, the reader will be able to: 1. Discuss the relationships among macrocytic anemia, megaloblastic anemia, and pernicious anemia, and classify anemias appropriately within these categories. 2. Discuss the physiologic roles of folate and vitamin B12 in DNA production and the general metabolic pathways in which they act. 3. Describe the absorption and distribution of vitamin B12, including carrier proteins and the biologic activity of various vitamin-carrier complexes. 4. Describe the biochemical basis for development of anemia with deficiencies of vitamin B12 and folate, and explain the cause of the accompanying megaloblastosis. 5. Recognize individuals at risk for megaloblastic anemia by virtue of age, dietary habits, physiologic circumstance such as pregnancy, drug regimens, or pathologic conditions. 6. Recognize complete blood count, reticulocyte count, red and white blood cell morphologies, and bone marrow findings consistent with megaloblastic anemia. 7. Given the results of tests measuring levels of serum vitamin B12, serum methylmalonic acid, serum folate, plasma or serum homocysteine, and antibodies to intrinsic factor and parietal cells, determine the likely cause of a patient’s deficiency. 8. Recognize results of bilirubin and lactate dehydrogenase tests that are consistent with megaloblastic anemia and explain why the test values are elevated in this condition. Because of these findings, additional tests were ordered, with the following results: 1. Which of the CBC findings led the physician to order the vitamin assays? 2. Is the patient’s reticulocyte response adequate to compensate for the anemia? 3. Based on the available test results, what can you conclude about the cause of the patient’s anemia? 4. What additional testing would be helpful to diagnose the specific cause of this patient’s anemia? The root cause of megaloblastic anemia is impaired DNA synthesis. The anemia is named for the very large cells of the bone marrow that develop a distinctive morphology (see section on laboratory diagnosis) due to a reduction in the number of cell divisions. Megaloblastic anemia is one example of a macrocytic anemia. Box 20-1 shows the classification of macrocytic anemias. Understanding the etiology of megaloblastic anemia requires a review of DNA synthesis with particular attention to the roles of vitamin B12 (cobalamin) and folic acid (folate). Vitamin B12 (cobalamin) is an essential nutrient consisting of a tetrapyrrole (corrin) ring containing cobalt that is attached to 5,6-dimethylbenzimidazolyl ribonucleotide (Figure 20-1). Vitamin B12 is a coenzyme in two biochemical reactions in humans. One is isomerization of methylmalonyl coenzyme A (CoA) to succinyl CoA, which requires vitamin B12 (in the adenosylcobalamin form) as a cofactor and is catalyzed by the enzyme methylmalonyl CoA mutase (Figure 20-2). In the absence of vitamin B12, the impaired activity of methylmalonyl CoA mutase leads to a high level of serum methylmalonic acid, which is useful for the diagnosis of vitamin B12 deficiency (discussed in the section on laboratory diagnosis). The second reaction is the transfer of a methyl group from 5-methyltetrahydrofolate (5-methyl THF) to homocysteine, which thereby generates methionine. This reaction is catalyzed by the enzyme methionine synthase and uses vitamin B12 (in the methylcobalamin form) as a coenzyme (discussed later in this section). Methylcobalamin is synthesized through reduction and methylation of vitamin B12. This reaction represents the link between folate and vitamin B12 coenzymes and appears to account for the requirement for both vitamins in normal erythropoiesis.1,2 Folate is the general term used for any form of the vitamin folic acid. Folic acid is the synthetic form in supplements and fortified food. Folates consist of a pteridine ring attached to para-aminobenzoate with one or more glutamate residues (Figure 20-3). The function of folate is to transfer carbon units in the form of methyl groups from donors to receptors. In this capacity, folate plays an important role in the metabolism of amino acids and nucleotides. Deficiency of the vitamin leads to impaired cell replication and other metabolic alterations. Folate circulates in the blood predominantly as 5-methyl THF.3 5-Methyl THF is metabolically inactive until it is demethylated to tetrahydrofolate (THF), whereupon folate-dependent reactions may take place. Folate has an important role in DNA synthesis. As seen in Figure 20-4, within the cytoplasm of the cell, a methyl group is transferred from 5-methyl THF to homocysteine, which converts it to methionine and generates THF. This reaction is catalyzed by the enzyme methionine synthase and requires vitamin B12 in the form of methylcobalamin as a cofactor. THF is then converted to 5,10-methylenetetrahydrofolate (5,10-methylene THF); the methyl group for this reaction comes