▪ 14A Biology of Bone Metastases in Men with Prostate Cancer
Philip J. Saylor
Matthew R. Smith
NORMAL BONE REMODELING
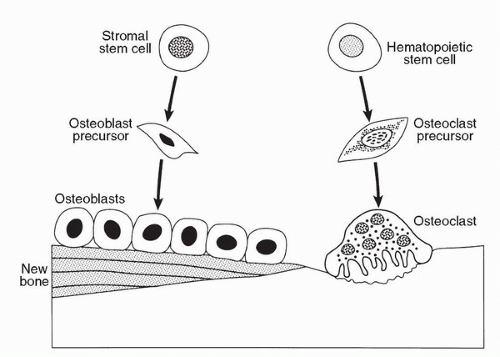
Bone is a rigid yet dynamic organ that is continuously shaped and repaired by a process termed remodeling. Bone remodeling is the predominant metabolic process regulating bone structure and function during adult life. Bone remodeling involves the breakdown and formation of bone by the coordinated action of osteoclasts and osteoblasts (Fig. 14A.1). Bone remodeling occurs in discrete microscopic units throughout the skeleton. Remodeling of each unit is geographically and temporally separated from other units. The process is initiated by migration of osteoclasts to these sites, resorption of bone, apoptosis of osteoclasts, and new bone formation by osteoblasts.
Osteoclasts are tissue-specific macrophages derived from hematopoietic stem cells from the monocyte/macrophage lineage. Bone resorption is a multistep process initiated by proliferation of immature osteoclast precursors, commitment of these cells to the osteoclast phenotype, and degradation of the bone matrix by mature multinucleated osteoclasts. Osteoclasts attach to the bone matrix and develop a specialized cytoskeleton or ruffled border. A process involving proton transport degrades the matrix in the isolated microenvironment at the ruffled border. The bone matrix is a storehouse of growth factors, and normal bone resorption releases a variety of factors that activate osteoblasts including transforming growth factor β(TGFβ), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), and insulin-like growth factor (IGF) I and II. Osteoblasts are derived from stromal stem cells. Osteoblasts synthesize and secrete the organic matrix. Mineralization begins soon after the organic matrix is secreted and is completed within weeks.
FIGURE 14A.1. Normal bone remodeling. Bone remodeling involves the breakdown and formation of bone by the coordinated action of osteoclasts and osteoblasts. Osteoclasts are tissue-specific macrophages derived from hematopoietic stem cells from the monocyte/macrophage lineage. Osteoblasts are derived from stromal stem cells.
The RANK signaling pathway regulates the activation, differentiation, proliferation, and apoptosis of osteoclasts (1). This pathway consists of receptor activator of NF-κB ligand (RANKL), its receptor RANK, and its decoy receptor osteoprotegerin (OPG). RANKL binds and activates RANK, a transmembrane receptor expressed on hematopoietic stem cells and osteoclasts. RANK expression on stem cells is required for osteoclast differentiation and activation. Hormones and other factors that stimulate bone resorption induce the expression of RANKL by bone stromal cells. RANKL expression by osteoblasts coordinates bone remodeling by stimulating bone resorption by osteoclasts that in turn stimulates new bone formation by adjacent osteoblasts in a process termed coupling. RANKL signaling pathway is negatively regulated by OPG. OPG is a soluble protein produced by osteoblasts in response to anabolic agents including estrogen and bone morphogenic proteins (BMPs). OPG acts as a decoy receptor that blocks RANKL binding to RANK. OPG also acts as a decoy receptor for TNF-related apoptosis-inducing ligand (TRAIL).
CLINICAL MANIFESTATIONS OF BONE METASTASES
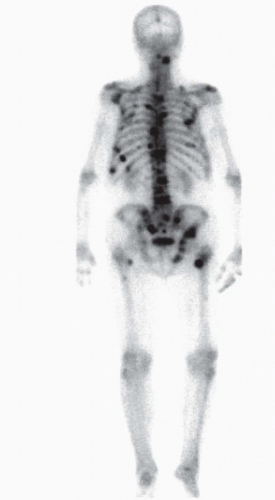
Prostate cancer preferentially spreads to the skeleton. More than 80% of men who die from prostate cancer have bone metastases at autopsy (2). In contrast to most other cancers, prostate cancer forms predominantly osteoblastic metastases. The vertebral column, pelvis, ribs, and proximal long bones are the most common sites of skeletal metastases (Fig. 14A.2). These sites correspond to the most abundant regions of bone marrow in the skeleton.
Pain is the most common symptom of metastatic prostate cancer. Metastases to the vertebrae may cause spinal cord compression, nerve root compression, or cauda equina syndrome. Metastases to the base of the skull can impinge on cranial nerves. Clinical fractures are common, with most fractures involving the vertebral bodies. In contrast to osteolytic bone disease, pathological fractures of long bones due to prostate cancer bone metastases are unusual. Hypocalcemia due to excess deposition of calcium in newly formed bone is commonly observed but rarely associated with symptoms. Most men with metastatic prostate cancer have normochromic normocytic anemia.
PATHOPHYSIOLOGY OF OSTEOBLASTIC METASTASES
Most bone metastases in men with prostate cancer appear osteoblastic by radiographic imaging. Osteolytic and osteoblastic lesions represent two extremes of a spectrum, however, and morphologic studies suggest that most bone metastases from prostate cancer are characterized by excess bone formation and excess bone resorption. This pathological acceleration of bone remodeling results in disorganized bone with impaired biomechanical properties.
FIGURE 14A.2. Bone metastases from prostate cancer. Radionuclide bone scan demonstrates multiple areas of increased tracer uptake at multiple sites in a man with progressive metastatic prostate cancer. Prostate cancer preferentially metastasizes to the vertebrae, pelvis, ribs, and proximal long bones. In contrast to most other malignancies, prostate cancer tends to form osteoblastic metastases.
Osteoblastic metastases from prostate cancer feature increased osteoblast number and activity, increased bone volume, and increased bone mineralization rate (3). Osteoclast number and activity are increased in osteoblastic metastases, at bone adjacent to metastases, and distant uninvolved bone (3,4). Biochemical markers of osteoclast activity are also elevated in men with osteoblastic metastases from prostate cancer (5). Although osteoclast activity is increased in men with prostate cancer, it is unclear whether osteoclast activation precedes bone formation as in normal bone remodeling or is secondary to excessive osteoblast activity in the metastases. Markers of osteoclast activity independently predict the risk for subsequent skeletal complications (6), suggesting that cancer-mediated osteoclast activation not only accompanies bone metastases but also contributes to the clinical complications of metastatic disease.
Hyperparathyroidism is common in men with metastatic prostate cancer. Serum concentrations of calcium are typically normal or low, suggesting that excessive parathyroid hormone secretion is secondary to increased calcium phosphate deposition in osteoblastic metastases (7). Secondary hyperparathyroidism may propagate a vicious cycle involving parathyroid hormone activation of osteoclasts, release of bone-derived growth factors, tumor cell proliferation, osteoblast activation, and further calcium phosphate deposition.
Androgen deprivation therapy, the cornerstone of treatment for recurrent and metastatic prostate cancer, also contributes to osteoclast activation in men with prostate cancer. Androgen deprivation therapy accelerates normal bone turnover and increases biochemical markers of osteoclast activity by approximately twofold in men without bone metastases (8). The unintended skeletal effects of androgen deprivation therapy increase fracture risk in men with prostate cancer (9). In addition, treatment-related increases in osteoclast activity may promote progression of bone metastases by increasing the release of bone-derived growth factors.
Zoledronic acid is a potent bisphosphonate that inhibits osteoclast attachment, differentiation, and survival (10). In a randomized placebo-controlled trial, zoledronic acid decreased the risk of skeletal complications in men with bone metastases and progressive disease after first-line hormonal therapy (11). The efficacy of zoledronic acid supports the concept that excessive osteoclast activity has a causal role in skeletal complications from metastatic prostate cancer.
MECHANISMS OF BONE METASTASES
Recent work has led to a growing understanding of the cellular changes that allow prostate cancer to travel to bone. Cancer cells must acquire the ability to detach from the primary tumor and survive outside of their native tissue, a process known as the epithelial-mesenchymal transition. PI3K/Akt, Wnt/β-catenin, Notch, Ras, integrin-linked kinase, and other integrin signaling pathways are all thought to be involved in cancer epithelial-mesenchymal transition (12).
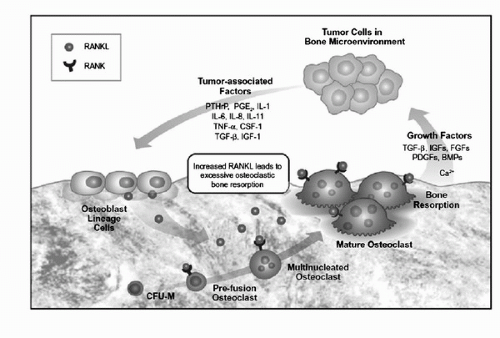
The concept that interactions between “seed” (circulating tumor cells) and “soil” (metastatic site) determine the metastatic potential of a cancer was first proposed by Stephen Paget more than a century ago (13). Consistent with the “seed and soil” hypothesis, reciprocal interactions between prostate cancer cells and bone stroma appear to account for both the predominant skeletal localization of metastases and the characteristic osteoblastic response (Fig.14A.3).
