Androgen Receptor Signaling in Castration-Resistant Prostate Cancer
Brett S. Carver
Charles L. Sawyers
The androgen receptor (AR) gene is located on chromosome Xq11-12 and encodes for a 110 kDa nuclear receptor protein that mediates the transcription of target genes that modulate growth, differentiation, and homeostasis of androgen dependent cells. AR signaling is crucial for the development and maintenance of male reproductive organs including the prostate gland.
Huggins and Hodges first demonstrated that the dependence of prostate cells on androgen signaling was maintained in prostate cancer by observing regression of disease following bilateral orchiectomy (1). Since that time, androgen deprivation therapy has been the treatment of choice for locally advanced and metastatic prostate cancer. Nevertheless, response to withdrawal of androgens is temporary, and the median time to castration-resistant disease is approximately 18 months (2,3). While a minority of castration-resistant prostate cancers bypass the requirement for AR signaling, the vast majority remain dependent on the androgen pathway. In this chapter, we discuss the molecular mechanisms of AR signaling in castration-resistant disease and therapies directed at further inhibiting AR function.
OBSERVATIONS IN THE CLINIC REVEAL THAT CASTRATION-RESISTANT PROSTATE CANCER MAINTAINS A FUNCTIONAL AR AXIS
Progression of metastatic prostate cancer following androgen deprivation (castration-resistant disease) was previously referred to as androgen-independent prostate cancer; however, it has long been observed clinically that this disease state is, in fact, still dependent on androgen signaling. First, prostate specific-antigen (PSA), an AR regulated gene and marker for disease activity as measured by serum levels, declines following androgen deprivation therapy and a subsequent rise in serum PSA levels is often the initial sign of the emergence of disease progression. This increase in serum PSA levels clearly indicates reactivation of the androgen axis in the emergence of castration-resistant disease. Secondly, after the development of castration-resistant disease, the administration of AR antagonists, such as flutamide or bicalutamide, has been demonstrated to elicit a therapeutic response associated with reduction in serum PSA levels (4,5). Lastly, patients demonstrating progression of castration-resistant disease following treatment with flutamide or bicalutamide, once again defined by increases in serum PSA levels, were observed to have declining serum PSA levels following antiandrogen withdrawal, indicating that the antiandrogens began working as agonists to AR (6,7). Collectively, these clinical observations led to the appreciation that castration-resistant prostate cancer remains dependent on AR signaling.
MOLECULAR MECHANISMS OF AR ACTIVATION IN CASTRATION-RESISTANT PROSTATE CANCER
Several molecular mechanisms have been implicated to promote reactivation of AR in the castrate environment, and many more may be identified in the future. Initial studies focused molecular analysis directly on AR in the castration-resistant setting. Mutations in AR that allow other steroids, such as corticosteroids, as well as antiandrogens to act as agonists are detected in approximately 10% of castration-resistant prostate cancers, though the actual incidence could be higher using newer, much more sensitive sequencing technologies (8,9). These mutations result in structural changes in the AR protein altering the specificity of ligand binding. Thus, the mutated AR may be activated by other steroid hormones such as progesterone or known AR antagonists such as flutamide.
Other mechanisms resulting in activation of AR include amplification of the AR gene, upregulation of AR expression, modulation of AR cofactors, and activation of kinase pathways promoting AR function. Figure 3.1 summarizes the potential mechanisms of AR activation in castration-resistant disease.
AR AMPLIFICATION AND OVEREXPRESSION IN CASTRATION-RESISTANT PROSTATE CANCER
Overexpression of AR is a frequent event in prostate cancer. Compared to hormone-naïve organ confined prostate cancer, castration-resistant prostate cancer is enriched for AR amplification and increased mRNA levels. While AR amplification has not been observed in primary prostate cancer, approximately 20% of castration-resistant metastases will demonstrate AR amplification (10,11,12). Furthermore, mRNA upregulation is observed in approximately 60% of castration-resistant prostate cancers (12,13). Studies have demonstrated that exogenous overexpression of AR is sufficient to induce castration-resistant disease in preclinical models (14). Intriguingly, overexpression of AR also resulted in reduced efficacy of the antiandrogen bicalutamide, converting this AR antagonist to a weak agonist in cell lines and xenograft models (14).
ANDROGEN PRODUCTION IN CASTRATION-RESISTANT PROSTATE CANCER
Androgen deprivation therapy does not completely eliminate circulating serum androgens. Serum testosterone levels
are reduced to a mean of 15 ng/mL from a normal range of >200 ng/mL, while serum levels of adrenal androgens such as androstenedione and dehydroepiandrosterone are unaffected following androgen deprivation therapy (15). Studies have demonstrated that adrenal androgens and downstream metabolites are capable of binding to and activating the AR in a castrate setting (16,17). While serum testosterone levels significantly decrease following androgen deprivation therapy, the decline in the intraprostatic concentration of androgens is much less dramatic (18). Although this level of reduction is sufficient to induce cellular response in hormone-naïve prostate cancer, the AR in residual cells has increased sensitivity to low levels of ligands.
are reduced to a mean of 15 ng/mL from a normal range of >200 ng/mL, while serum levels of adrenal androgens such as androstenedione and dehydroepiandrosterone are unaffected following androgen deprivation therapy (15). Studies have demonstrated that adrenal androgens and downstream metabolites are capable of binding to and activating the AR in a castrate setting (16,17). While serum testosterone levels significantly decrease following androgen deprivation therapy, the decline in the intraprostatic concentration of androgens is much less dramatic (18). Although this level of reduction is sufficient to induce cellular response in hormone-naïve prostate cancer, the AR in residual cells has increased sensitivity to low levels of ligands.
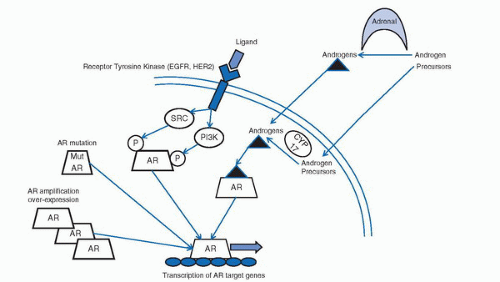 FIGURE 3.1. Molecular mechanism of AR signaling in castration-resistant prostate cancer. In the castration-resistant environment, AR activity may be restored through (a) the production of adrenal or intratumoral androgens, (b) the activation of AR in a ligand independent fashion through receptor tyrosine kinase regulation, (c) mutation of AR leading to alternative steroid binding or antiandrogen switch, and (d) amplification or overexpression of AR with increased sensitivity to low levels of androgen or cofactor imbalance.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|