from serine as it is converted to glycine. The methyl group of 5,10-methylene THF is then transferred to deoxyuridine monophosphate (dUMP), which converts it to deoxythymidine monophosphate (dTMP). This reaction is catalyzed by thymidylate synthetase and results in the conversion of 5,10-methylene THF to dihydrofolate (DHF). Deoxythymidine monophosphate is a precursor to deoxythymidine triphosphate (dTTP), which, like the other nucleotide triphosphates, is a building block of the DNA molecule. THF is regenerated by the conversion of DHF to THF by the enzyme DHF reductase. Because some of the folate is catabolized during the cycle, the regeneration of THF also requires additional 5-methyl THF from the plasma. It is important to note that once in the cell, folate is rapidly polyglutamated by the addition of one to six glutamic acid residues. This conjugation is required for retention of THF in the cell and it also promotes attachment of folate to enzymes.4 When either folate or vitamin B12 is missing, thymidine nucleotide production for DNA synthesis is impaired. Folate deficiency has the more direct effect, ultimately preventing the methylation of dUMP. The effect of vitamin B12 deficiency is more indirect, preventing the production of THF from 5-methyl THF. When vitamin B12 is deficient, progressively more and more of the folate becomes metabolically trapped as 5-methyl THF. This constitutes what has been called the folate trap as 5-methyl THF accumulates and is unable to supply the folate cycle with THF. Some 5-methyl THF also leaks out of the cell if it is not readily polyglutamated. This results in a decrease in intracellular folate.5 In addition, when either folate or vitamin B12 is deficient, homocysteine accumulates, because vitamin B12 is unable to convert it to methionine (see Figure 20-4). In this state of diminished thymidine availability, uridine is incorporated into DNA.6,7 The DNA repair process can remove the uridine, but without available thymidine, the repair process is unsuccessful. Although the DNA can unwind and replication can begin, at any point where a thymidine nucleotide is needed, there is essentially an empty space in the replicated DNA sequence, which results in many single-strand breaks. When excisions at opposing DNA strand sites coincide, double-strand breaks occur. Repeated DNA strand breaks lead to fragmentation of the DNA strand.4 The resulting DNA is nonfunctional, and the DNA replication process is incomplete. Cell division is halted, which results in either cell lysis or apoptosis8 of many erythroid cells. Cells that do not lyse are released into the circulation. The dependency of DNA production on folates has been used in cancer chemotherapy (Box 20-2). In the case of red blood cell (RBC) development, these vitamin deficiencies result in ineffective hematopoiesis with increased apoptosis of erythroid progenitor cells. The remaining erythroid cells are larger than those cells that are normally seen during the final stages of erythropoiesis and their nuclei are immature-appearing compared with the cytoplasm (asynchrony). In contrast to the normally dense chromatin of comparable normoblasts, megaloblastic erythroid precursors have an open, finely stippled, reticular pattern.5 The nuclear changes seen in the megaloblastic cells are related to cell cycle delay, prolonged resting phase, and arrest in nuclear maturation. Electron microscopy has revealed that reduced synthesis of histones is also responsible for morphologic changes in the chromatin of megaloblastic cells, regardless of the causes.9 Because ribonucleic acid (RNA) contains uracil instead of thymidine residues, RNA function is not affected by vitamin B12 or folate deficiency. Thus cytoplasmic development is not generally disturbed in these cells, which results in the asynchrony between cytoplasm and nucleus. Together, the accumulation of cells at earlier stages of differentiation and cells with increased size and immaturity result in the appearance of erythroid cells in the bone marrow that are pathognomonic of megaloblastic anemia.8 Due to the ineffective hematopoiesis, pancytopenia is also evident, with certain distinctive cellular changes (see section on laboratory diagnosis). Vitamin B12 and folate deficiency are not the only causes of megaloblastic erythrocytes. Dysplastic erythroid cells in myelodysplastic syndrome (MDS) can also have megaloblastoid features (see Chapter 35). In MDS, the macrocytic erythrocytes and their progenitors characteristically show delayed cytoplasmic and nuclear maturation, including cytoplasmic vacuole formation, nuclear budding, multinucleation, and nuclear fragmentation, and thus may be distinguished from the megaloblastic RBCs seen in the vitamin deficiencies. In addition, nuclear-cytoplasmic asynchrony and megaloblastic RBCs may be seen in congenital dyserythropoietic anemia (CDA) types I and III (see Chapter 21). The CDAs are rare conditions that usually manifest in childhood and may be distinguished from the acquired causes of megaloblastosis by clinical history and morphologic differences. In CDA I internuclear chromatin bridging of erythroid cells or binucleated forms are observed, and in CDA III giant multinucleated erythroblasts are present. Another rare condition in which RBC precursors have a megaloblastic appearance is acute erythroleukemia, previously classified as FAB M6 (see Chapter 36). In this condition, the cells are macrocytic, and the immature appearance of the nuclear chromatin is similar to the more open appearance of the chromatin in megaloblasts. There are usually other aberrant findings in erythroleukemia, including an increase of myeloblasts in the bone marrow; however, an experienced morphologist can discern the subtle differences. Reverse transcriptase inhibitors, used to treat human immunodeficiency virus (HIV) infections, interfere with DNA production and may also lead to megaloblastic changes.10 Although the blood pictures seen with the two vitamin deficiencies are indistinguishable, the clinical presentations vary. In vitamin B12 deficiency, neurologic symptoms may be pronounced and may even occur in the absence of anemia.5 These include memory loss, numbness and tingling in toes and fingers, loss of balance, and further impairment of walking by loss of vibratory sense, especially in the lower limbs.11 Neuropsychiatric symptoms may also be present, including personality changes and psychosis. These symptoms seem to be the result of demyelinization of the spinal cord and peripheral nerves, but the relationship of this demyelinization to vitamin B12 deficiency is unclear. The role of increases in tumor necrosis factor-α, a neurotoxic agent, and decreases in epidermal growth factor, a neurotrophic agent, in the development of neurologic symptoms in vitamin B12-deficient patients is being researched.12,13 At one time, folate deficiency was believed to be more benign clinically than vitamin B12 deficiency. Later research suggested that low levels of folate and the resulting high homocysteine levels were risk factors for cardiovascular disease.14 More recent research has not substantiated this association,15–17 although some point out that high folate levels provide a cardioprotective effect in diabetic patients and certain ethnic populations.18,19 The evidence at this time is unclear as to whether persistent suboptimal folate status may have a significant long-term health impact. In addition, there is evidence of depression, peripheral neuropathy, and psychosis related to folate deficiency.20–22 Folate levels appear to influence the effectiveness of treatments for depression.23 Folate deficiency during pregnancy can result in impaired formation of the fetal nervous system, resulting in neural tube defects such as spina bifida,24 despite the fact that the fetus accumulates folate at the expense of the mother. Pregnancy requires a considerable increase in folate to fulfill the requirements related to rapid fetal growth, uterine expansion, placental maturation, and expanded blood volume.3 Ensuring adequate folate levels in women of childbearing age is particularly important because many women are likely to be unaware of their pregnancy during the first crucial weeks of fetal development. Fortification of the U.S. food supply with folic acid in grain and cereal products was mandated by the Food and Drug Administration in 1998 to lower the risk of neural tube defects in the unborn. The most common cause of folate deficiency is inadequate dietary intake.25 Folate is ubiquitous in foods, but a generally poor diet can result in deficiency. Good sources of folate include leafy green vegetables, dried beans, liver, beef, fortified breakfast cereals, and some fruits, especially oranges.26 Folates are heat labile, and overcooking of foods can diminish their nutritional value.3
Anemias Caused by Defects of DNA Metabolism
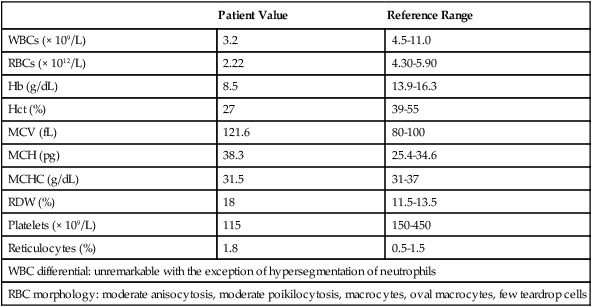
Case Study
Patient Value
Reference Range
WBCs (× 109/L)
3.2
4.5-11.0
RBCs (× 1012/L)
2.22
4.30-5.90
Hb (g/dL)
8.5
13.9-16.3
Hct (%)
27
39-55
MCV (fL)
121.6
80-100
MCH (pg)
38.3
25.4-34.6
MCHC (g/dL)
31.5
31-37
RDW (%)
18
11.5-13.5
Platelets (× 109/L)
115
150-450
Reticulocytes (%)
1.8
0.5-1.5
WBC differential: unremarkable with the exception of hypersegmentation of neutrophils
RBC morphology: moderate anisocytosis, moderate poikilocytosis, macrocytes, oval macrocytes, few teardrop cells
Etiology
Physiologic Roles of Vitamin B12 and Folate
Defect in Megaloblastic Anemia due to Deficiency in Folate and Vitamin B12
Other Causes of Megaloblastosis
Systemic Manifestations of Folate and Vitamin B12 Deficiency
Causes of Vitamin Deficiencies
Folate Deficiency
Inadequate Intake
Anemias Caused by Defects of DNA Metabolism