The bone extracellular matrix supports cellular adhesion and may contribute to the preferential skeletal localization of prostate cancer metastases. Integrin-mediated adhesion to the extracellular matrix increases androgen responsiveness and increased expression of androgen-responsive genes (14,15). In addition, adhesion to the extracellular matrix increases androgen-independent expression of some androgen-responsive homeobox genes (16), suggesting that interactions with the extracellular matrix contribute to androgen-independent growth.
Several bone-derived growth factors, including TGFβ, epidermal growth factor (EGF), and bFGF, promote prostate cancer growth and differentiation. Bone-derived growth factors may also contribute to the preferential skeletal localization of prostate cancer metastases. TGFβ facilitates adhesion to the extracellular matrix (17). EGF promotes migration of prostate cancer cells (18). Stromal cell-derived factor-1 (SDF-1) can bind to its receptor CXCR4 on prostate cancer cells to promote both adhesion and migration (19).
Many tumor-derived factors have been implicated in the pathogenesis of osteoblastic metastases and have led to the concept that prostate cancer cells acquire a bone-like phenotype. Cancer-produced factors include endothelin-1 (ET-1), BMPs, IGFs, OPG, TGFβ, and serine proteases prostate-specific antigen (PSA) and urokinase-type plasminogen activator (uPA).
FIGURE 14A.3. Osteoclast and tumor growth in the bone microenvironment. RANK ligand is a central mediator of bone pathology due to metastatic cancer. Tumor cells localize to bone and release or cause stromal release of tumor-associated factors. Among other downstream effects of these tumor-associated factors, they cause osteoblasts to express and secrete RANKL. RANKL stimulates osteoclast precursors to grow and differentiate. Additionally, RANKL activates mature osteoclasts to resorb bone and to secrete growth factors that stimulate cancer growth and perpetuate the cycle. (Reproduced from Roodman GD, Dougall WC. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat Rev 2008;34(1):92-101, with permission.)
Endothelin-1
The endothelin family consists of three paracrine/autocrine peptide factors (ET-1, ET-2, and ET-3) that act as modulators of vasomotor tone, nociception, hormone production, and cell proliferation (20). Endothelins are produced by a variety of normal tissues. ET-1 is the predominant circulating endothelin and is uniformly produced by endothelial cells and normal prostate epithelium. Two endothelin receptors have been identified: the ETA receptor has high affinity for ET-1 and ET-2 and the ETB has receptor has equal affinity for all the endothelin-related polypeptides. The growth-promoting effects of ET-1 are mediated by the ETA receptor (21). The ETB receptor attenuates the activity of ET-1 by decreasing ET-1 secretion, increasing ET-1 clearance, and activating inhibitory pathways.
ET-1 has important roles in both the prostate and the skeleton. ET-1 is produced by normal and malignant prostate epithelium and has been implicated in prostate cancer progression and bone metastases. ET-1 inhibits osteoclastmediated bone resorption and promotes the proliferation of osteoblasts (22), effects that may be mediated by its activation of the Wnt signaling pathway (23). Prostate-derived ET-1 promotes new bone formation (24) suggesting that ET-1 contributes to the osteoblastic phenotype of prostate cancer metastases.
Atrasentan and zibotentan (ZD4054) are orally available ETA receptor antagonists that block the effects of ET-1. Atrasentan selectively blocks ETA but has some activity at the growth inhibitory ETB. Zibotentan has no detectable activity at ETB. Two phase III studies of atrasentan in men with castration-resistant prostate cancer reported negative results (25,26). Three phase III studies of zibotentan for prostate cancer are ongoing and together are planned to enroll more than 3,000 subjects (27). Among other contrasts to the atrasentan trials, the zibotentan trials feature longterm follow-up and do not allow crossover from placebo to treatment.
Parathyroid hormone-related protein (PTHrP) shares some sequence homology with ET-1 and is also present at elevated concentration within prostate cancer bone metastases (28).
Bone Morphogenic Proteins and Other Growth Factors
BMPs are members of the TGFβ superfamily of developmentally regulated proteins involved in mesoderm formation and organ patterning. Both normal human prostate and prostate cancer cell lines express a variety of BMPs (29). Expression of BMP-6 by primary tumors is also associated with radiographic evidence of bone metastases, suggesting a potential role in the pathogenesis of metastases (30). Fibroblast growth factors (FGFs) and vascular endothelial growth factor (VEGF) are also expressed by prostate cancer cells and stimulate osteoblast activity.
Insulin-like Growth Factors
IGFs promote the proliferation of both prostate cancer cells and osteoblasts by interactions with specific IGF receptors (31). The biologic activity of IGFs appears to be regulated at the level of IGF-binding proteins (IGFBPs) and IGF receptors of target cells. Men with metastatic prostate cancer have elevated serum levels of IGFs (32). Cleavage of IGFBPs by several IGFBP proteases may contribute to the elevated expression of IGFs in men with prostate cancer. In addition, PSA specifically cleaves IGFBP-3 (33).
RANK and Osteoprotegerin
The RANK signaling pathway regulates osteoclast activation, differentiation, proliferation, and apoptosis. RANK, a cell surface receptor, is present on both immature and mature osteoclasts. RANK ligand (RANKL) is stimulatory to osteoclasts and is expressed on the surface of bone marrow stromal cells, T cells, and osteoblast precursors under usual circumstances (34). Prostate cancer bone metastases but not primary prostate tumors consistently express RANKL and its decoy receptor OPG (35). As prostate cancer metastatic to bone expresses RANKL, the local abundance of RANKL rises relative to OPG and enhances osteoclast activity (36). In patients with prostate cancer, baseline markers of enhanced osteoclast activity are correlated with disease progression and skeletal complications. RANKL is therefore a rational treatment target.
Denosumab is a fully human monoclonal antibody that binds to RANKL with high avidity and specificity. Its administration mimics the effects of endogenous OPG and potently suppresses osteoclast activity. Denosumab is in broad clinical development for prevention and treatment of benign and malignant bone disease. In a phase III study men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab significantly increased bone mineral density and decreased new vertebral fractures (37). An ongoing phase III study compares denosumab with zoledronic acid for the prevention of skeletalrelated events in men with castration-resistant prostate cancer and bone metastases (38). An ongoing placebo-controlled phase III study evaluates denosumab for prevention of bone metastases in men with castration-resistant nonmetastatic prostate cancer.
Transforming Growth Factor β
TGFβ is secreted by osteoblasts in a latent inactive form that is incorporated into the extracellular matrix. Latent TGFβ is a composed of TGFβ, the amino terminal part of the TGFβ precursor, and latent TGFβ binding protein. Latent TGFβ can be activated by proteolytic cleavage. TGFβ promotes new bone formation by a variety of mechanisms including osteoclast differentiation and maturation. Primary human prostate cancer and prostate cancer cell lines express TGFβ (39,40). The importance of TGFβ in normal bone physiology suggests that tumor-derived TGFβ promotes angiogenesis (41) and may have a central role in the development and progression of osteoblastic metastases.
Serine Proteases
Most tumor-derived factors implicated in the pathogenesis of osteoblastic metastases directly stimulate osteoblast activity. In contrast, proteases activate osteoblasts by indirect mechanisms. PSA and uPA are serine proteases expressed in human prostate cancers. Both PSA and uPA may activate TGFβ and IGF-1 by cleaving them from their binding proteins (42). These proteases may also contribute to the osteoblastic phenotype by cleavage of tumor-derived PTHrP.
SRC
Though not a tumor-derived factor, SRC is also important to the pathogenesis of bone metastases. SRC is a nonreceptor tyrosine kinase involved in signaling that leads to normal bone remodeling, tumor growth, and cancer metastasis. SRC is necessary for osteoclast function under normal circumstances. In the setting of malignancy, SRC mediates signals from several important cell surface receptors (epidermal growth factor receptor, HER2, VEGF receptor, and IGF receptors) (43). Further, in vitro studies have shown that SRC activity mediates prostate cancer cell growth in response to androgens (44) and contributes to metastatic potential (45). Several drugs that inhibit SRC are currently in clinical development for the treatment of prostate and breast cancer (43). Notably, dasatinib suppressed markers of osteoblast and osteoclast activity in a phase II trial (46) and is under phase III study in combination with docetaxel for the treatment of castration-resistant prostate cancer.
CONCLUSIONS
Bone metastases are a major cause of morbidity from prostate cancer. Prostate cancer spreads preferentially to the skeleton. Although most bone metastases in men with prostate cancer appear osteoblastic by radiographic imaging, “osteoblastic metastases” are characterized by abnormal activation of both osteoblasts and osteoclasts. Pathological acceleration of bone remodeling results in disorganized bone with impaired biomechanical properties. Complex reciprocal interactions between tumor and bone account for both the preferential spread to skeleton and the characteristic osteoblastic phenotype. Several tumor-derived factors have been implicated in the pathogenesis of osteoblastic metastases, including ET-1, BMPs, IGFs, OPG, and TGFβ. Several bone-derived growth factors, including TGFβ, EGF, bFGF, and RANKL, appear to promote prostate cancer growth. Greater understanding of these tumor-bone interactions may provide future therapeutic opportunities.
2. Harada M, Iida M, Yamaguchi M, et al. Analysis of bone metastasis of prostatic adenocarcinoma in 137 autopsy cases. Adv Exp Med Biol 1992;324:173-182.
3. Clarke NW, McClure J, George NJ. Osteoblast function and osteomalacia in metastatic prostate cancer. Eur Urol 1993;24(2):286-290.
4. Clarke NW, McClure J, George NJ. Morphometric evidence for bone resorption and replacement in prostate cancer. Br J Urol 1991;68(1):74-80.
5. Garnero P, Buchs N, Zekri J, et al. Markers of bone turnover for the management of patients with bone metastases from prostate cancer. Br J Cancer 2000;82(4):858-864.
6. Berruti A, Dogliotti L, Bitossi R, et al. Incidence of skeletal complications in patients with bone metastatic prostate cancer and hormone refractory disease: predictive role of bone resorption and formation markers evaluated at baseline. J Urol 2000;164(4):1248-1253.
7. Murray RM, Grill V, Crinis N, et al. Hypocalcemic and normocalcemic hyperparathyroidism in patients with advanced prostatic cancer. J Clin Endocrinol Metab 2001;86(9):4133-4138.
8. Smith MR, McGovern FJ, Zietman AL, et al. Pamidronate to prevent bone loss in men receiving gonadotropin releasing hormone agonist therapy for prostate cancer. N Engl J Med 2001;345(13):948-955.
9. Shahinian VB, Kuo YF, Freeman JL, et al. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 2005;352(2):154-164.
10. Green JR. Preclinical pharmacology of zoledronic acid. Semin Oncol 2002;29(6 Suppl 21):3-11.
11. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst 2002;94(19):1458-1468.
12. Ibrahim T, Flamini E, Mercatali L, et al. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010; 116(6):1406-1418.
13. Paget S. The distribution of secondary growths of cancer in cancer of the breast. Lancet 1889;1:571-573.
14. Fong CJ, Sherwood ER, Braun EJ, et al. Regulation of prostatic carcinoma cell proliferation and secretory activity by extracellular matrix and stromal secretions. Prostate 1992;21(2):121-131.
15. Murphy BC, Pienta KJ, Coffey DS. Effects of extracellular matrix components and dihydrotestosterone on the structure and function of human prostate cancer cells. Prostate 1992;20(1):29-41.
16. Robbins SE, Shu WP, Kirschenbaum A, et al. Bone extracellular matrix induces homeobox proteins independent of androgens: possible mechanism for androgen-independent growth in human prostate cancer cells. Prostate 1996;29(6):362-370.
17. Kostenuik PJ, Singh G, Orr FW. Transforming growth factor beta upregulates the integrin-mediated adhesion of human prostatic carcinoma cells to type I collagen. Clin Exp Metastasis 1997;15(1):41-52.
18. Rajan R, Vanderslice R, Kapur S, et al. Epidermal growth factor (EGF) promotes chemomigration of a human prostate tumor cell line, and EGF immunoreactive proteins are present at sites of metastasis in the stroma of lymph nodes and medullary bone. Prostate 1996;28(1):1-9.
19. Sun YX, Wang J, Shelburne CE, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem 2003;89(3):462-473.
20. Battistini B, Chailler P, D’Orleans-Juste P, et al. Growth regulatory properties of endothelins. Peptides 1993;14(2):385-399.
21. Nelson JB, Chan-Tack K, Hedican SP, et al. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res 1996;56(4):663-668.
22. Nelson JB, Carducci MA. The role of endothelin-1 and endothelin receptor antagonists in prostate cancer. BJU Int 2000;85(Suppl 2):45-48.
23. Clines GA, Mohammad KS, Bao Y, et al. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol 2007;21(2):486-498.
24. Nelson JB, Nguyen SH, Wu-Wong JR, et al. New bone formation in an osteoblastic tumor model is increased by endothelin-1 overexpression and decreased by endothelin A receptor blockade. Urology 1999;53(5):1063-1069.
25. Nelson JB, Love W, Chin JL, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 2008;113(9):2478-2487.
26. Carducci MA, Saad F, Abrahamsson PA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer 2007;110(9):1959-1966.
27. Fizazi K, Miller K. Specific endothelin-A receptor antagonism for the treatment of advanced prostate cancer. BJU Int 2009;104(10):1423-1425.
28. Liao J, McCauley LK. Skeletal metastasis: established and emerging roles of parathyroid hormone related protein (PTHrP). Cancer Metastasis Rev 2006;25(4):559-571.
29. Harris SE, Harris MA, Mahy P, et al. Expression of bone morphogenetic protein messenger RNAs by normal rat and human prostate and prostate cancer cells. Prostate 1994;24(4):204-211.
30. Bentley H, Hamdy FC, Hart KA, et al. Expression of bone morphogenetic proteins in human prostatic adenocarcinoma and benign prostatic hyperplasia. Br J Cancer 1992;66(6):1159-1163.
31. Peehl DM, Cohen P, Rosenfeld RG. The insulin-like growth factor system in the prostate. World J Urol 1995;13(5):306-311.
32. Kanety H, Madjar Y, Dagan Y, et al. Serum insulin-like growth factor-binding protein-2 (IGFBP-2) is increased and IGFBP-3 is decreased in patients with prostate cancer: correlation with serum prostate-specific antigen. J Clin Endocrinol Metab 1993;77(1):229-233.
33. Cohen P, Graves HC, Peehl DM, et al. Prostate-specific antigen (PSA) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J Clin Endocrinol Metab 1992;75(4):1046-1053.
34. Martin TJ. Paracrine regulation of osteoclast formation and activity: milestones in discovery. J Musculoskelet Neuronal Interact 2004;4(3):243-253.
35. Brown JM, Corey E, Lee ZD, et al. Osteoprotegerin and rank ligand expression in prostate cancer. Urology 2001;57(4):611-616.
36. Hofbauer LC, Neubauer A, Heufelder AE. Receptor activator of nuclear factor-kappaB ligand and osteoprotegerin: potential implications for the pathogenesis and treatment of malignant bone diseases. Cancer 2001;92(3):460-470.
37. Smith MR, Egerdie B, Hernandez Toriz N, et al. Denosumab in men receiving androgendeprivation therapy for prostate cancer. N Engl J Med 2009;361(8):745-755.
38. Saylor PJ, Smith MR. Bone health and prostate cancer. Prostate Cancer Prostatic Dis 2010;13(1):20-27.
39. Marquardt H, Lioubin MN, Ikeda T. Complete amino acid sequence of human transforming growth factor type beta 2. J Biol Chem 1987;262(25):12127-12131.
40. Muir GH, Butta A, Shearer RJ, et al. Induction of transforming growth factor beta in hormonally treated human prostate cancer. Br J Cancer 1994;69(1):130-134.
41. Yang F, Tuxhorn JA, Ressler SJ, et al. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res 2005;65(19):8887-8895.
42. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002;2(8):584-593.
43. Saad F, Lipton A. SRC kinase inhibition: Targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev 2010;36(2):177-184.
44. Asim M, Siddiqui IA, Hafeez BB, et al. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene 2008;27(25):3596-3604.
45. Boyer B, Bourgeois Y, Poupon MF. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 2002;21(15):2347-2356.
46. Yu EY, Wilding G, Posadas E, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res 2009;15(23):7421-7428.
▪ 14B Initial Management of Metastatic Prostate Cancer
Ajjai S. Alva
Maha Hussain
INTRODUCTION
Androgen deprivation therapy (ADT) is well established as the initial treatment of metastatic prostate cancer. Surgical bilateral orchiectomy was the earliest method of ablating the production of androgens. John Hunter noted in 1786 that castration decreased prostate mass in bulls (1). Castration was later utilized as a therapy for benign prostatic hypertrophy causing urinary obstruction (2). In 1941, Huggins and Hodges reported on the effects of castration on prostate cancer metastatic to the bone while delineating the role of testosterone in promoting prostate cancer (3). They also noted symptomatic improvements and corresponding decreases in prostatic acid phosphatase and prostatic alkaline phosphatase in the serum of such patients and thus established the role of hormone therapy in the management of metastatic prostate cancer. Castration-resistant prostate cancer (CRPC) is inevitable after a variable period of response to ADT. Strategies to delay CRPC are an active area of investigation with promising novel agents emerging as candidates for trials in first-line therapy of metastatic cancer.
PATHOPHYSIOLOGY
A review of the hypothalamic-pituitary-gonadal axis as it pertains to the regulation of the synthesis of androgens from the testes, the major site of androgen production, is in order (4). GnRH (gonadotropin-releasing hormone) secreted by the hypothalamus is transported to the anterior pituitary by the hypophyseal portal system where GnRH acts on specific GnRH receptors to stimulate secretion of LH (luteinizing hormone) and FSH (follicle stimulating hormone). GnRH is therefore also known as LHRH (luteinizing hormone-releasing hormone). LH and FSH are then secreted into the systemic circulation. In the testes, LH stimulates testosterone production by Leydig cells, while FSH causes inhibin (important in feedback control of FSH production) secretion from Sertoli cells. The HPG axis is regulated by negative feedback loops of testosterone and inhibin on the anterior pituitary and hypothalamus.
Testosterone is converted by the enzyme 5-alpha reductase in tissues to a more potent androgen, dihydrotestosterone (DHT). DHT has greater affinity for the androgen receptor (AR) in target organs than testosterone itself. AR is kept inactive in the cytoplasm of normal prostate cells by heat shock proteins (HSP). The binding of DHT to AR frees it from HSP. The DHT-AR complex translocates to the nucleus and forms a dimer with another DHT-AR complex. The dimerized molecule binds to androgen-response elements on target genes. AR thus modulates expression of several downstream genes (including PSA) and pathways in the prostate cell that promote survival and growth.
MODALITIES OF ANDROGEN BLOCKADE
Orchiectomy is a relatively simple surgical procedure with the resultant advantages of low cost and immediacy of attaining castrate testosterone levels (5). These merits are however counterbalanced by the psychological impact of the surgery and the irreversibility particularly given the younger age at diagnosis in the PSA era and the options for intermittent therapy. In a questionnaire-based study, only 24% of eligible men opted surgical castration compared to medical alternatives (6). Advancements such as subcapsular orchiectomy and testicular prostheses have not appeared to change this scenario.
Estrogens: The earliest means of medical castration were synthetic estrogens that blocked the hypothalamic-pituitarygonadal axis via feedback inhibition. The Veterans Administrative groups conducted the initial trials in metastatic prostate cancer with the nonsteroidal estrogen, diethylstilbestrol (DES) (7). DES causes peripheral blockade of the AR and direct cytotoxic effects on the cancer cells in addition to blocking LHRH release. Prostate cancer-specific survival was equivalent in patients receiving 1 mg DES and 5 mg DES. However, the 5-mg dose was associated with markedly increased cardiovascular deaths. Cardiovascular toxicity was particularly prominent at daily doses >3 mg that are needed for reliable castrate levels of testosterone and in older men with cardiovascular risk factors. Conjugated estrogens, ethinyl estradiol, and medroxyprogesterone acetate have all been tested and are occasionally used in the second-line setting but seldom in the initial management of prostate cancer (8).
LHRH (GnRH) agonists (9) bind to GnRH receptors in the anterior pituitary, causing an initial transient increase in LH release and rise in testosterone levels. Subsequently, GnRH receptors are downregulated in the second week resulting in a decline in testosterone levels to the castrate range over the next 3 to 4 weeks. LHRH agonists include leuprolide, goserelin, buserelin, and triptorelin. Depot formulations of leuprolide are available as intramuscular injections every 1 month (7.5 mg), 3 months (22.5 mg), 4 months (30 mg), or 6 months (45 mg) though an even longer-acting formulation, leuprolide acetate, is available as a 1-year subcutaneous implant (72 mg). Goserelin is given as a 3.6-mg subcutaneous injection every month or 10.8-mg subcutaneous (sc) injections every 3 months. Buserelin is available as nasal spray or subcutaneous injections given every 8 hours. Triptorelin is available as 3.75-mg intramuscular injection every month and 11.5-mg injection every 3 months. The different LHRH formulations are considered equivalent in efficacy. In the initial studies, leuprolide was administered as daily subcutaneous injections of 1 mg and compared to oral DES dosed at 3 mg daily in the first-line treatment of metastatic prostate cancer (10). 86% of patients in the leuprolide group and 85% of patients treated with DES achieved objective responses. Moreover, side effects including major cardiac events occurred more frequently in the DES patients, although cardiovascular complications were not reached statistically higher after DES. Long-term administration of leuprolide either as daily doses or as long-acting depot preparations achieves comparable testosterone suppression and objective responses (11,12). Similar efficacy of other LHRH agonists such as goserelin has been demonstrated in clinical trials (13,14) and in meta-analyses (15).Other than the injection-related issues, side effects from LHRH agonists are from testosterone deprivation and include impotence, loss of libido, metabolic side effects that are increasingly being recognized such as dyslipidemia including reduction in HDL-cholesterol with elevation in cardiovascular risk, changes in body habitus due to altered fat distribution, gynecomastia both from breast tissue increase and from increased fat deposition on the chest, hair loss, loss of muscle mass and bone loss, hot flashes, and night sweats (Table 14B.1). The obvious advantages of LHRH agonists are ability to achieve effective castrate levels of testosterone, reversibility, and ease of administration though the cumulative economic costs over a lifetime are substantial. A unique phenomenon stemming from their LHRH agonist activity is the initial and transient upsurge in LHRH activity and consequent rise in testosterone effect. In situations where such a “flare response” could have deleterious effects clinically such as cord compression from substantial spinal metastases or ureteric or urethral outflow obstruction from enlarged lymph nodes or prostate, initiation of an AR blocker before an LHRH agonist is important. No studies have been done to determine the duration of the lead in therapy; however, it is reasonable to consider a 7 to 10 day therapy with an antiandrogen alone and continuing for at least 2 weeks after the LHRH agonist has been started.
TABLE 14B.1 ADVERSE EFFECTS OF ADT
System
Side Effect
Strategies to Alleviate/Manage
Sexual
Impotence Loss of libido
Erectile dysfunction therapy Counseling
Metabolic
Dyslipidemia Diabetes mellitus
Diet, exercise Statins
Body habitus
Obesity Gynecomastia, breast tenderness
Diet, exercise, nutrition consult Statins
Hair loss Loss of muscle mass Loss of bone mass
Calcium, vitamin D Bone mineral density surveillance, exercise against gravity, bisphosphonates
Systemic
Hot flashes Night sweats Fatigue Anemia
Intermittent androgen deprivation Erythropoietin
Cognitive/Psychosocial
Depression Lack of initiative Emotional lability
Antidepressants Counseling
LHRH antagonists are not associated with the flare response. Abarelix was withdrawn from the market in 2005 following reports of acute onset allergic reactions including hypotension and syncope following intramuscular administration. Degarelix has recently been approved (240 mg sc, given as 2 injections of 120 mg each followed by maintenance of 80 mg sc every 28 days) in advanced prostate cancer based on a randomized study comparing degarelix to monthly leuprolide for a duration of 1 year (16). The primary end point of percentage of patients who achieved castrate levels of testosterone maintained for 1 year was similar in both groups. Declines in testosterone to castrate range were faster (>95% of patients at 3 days) and PSA levels were lower (at 14 and 28 days) with degarelix. There was a higher incidence of injection site reactions with degarelix but no systemic allergic or anaphylactic reactions.
Antiandrogen molecules were developed in the late 1960s to specifically block the DHT receptor (17). They block the effect of gonadal and extragonadal androgens (adrenal) at the target tissue level. As a result, LH and testosterone levels increase and sexual function and libido are preserved more commonly than with ADT. Based on molecular structure, antiandrogens are classified as steroidal or nonsteroidal. Nonsteroidal antiandrogens include flutamide, bicalutamide, and nilutamide. Flutamide has a short half-life of 5 hours and is usually dosed at 250 mg every 8 hours. Once a day dosing of flutamide at 500 mg has been demonstrated to be equally efficacious as thrice daily dosing. Half-lives of bicalutamide and nilutamide are longer (˜7 and 2 days) and are dosed once a day (bicalutamide at usual daily dose of 50 mg and nilutamide at 300 mg for 30 days followed by 150 mg daily). Side effects of nonsteroidal antiandrogens (Table 14B.2) as a class include nausea, vomiting, diarrhea, breast growth or enlargement with or without tenderness (due to higher testosterone levels and with incidence up to 74% associated with bicalutamide), and potentially fatal hepatotoxicity (most with flutamide and least with bicalutamide but requires periodic liver function tests with use of any antiandrogen). Serum transaminase levels should be measured prior to starting treatment, monthly for the first 4 months of therapy, and periodically thereafter. Flutamide should not be started or continued in patients whose ALT values exceed twice the upper limit of normal. Nilutamide has, in addition, specific toxicities of decreased dark adaptation, rare but serious interstitial pneumonitis and alcohol intolerance. In terms of efficacy, the nonsteroidal antiandrogens are considered equivalent. For example, a four-armed randomized trial of an antiandrogen (bicalutamide or flutamide) in combination with an LHRH-agonist (leuprolide or goserelin) showed equivalent overall survival (18).
Steroidal antiandrogens (not generally used in North America) such as cyproterone acetate, megestrol acetate, and medroxyprogesterone acetate also have the progestational adverse effects of loss of libido and impotence and the advantage of reducing hot flashes from ADT. Several randomized trials used cyproterone acetate (not approved in USA) as part of combined androgen deprivation (CAD). A meta-analysis (PCTCG) suggested a trend toward decreased overall survival with cyproterone acetate due to a greater number of nonprostate cancer deaths (19).
TABLE 14B.2 ANTIANDROGENS
Antiandrogen
Dose
Side-Effects
As a class
Nausea, vomiting, diarrhea, breast enlargement, breast tenderness, hepatotoxicity
Flutamide
250 mg tid
Hepatotoxicity (in particular)
Nilutamide
300 mg/d for 30 d and then 150 mg/d
Decreased dark adaptation, interstitial pneumonitis (rare), alcohol intolerance
Bicalutamide
50 mg/d
Breast enlargement, breast tenderness (in particular)
COMBINED ANDROGEN DEPRIVATION VERSUS MONOTHERAPY WITH PRIMARY GONADAL SUPPRESSION ALONE
Preclinical and Early Clinical Data
The theoretical benefit of combining primary gonadal androgen suppression (LHRH agonist or orchiectomy) and an antiandrogen (so-called combined androgen deprivation or CAD) is in abrogating extragonadal sources of androgens. Adrenal androgens could account for 5% to 45% of postorchiectomy androgens. Adrenal production of testosterone can amount to 400 μg daily (20). Preclinical studies indicate that low levels of androgens (such as detected following castration) do exert stimulatory effects on protein synthesis in prostate cancer cells (21,22). A threshold level of DHT may be required to promote prostate cancer growth (23,24,25). Support for this hypothesis comes from the Dunning rat model. After orchiectomy, animals without prostate cancer were treated with varying concentrations of testosterone and tumor growth was assessed. Testosterone increased tumor mass only at levels above a threshold. However, in Dunning rats with prostate cancer, combined androgen blockade has not been shown to decrease tumor mass or prolong survival contrary to effects on normal prostates (18,19). The theory underpinning suppression of circulating androgens is further complicated by recent evidence showing tumor cells can synthesize androgens (26).
Clinical utility of CAD was tested in a study utilizing an LHRH agonist (buserelin) in combination with nilutamide in therapy of advanced prostate cancer (27) in a small phase II trial. A higher than previously noted response rate, avoiding flare, symptomatic improvements in pain, and no apparent increase in adverse effects were observed. The authors recommended CAD as first-line therapy to accomplish “complete androgen withdrawal” (28). They also hypothesized that exposure to low levels of adrenal androgens in addition to promoting survival of a fraction of the cells promoted evolution to androgen resistance in a manner similar to antibiotic resistance (28).
Several randomized trials were subsequently conducted to compare CAD with gonadal suppression alone (Table 14B.3) and the larger trials will be discussed below. Heterogeneity in trial designs and dosing schedules used, use of orchiectomy versus LHRH agonists, and nonsteroidal versus steroidal antiandrogens make a straightforward comparison between trials difficult.
Evidence Favoring CAD over Monotherapy
Four independent randomized studies have shown significant benefits for CAD over castration alone in one or more of the following outcomes: time to progression, progression-free survival or overall survival (23,24,25,26).
TABLE 14B.3 CAD VERSUS PRIMARY GONADAL SUPPRESSION ALONE
OS, overall survival; PFS, progression free survival; TTF, time to treatment failure; TTP, time to progression.
In the INT 0036 study, 603 patients with new metastatic prostate cancer were randomized to either CAD with daily leuprolide and flutamide or to daily leuprolide and placebo (23). CAD was associated with significantly longer pro-gression-free survival (16.5 vs. 13.9 months, p = 0.039) and overall survival (35.6 vs. 28.3 months, p = 0.035). The only adverse event significantly worse in the CAD group was diarrhea (13.6% vs. 4.9%, p ≤ 0.001). The conclusion of the INT 0036 study was that leuprolide combined with flutamide was superior to leuprolide alone and thus the extrapolation that CAD was superior to monotherapy. Several alternate explanations could be advanced to explain the observed benefit of CAD over monotherapy other than intrinsic superiority of the combination. Addition of an antiandrogen possibly prevented the “flare” response to LHRH agonist. Even a short duration of testosterone upsurge during the flare could be hypothesized to accelerate tumor cell growth that could be subsequently maintained. Indeed, there were favorable trends in pain intensity, performance status, and prostatic acid phosphatase in the CAD arm during the initial phase of treatment. A more important factor could be the noncompliance with daily leuprolide injections, causing incomplete gonadal suppression with leuprolide alone. However, testosterone measurements were not mandated as part of the trial and therefore neither hypothesis could be verified. An unplanned subset analysis of INT 0036 revealed particularly striking benefit to CAD in patients with minimal metastatic disease (axial skeleton with/without nodal disease without visceral or appendicular skeleton involvement) with PFS of 48 months versus 19 months in the leuprolide + placebo arm. In multivariate analysis, extent of disease was a prognostic factor for survival.
EORTC 30853 entered 327 patients to a randomized phase III study that compared CAD (goserelin acetate, 3.6 mg subcutaneously every 4 weeks, plus flutamide, 250 mg three times a day orally) with bilateral orchiectomy (24). With 5 years of follow-up, there was a statistically significant difference in favor of CAD with respect to objective progression (46% vs. 52%, hazard ratio [HR] = 0.64, 95% confidence interval [CI] = 0.46-0.89, p = 0.008), median time to objective progression (133 weeks vs. 85 weeks, p = 0.008), overall survival (34.4 months vs. 27.1 months, HR = 0.73, 95% CI = 0.56-0.95, p = 0.02), and death due to malignant causes (28.8 vs. 43.9 months, HR = 0.67, 95% CI = 0.49-0.90, p = 0.007). With longer follow-up (27), these differences were maintained for overall survival (p = 0.04), time to death due to malignant disease (p = 0.008), time to first progression (p = 0.009), and progression-free survival (p = 0.02). Analysis of a minimal disease subset in EORTC 30853 (defined by number of bone metastases alone and irrespective of visceral involvement, differently from INT0036) also showed a marked benefit to CAD over orchiectomy alone. Hot flushes were reported for 58% of patients who underwent orchiectomy and 68% of patients treated with CAD. Gynecomastia was noted in 8% and 19% (4% painful) of orchiectomy patients and CAD patients, respectively. Differences in ALT elevations (Grade ≥2) were not significant (3% vs. 9%, p = 0.07).
The International Anandron Study Group compared bilateral orchiectomy and placebo versus bilateral orchiectomy and 300 mg per day of nilutamide in 457 patients (29). With 8.5 years of follow-up, CAD was associated with prolongation of median PFS by 6.5 months (21.2 months vs. 14.7 months, a 44% relative increase, p = 0.002) and a 5-year PFS of 20% versus 12%. Median overall survival was also significantly longer with CAD (37 months vs. 29.8 months, p = 0.013); 6-year survival was 32% in the CAD group versus 21%. The overall response rate and pain improvement was significantly better in the CAD group. 15.5% of the patients in the nilutamide group and 9% in the placebo group were withdrawn from the study prematurely for adverse or intercurrent events. Interstitial pneumonitis (1 of 225 patients), reversible visual disturbances (difficulty with light-to-dark adaptation or blurred vision) in 27% (leading to withdrawal from study in 2%), and transient increases in liver enzyme levels were reported more commonly in the nilutamide group.
In a Japanese trial, 205 prostate cancer patients (87 patients with metastatic disease) were randomized to LHRH agonist + bicalutamide 80 mg daily versus LHRH monotherapy. In the entire population, there was a significant prolongation of time to progression and time to treatment failure in the combined therapy arm (24). In an updated report with a median follow-up of 5.2 years, overall survival was significantly prolonged in CAD versus monotherapy (HR = 0.78; 95% CI = 0.60-0.99; p = 0.0498) (25).
Evidence Against a Benefit to CAD over Monotherapy
Contrary to the four studies presented above, several other trials did not show a benefit to CAD (30,31,32,33,34,35,36,37,38,39,40,41,42). The single largest study to date was INT-0105 and has been reported by Eisenberger et al. (34). Thousand three-hundred and eightyseven men with untreated metastatic prostate adenocarcinoma were randomized to either orchiectomy and placebo or orchiectomy and flutamide (250 mg three times daily). The study was designed to detect an increase in overall survival (the primary end point) with flutamide versus placebo of 25% with power of 90% and an anticipated survival of 28.3 months in the placebo arm. The primary end point of an overall survival benefit of 25% in the CAD group was not met (HR = 0.91, 90% CI = 0.81-1.01, p = 0.14) at a median follow-up of 50 months. The median overall survival was 33.5 months versus 29.9 months, respectively. PSA responses to ≤4 ng/mL were significantly better in the CAD arm (74% vs. 61.5%, p < 0.001). PFS was no different between the 2 arms either (20.4 months vs. 18.6 months, p = 0.26). However, there was a statistically significant difference in the normalization of serum PSA (81% vs. 69%) in favor of the combination arm. INT-0105 study was adequately powered, enrollment exceeded the target, follow-up was prolonged in the context of metastatic disease, and the use of orchiectomy avoided potential confounding effect of incomplete gonadal suppression. The authors concluded that the benefit of CAD over orchiectomy alone is negligible if at all. However, it should be noted that a survival benefit to CAD over orchiectomy may exist but is less in magnitude than the 25% that INT-0105 was powered to detect. In contrast to INT 0036, INT-0105 did not confirm the benefit of CAD in the minimal disease subset in either PFS or overall survival (only 147 of 1387 patients enrolled in both arms in INT-0105 were classified as minimal disease). The only significantly worse Grade 2 adverse events in the CAD arm were diarrhea (6.3% vs. 2.7%, respectively) and anemia (8.5% vs. 5.4%). INT-0105 also enrolled patients in a quality of life (QoL) protocol (43). Patients completed a questionnaire on five primary QoL parameters (diarrhea, gas pain, body image, physical functioning, and emotional functioning) at baseline and subsequently at 1, 3, and 6 months on treatment. Crosssectional analyses demonstrated that patients in the flutamide arm had significantly more episodes of diarrhea and worse overall emotional functioning than on the placebo arm. Most patients on both arms showed improvement in QoL with time but less so for the flutamide arm. Those receiving flutamide also discontinued the treatment more often due to drug toxicity (33 vs. 10, p = 0.002).
The Danish Prostatic Cancer Group DAPROCA trial (37) (orchiectomy and placebo versus goserelin acetate and flutamide), a French multicenter study (36) (goserelin acetate and placebo versus goserelin acetate and flutamide), and the International Prostate Cancer Study Group trial (goserelin with or without flutamide) (41) did not detect any differences in overall survival or time to progression.
META-ANALYSES OF CONTROLLED STUDIES
Several large meta-analyses have been conducted to address the issue (19,44,45,46,47). The Prostate Cancer Trialists’ Collaborative Group (PCTCG) published a meta-analysis using individual patient data from 8,275 men from 27 randomized trials in an intention-to-treat fashion. The data included 98% of all patients registered in randomized trials of prostate cancer to compare CAD versus monotherapy to that point in time (19). Of the 27 included trials, 8 studied nilutamide, 12 flutamide, and 7 cyproterone acetate. Eighty-eight percent of the patients had metastatic disease, fifty percent were older than 70 years, and average follow-up was 5 years. At the time of analysis, 70% had died (80% of deaths attributable to prostate cancer). Five-year survival was 25.4% with CAD versus 23.6% with monotherapy alone (p = 0.11). When the cyproterone acetate trials (20% of the patients) were excluded, there was an overall survival benefit to CAD (nilutamide or flutamide) at 5 years of 27.6% versus 24.7% (p = 0.005). For cyproterone acetate trials, overall survival was slightly but significantly worse with CAD with 5-year overall survival 15.4% versus 18.1% (p = 0.04). Non-prostate cancer deaths (did not reach statistical significance) seemed to explain the worse outcome with cyproterone acetate. The authors concluded that there was probably a 2% to 3% overall survival benefit to CAD (excluding cyproterone acetate) although the true benefit statistically could range from 0% to 5% at 5 years. Criticism of the metaanalysis included the fact that control groups in some of the included trials used short-term antiandrogens to prevent the flare response.
A Cochrane review analyzed 6,320 patients from 20 trials comparing combined therapy to monotherapy (45). Odds ratio (OR) for overall survival with combined androgen blockade was 1.29 (95% CI = 1.11-1.50; n = 3,550 from seven trials) at 5 years. The authors estimated a 5% improvement in 5-year survival rate (30% vs. 25%) with CAD. PFS was improved at 1 year (OR = 1.38; 95% CI = 1.15-1.67; n = 2278 from seven trials). Cancer-specific survival was improved at 5 years (OR = 1.58; 95% CI = 1.05-2.37; n = 781 from two trials). However, when analysis was limited to studies identified as being of high quality, the OR was not significant at any follow-up interval (1, 2, or 5 years).
The individual trials and meta-analyses have been criticized for several reasons. Most of the studies included were early incomplete looks at the data or contained too few patients to reach statistical significance on their own. The lack of statistical power in several of the reported “negative trials” leading to incomplete information to the reader and consequent misinterpretation of “inconclusive trials” as “negative trials” has been highlighted (48). The trials also included various antiandrogens and modalities of gonadal suppression. In particular, few of the trials evaluated bicalutamide as the antiandrogen.
While it is likely that a small overall survival benefit indeed exists to CAD (2%-3% at 5 years) but is not certain, data on improvements in other outcomes such as TTP and PFS are not conclusive either. Data on quality of life have been assessed in some of the trials (including the Japanese trial and INT-0105) with conflicting results. The Japanese trial showed a trend toward improved QoL parameters while INT-0105 showed worse QoL in the CAD group.
Although there is no conclusive evidence-based consensus about the advantage of CAD as first-line therapy for metastatic prostate cancer over castration alone, treatment decisions have to be individualized, as always in medicine, weighing potential benefit with risk of increased toxicity.
PERIPHERAL ANDROGEN BLOCKADE
Antiandrogens as monotherapy for initial hormonal therapy have the theoretical benefit of avoiding androgen deprivation and consequent side effects while possibly controlling disease from metastatic prostate cancer. Direct inhibition of the DHT receptor would allow for some hormonal effects of circulating testosterone such as the maintenance of sexual interest and function. Initial comparisons of low-dose bicalutamide (50 mg daily) with castration showed inferior response and survival outcomes with antiandrogen monotherapy but improved QoL as expected (49,50,51,52). Large randomized trials with higher doses of bicalutamide have provided mixed results in patients with metastatic and locally advanced T3/4 disease (53,54). Tyrrell et al. used optimal-dose estimation arms and demonstrated improved PSA response at a dose of 150 mg (vs. 100 mg); thus, the higher dose was used for the remainder of the study. With over 800 patients enrolled with metastatic disease (of the total of 1,453 patients in the study), the study was stopped due to a 6-week median survival benefit to castration at a median follow-up period of approximately 100 weeks. Thus, bicalutamide at 150 mg daily was associated with poorer survival (HR of 1.30 for time to death). In patients with symptomatic metastases, bicalutamide was associated with a statistically significant improvement in subjective response (70%) compared with castration (58%). Analysis of a QoL questionnaire demonstrated an advantage for bicalutamide in sexual interest and physical capacity. Bicalutamide had a substantially lower incidence of hot flushes compared to castration (6%-13% compared with 39%-44%), but significantly more breast tenderness that did not lead to withdrawal from therapy. The authors concluded that bicalutamide monotherapy could be an option in a well-counseled patient with metastatic disease who wishes to avoid certain side effects of castration. A secondary analysis of the data suggested that patients with PSA <400 ng/mL or less than six metastatic sites had equivalent outcomes with castration or antiandrogen monotherapy (55). A larger total number of ARs in patients with extensive disease might explain the worse outcomes in that subset. Two smaller underpowered trials in Europe also evaluated bicalutamide at 150 mg against CAD. No significant outcome differences were noted though these trials are limited by small sample size and inclusion of nonmetastatic patients in one trial (53,56).
Conclusions regarding flutamide as monotherapy are somewhat more difficult to make. Flutamide has been randomized against CAD, orchiectomy, and DES (57,58,59). Survival time in the DES study with 92 patients only was much shorter in the flutamide group (43.2 months vs. 28.5 months) while survival was equivalent in the other two studies (against CAD with 319 patients and against orchiectomy in 104 patients). Thorpe et al. examined CPA monotherapy in a three-armed study of 525 patients comparing monotherapy with CPA versus goserelin versus combination therapy with both (40). CPA performed slightly worse than LHRH-agonist in time to progression. Two other trials with a total of 175 patients have examined CPA against DES (3 mg) and orchiectomy and found no difference in overall survival at up to 5 years (60,61). The combination of an antiandrogen with a 5-alpha reductase inhibitor has been tested but with very few metastatic patients among those enrolled (62).
Currently available data do not support the routine use of peripheral blockade with current agents in the first-line treatment of metastatic disease.
EARLY VERSUS DELAYED HORMONE THERAPY
ADT has well-accepted benefits in palliating painful bone metastases and urinary tract obstruction in patients with metastatic disease. However, ADT is not considered curative, and the response to therapy is temporary, with most patients progressing within 2 years (63). Since the introduction of ADT, controversy has existed over optimum timing. Many have advocated initiating treatment at the time of diagnosis with the aim of delaying disease progression and possibly prolonging survival. Others have argued that survival is not prolonged and that the treatment may be deferred until symptoms develop.
Some experimental evidence supports the early treatment of prostate cancer (not necessarily in the metastatic setting). Using the Dunning R3327 rat prostatic adenocarcinoma model, studies showed increased regression and survival after castration if the tumor volume was relatively small as opposed to when the tumor was allowed to grow larger (64). Using the same model, researchers were able to show that the mechanism responsible for the emergence of androgen resistance was the intrinsic heterogeneity of the tumor and not castration (65). They also demonstrated that poorly differentiated, fastgrowing androgen-sensitive tumors respond less to androgen withdrawal later in their lifespan.
Some nonrandomized data suggest a potential benefit from earlier therapy (66,67). On the other hand, the Veterans Administration Cooperative Urological Research Group study demonstrated that delaying hormonal therapy did not compromise overall survival (5,7,68,69) and demonstrated that many of the patients died of other causes while only 41% died from prostate cancer.
The Medical Research Council in the UK randomized 938 patients (934 patients had data available for analysis) with locally advanced prostate cancer or asymptomatic metastasis to either immediate treatment (orchiectomy or LHRH analog) or treatment deferred until symptoms occurred (70). Eighty patients died before treatment commenced (29 from prostate cancer and 51 from other causes). Progression from M0 to M1 disease (p < 0.001) and development of metastatic pain occurred more rapidly in deferred patients. One-hundred and forty-one deferred patients needed transurethral resection for local progression compared with sixty-five treated immediately (p < 0.001). Pathological fracture, spinal cord compression, ureteric obstruction, and development of extraskeletal metastases were twice as common in deferred patients. Three-hundred and sixty-one patients died in the deferred arm compared with three-hundred and twenty-eight in the immediate arm (p = 0.02). Sixty-seven of all deaths were from prostate cancer. Two-hundred and fifty-seven and two-hundred and three deaths were from prostate cancer in the deferred versus immediate treatment arms, respectively (p = 0.001). The difference was seen largely in locally advanced nonmetastatic patients, with 119 and 81 deaths from prostate cancer, respectively (p < 0.001).
The Cochrane databases compiled a meta-analysis of data from four randomized controlled trials in 2001 (71). The trials included were VACURG-I, VACURG-II, the MRC trial, and an ECOG trial studying early versus delayed androgen deprivation as adjuvant therapy in patients with pathologically involved lymph nodes after radical prostatectomy and lymph node dissection. The authors concluded that early hormonal therapy definitively reduces disease progression and the subsequent complications with a small improvement (6%) in overall survival at 10 years. Of note, 97% of the patients included in the 10-year analysis were extracted from the VACURG-I data, which makes this result sensitive to the weaknesses of that trial.
The clearest indication for immediate therapy for patients with new M1 disease is in the context of symptoms. Data from the MRC trial showing that early hormonal therapy can lower risks of significant complications such as cord compression and urinary tract obstruction and other data discussed above collectively provide the clinical rationale to treat early in the context of the commonly encountered scenario of the patient with asymptomatic metastasis.
NOVEL AGENTS AND APPROACHES
The better understanding of the biology of progression from hormonal dependence to castration resistance has led to a variety of novel agents and experimental approaches intended to enhance the effectiveness of therapy for patients with metastatic disease. Several such approaches/agents, discussed below, are in different phases of development at this time.
Mechanisms of Castration Resistance
Almost all patients with metastatic prostate cancer treated with androgen-withdrawal therapy eventually exhibit uncontrolled growth of CRPC. With a growing knowledge of tumor biology and apoptosis, there is a better understanding of the mechanisms responsible for the progression to castration-resistant cancer (72). Resistance is postulated to occur via cellular pathways that may or may not involve the AR. AR may be amplified (additional copies of the gene) thereby increasing sensitivity of the cell to low levels of testosterone (73). AR may be mutated to be constitutively activated mediated by the N terminal of the AR or “promiscuously” activated by nonandrogen ligands. Recently, splice variants of AR have been identified that are overexpressed in CRPC (74). Novel mechanisms of resistance involving AR include overexpression of AR coactivators including BRCA-2, intracrine synthesis of androgens in the tumor cell itself, and parallel pathways of activation of downstream targets mediated by down regulation of molecules such as TRADD (75). Other pathways for androgen resistance bypass the AR. Loss of PTEN permits Akt to phosphorylate the molecule Bad followed by the release of Bcl-2, an antiapoptotic molecule. Androgen-resistant cells may overexpress Bcl-2. In addition, neuropeptides like bombesin from adjacent cells could induce growth of prostate cancer cells in a metacrine fashion (72).
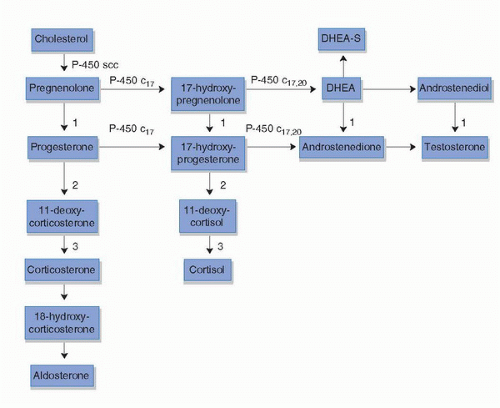
FIGURE 14B.1. Adrenal steroid hormone synthesis pathway. Enzyme 1 is 3β-hydroxysteroid dehydrogenase; Enzyme 2 is 21-hydroxylase; and Enzyme 3 is 11β-hydroxylase. (C17, 17-hydroxylase; C17,20, 17,20 lyase; DHEA, dehydroepiandrosterone; DHEA-S, dehydroepiandrosterone sulfate; SCC, cholesterol side-chain cleavage enzyme.)
Adrenal and Intratumoral Androgens
A potentially important source of extragonadal androgens is the innermost layer of the adrenal medulla called the zona reticularis that synthesizes the adrenal androgens, dehydroepiandrosterone (DHEA), DHEA-sulfate, and androstenedione (Fig. 14B.1). The adrenal gland as a source of androgens could partially account for the fact that measurable levels of DHT continue to be detected in castrated subjects. The contribution of androgens produced by the adrenal glands to CRPC was suggested in early adrenalectomy studies (76,77). However, surgical adrenalectomy is associated with unacceptable mortality and morbidity and has been abandoned as a therapeutic modality. Aminoglutethimide inhibits several enzymes in the steroidogenesis pathway (“medical adrenalectomy”), and initial studies showed response in a subset of metastatic CRPC patients refractory to castration (78,79). Significant side effects including adrenal insufficiency limit use of aminoglutethimide.
Ketoconazole, which inhibits several cytochrome P450 enzymes including CYP17, 20 lyase that is required for adrenal androgen synthesis, has reported response rates in CRPC ranging from 20% to 75% (80). The PSA response rate in the largest study of ketoconazole with hydrocortisone (with simultaneous or sequential antiandrogen withdrawal) was 32% when used after antiandrogen withdrawal with a 7% objective response rate (81). Ketoconazole causes adrenal insufficiency among other adverse effects and is typically reserved for second-line hormonal therapy in prostate cancer.
Novel Antiandrogens
Even in CRPC, the AR remains biologically involved including mediating resistance to ADT as discussed above (82). In particular, recent research has demonstrated the production of androgens within the tumor cells by upregulation of several enzymes involved in steroid synthesis as well as conversion of weaker adrenal steroids to testosterone/DHT (26,83,84), thus suggesting continued relevance of the androgen pathway in CRPC.
MDV 3100 is a small molecule antagonist of the AR (85). In hormone refractory prostate cancer xenograft models, increases in AR mRNA were consistently associated with development of resistance to androgen deprivation. MDV 3100 is a pure antagonist of the AR while current antiandrogens could function as agonists. MDV 3100 has greater affinity for the AR than bicalutamide, reduces AR translocation to the nucleus, impairs the binding of AR to androgen response elements on DNA, and also interferes with recruitment of coactivators to the response elements. Preclinical testing of MDV 3100 in LNCap-AR cells derived from a patient with CRPC showed a dose-dependent suppression of PSA (86). In phase I and II studies of MDV 3100 in patients with and without prior docetaxel therapy, impressive PSA responses were noted (85). A multinational phase III trial evaluating MDV 3100 at 160 mg daily oral dose versus placebo in postdocetaxel CRPC patients with overall survival as the end point is ongoing.
Although MDV3100 is currently being evaluated in the setting of CRPC, it is clearly an agent that has a potential role in the setting of hormone sensitive disease and first-line therapy of metastatic disease in the future.
Novel Steroid Synthesis Enzyme Inhibitors
Abiraterone acetate (CB7630) is an oral compound that specifically inhibits the 17-alpha hydroxylase and the 17,20 lyase (CYP450c17) enzymes within the adrenal steroid synthetic pathway. With an inhibition constant of <1 nM, it is a manyfold potent inhibitor of CYP450c17 than ketoconazole. Phase I and phase II studies show clinical activity of abiraterone in CRPC in predocetaxel and postdocetaxel settings (87,88). The response rates range from 75% in patients without extensive secondary hormonal manipulations or chemotherapy to 45% in heavily pretreated patients. Prior treatment with ketoconazole did not adversely affect treatment response to abiraterone in a phase I dose escalation study (89). Treatment with abiraterone results in ablation of adrenal androgens. Circulating testosterone levels even if already in the castrate range (<50 ng/dL) before treatment become undetectable (<1 ng/dL) with abiraterone. Results of two randomized phase III studies evaluating abiraterone acetate and prednisone versus placebo and prednisone—one in the minimally symptomatic/asymptomatic predocetaxel setting and another in postdocetaxel metastatic CRPC—are awaited.
Another compound, TAK-700, an oral selective, nonsteroidal inhibitor of the CYP 17,20 lyase, is being studied in trials in several clinical contexts. The phase I data from a phase I/II trial of TAK-700 with or without prednisone in metastatic CRPC patients as second-line therapy were recently presented (90). In the Phase I portion of the trial, 26 patients were studied. Secondary objectives include assessment of efficacy, as shown by PSA response and reduction of hormone levels. Patients received one of five dose levels (100-600 mg) orally twice daily with one additional group of five patients receiving prednisone too in combination with TAK-700 at 400 mg bid. All patients (n = 15) who received TAK-700 at doses >300 mg twice daily for three or more cycles experienced a decrease in PSA levels associated with reduction in testosterone levels. At 4 weeks, median testosterone and DHEA-sulfate levels had decreased from 4.9 to 0.6 ng/dL and 53.8 to <0.1 ng/dL, respectively, in patients treated with 400 mg BID. Eleven patients had PSA declines of >50%. There were no dose-limiting toxicities experienced during the Phase I portion. The recommended phase II dose is 400 mg bid. A phase I/II study of TAK-700 in combination with docetaxel in metastatic CRPC is underway and a phase II study is set to commence in nonmetastatic CRPC patients with a rising PSA.
Novel antiandrogens and drugs directed against enzymes in the steroid synthesis pathway clearly have a potential role in first-line hormonal therapy of metastatic prostate cancer in the future.
Intermittent Androgen Deprivation (IAD)
The established relationship between permanent androgen deprivation and the development of castration resistance provided the foundation for intermittent therapy. The underlying hypothesis is that permanent androgen withdrawal promotes progression to independence and reintroduction of androgens before progression is established will maintain the hormonal dependence thus response to androgen withdrawal.
Preclinical evidence, discussed below, suggests that hormonal resistance may be delayed with intermittent AD, providing enthusiasm for this approach as a possible strategy to improve the side effect profile, enhance antitumor efficacy and prolong time to androgen independence.
Preclinical models: Bruchovsky et al. used the androgen-dependent Shionogi cancer cell line to demonstrate that continuous androgen exposure altered the ratio of stem cells in the tumor population (91). There was a 20-fold increase in the proportion of total stem cells and a 500-fold increase in the androgen-independent stem cell population. The investigators hypothesized that progression to “hormone independence” is associated with the activation of previously androgen-repressed genes, some of which may code for autocrine or paracrine growth factors that substitute for androgens in maintaining the viability of the surviving tumorigenic stem cells. Using the Shionogi mammary cancer cell line, the interval to androgen independence was increased threefold using intermittent androgen therapy as compared with continuous androgen withdrawal (91). Using the human prostate xenograft LNCaP model, intermittent androgen suppression led to the delayed development of androgen-independent regulation of PSA expression, but not survival (92). These experimental models, which demonstrate the recovery of androgen dependence and apoptotic potential, provide the rationale for intermittent androgen-deprivation treatment.
Clinical trials of IAD: The clinical objective of IAD would be to prolong the overall duration of effective ADT by preserving the dependence of cancer cells on androgens for growth and proliferation. Preliminary reports from several clinical trials demonstrated feasibility of intermittent ADT (93,94,95,96,97,98,99). The effectiveness of reinstitution of AD in prior responders has been observed in the majority of patients on IAD including those with biochemical recurrence alone, localized or metastatic disease. In general, the duration of successive offtreatment periods gets progressively shorter until the ultimate emergence of androgen resistance. Data from these trials suggest improvements in sexual function, energy level, and libido during the off-treatment periods. Moreover, loss of bone mineral density was noted to slow down during off-therapy periods.
Several large phase III trials of continuous versus intermittent androgen in metastatic prostate cancer have completed accrual, of those SWOG 9346 (INT-0162) evaluated this approach in patients with new M1 prostate cancer and the South European Urological Group (SEUG) trial included both metastatic and PSA-only disease patients.
SWOG 9346 (INT-0162): 3,038 patients with newly diagnosed prostate cancer patients with bone metastases and PSA ≥5 ng/mL were enrolled. Patients who achieve PSA ≤4 ng/mL after 7 months of ADT (1,750 patients) have been randomized to continuous or intermittent androgen deprivation with goserelin and bicalutamide. The study has a 90% power with 90% confidence levels to assess noninferiority of IAD compared to continuous androgen deprivation in terms of overall survival. Secondary outcomes to be analyzed include time to development of androgen independence, quality of life, PSA dynamics, and various sociologic (race, socioeconomic background, etc.) and biologic correlates.
The SEUG study data have been reported (100). Patients with locally advanced or metastatic prostate cancer not previously treated with hormonal therapy or chemotherapy were treated with cyproterone acetate (a steroidal antiandrogen) and an LHRH agonist for 12 to 14 weeks. Patients who experienced a >80% decline in PSA or whose PSA declined to <4 ng/mL (626 of 766 patients achieved one of these criteria) were randomized to continuous versus intermittent androgen deprivation. Around 30% of the 626 randomized patients had metastatic disease. For those who qualified for randomization by a PSA <4 ng/mL, therapy was resumed in the intermittent arm when PSA level increased to ≥10 ng/ml with symptoms or ≥20 ng/mL without symptoms. For those who qualified by a >80% PSA decline, therapy was restarted when the PSA increased to ≥20% above the nadir value. The primary end point was time to subjective or objective progression. Secondary outcomes were survival, quality of life (QoL) using EORTC QLQ C30 QoL questionnaire, and time off therapy. The study was powered to detect a 30% decrease in median time to progression in the intermittent arm compared with the continuous arm. Median follow-up was 51 months from the date of randomization.
HR for progression in the continuous therapy arm (107 patients with progression) versus intermittent arm (127 patients with progression) was 0.81 (95% CI= 0.63-1.05, p = 0.11). There was thus a trend to longer time to progression in the continuous arm although it was not statistically significant. There was no difference in overall survival with an HR of 0.99 (95% CI = 0.80-1.23). Cancer-specific mortality was lower in continuous therapy arm (84 vs. 106; HR = 0.80, 95% CI = 0.60-1.06) but was balanced by a higher death rate from cardiovascular events in the continuous arm (52 vs. 41; HR = 1.27; 95% CI = 0.84-1.91). It must be noted that around 15% of patients in each arm were lost to follow-up. Men treated with intermittent therapy reported better sexual function. Median time of therapy for the intermittent patients was 52 weeks (95% CI = 39.4-65.7). From month 3 of follow-up, the mean testosterone level was significantly higher in the intermittent group compared with the continuous group (p < 0.001).
Side effects including hot flushes, gynecomastia, headaches, and skin complaints were more frequent in continuous arm group. The differences in QoL features were as follows: emotional domain was better in continuous arm (p = 0.01), nausea and vomiting was less in the continuous arm (p = 0.01), as was severity of insomnia (p = 0.03). Global QoL was better in continuous arm (p = 0.05). The authors commented that though statistically significant the differences were not clinically significant. Sexual function was significantly better in the intermittent arm as reflected by greater sexual activity and fewer dysfunctions reported. Sexual activity correlated to whether patients were on or off therapy.
Biomarkers of Response to ADT
The authors of the INT-0105 argued that the failure to translate a significantly improved PSA response to an overall survival or PFS benefit called into question the utility of PSA in metastatic disease as a prognostic marker (35). However, as noted in several studies, PSA-based criteria continue to be of prognostic utility irrespective of treatment arm (101,102,103). The best evidence for PSA response to ADT as a predictor of survival comes from the induction phase of a large phase III trial (SWOG 9346/INT-0162) of D2 patients. Compared to patients who did not achieve a PSA ≤4 ng/mL at the end of a 7-month induction phase, patients who did achieve PSA ≤4 ng mL (PSA-n) and those who achieved PSA ≤0.2 ng/mL (PSA-u) had much lower risk of death (104). Patients with PSA-n versus those not achieving it had a HR = 0.41 (95% CI = 0.32, 0.54, p < 0.001) for risk of death. Patients with PSA-u versus those who did not achieve PSA-n HR: 0.26 (95% CI = 0.20, 0.35, p < 0.001). After adjustment for covariates, patients with PSA-u had significantly better survival than those with only PSA-n at end of induction (p < 0.001).
Ongoing Experimental Strategies
Based on the increasing understanding of mechanisms of progression to castration resistance and the significant relationship of PSA levels after 7 months of ADT with survival, several proposals aimed at improving the efficacy of ADT are in progress utilizing PSA end points. These include increasing the proportion of patients who achieve a PSA < 4 ng/mL or undetectable PSA at the end of the initial androgen deprivation cycle by adding Bcl2 antagonists (AT101) (NCT00666666) or IMC-A12 (Anti-IGF) (SWOG 0925) to ADT. Another approach to be evaluated in SWOG 0822 is to test the ability of Abiraterone acetate to reinduce a response in patients with suboptimal response (patients who do not achieve PSA <4 ng/mL) after 7 months of androgen deprivation.
Chemotherapy has also been investigated in combination with ADT in metastatic prostate cancer (105,106). Since the approval of docetaxel as the first chemotherapeutic agent with a survival benefit in metastatic prostate cancer (107,108), there is renewed interest in investigating docetaxel-based chemotherapy in the first-line treatment of newly diagnosed metastatic prostate cancer. An ongoing Intergroup ECOG trial (CHAARTED: ChemoHormonal Therapy Versus Androgen Ablation Randomized Trial for Extensive Disease in Prostate Cancer; NCT00309985) is a randomized phase III trial testing ADT alone or in combination with chemotherapy (up to six cycles of docetaxel). Overall survival is the primary end point; secondary outcomes include PSA response, time to CRPC, and time to clinical and PSA progression. QoL is also assessed as part of the study.
CONCLUSIONS AND FUTURE DIRECTIONS
ADT is the standard first-line therapy for metastatic prostate cancer. The recently developed novel potent inhibitors of enzymes involved in androgen synthesis and newer antiandrogens hold great promise as potential components of future first-line therapy in metastatic prostate cancer. Improving understanding of the molecular and cellular biology of development of androgen resistance should permit strategies to prolong duration of sensitivity to androgen deprivation and prevention of castration androgen resistance. Ongoing trials will demonstrate any potential value in adding chemotherapy to hormonal therapy in hormone sensitive metastatic disease while novel biomarkers in development such as circulating tumor cells or PSA will assist in individualizing therapy with the goal of maximizing benefit while minimizing toxicities. Critical to progress is the timely implementation and accrual to clinical trials in this disease.
References
1. Murphy L. The history of urology. Springfield, IL: 1972:68-386.
2. White J. The results of double castration in hypertrophy of the prostate. Ann Surg 1895;22:1-8.
3. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol 2002;168(1):9-12.
4. Hall JE. Guyton and Hall Textbook of medical physiology, 12th ed. Saunders, Elsevier. Philadelphia, PA:2010.
5. Blackard CE, Byar DP, Jordan WP Jr. Orchiectomy for advanced prostatic carcinoma. A reevaluation. Urology 1973;1(6):553-560.
6. Nyman CR, Andersen JT, Lodding P, et al. The patient’s choice of androgen-deprivation therapy in locally advanced prostate cancer: bicalutamide, a gonadotrophin-releasing hormone analogue or orchidectomy. BJU Int2005;96(7):1014-1018.
7. Byar DP, Corle DK. Hormone therapy for prostate cancer: results of the Veterans Administration Cooperative Urological Research Group studies. NCI Monogr 1988(7):165-170.
8. Pomerantz M, Manola J, Taplin ME, et al. Phase II study of low dose and high dose conjugated estrogen for androgen independent prostate cancer. J Urol 2007;177(6):2146-2150.
9. Moreau JP, Delavault P, Blumberg J. Luteinizing hormone-releasing hormone agonists in the treatment of prostate cancer: a review of their discovery, development, and place in therapy. Clin Ther 2006;28(10):1485-1508.
10. Leuprolide versus diethylstilbestrol for metastatic prostate cancer. The Leuprolide Study Group. N Engl J Med 1984;311(20):1281-1286.
11. Sharifi R, Knoll LD, Smith J, et al. Leuprolide acetate (30-mg depot every four months) in the treatment of advanced prostate cancer. Urology 1998;51(2):271-276.
12. Vance MA, Smith JA Jr. Endocrine and clinical effects of leuprolide in prostatic cancer. Clin Pharmacol Ther 1984;36(3):350-354.
13. Soloway MS, Chodak G, Vogelzang NJ, et al. Zoladex versus orchiectomy in treatment of advanced prostate cancer: a randomized trial. Zoladex Prostate Study Group. Urology 1991;37(1):46-51.
14. Debruyne FM, Denis L, Lunglmayer G, et al. Long-term therapy with a depot luteinizing hormone-releasing hormone analogue (Zoladex) in patients with advanced prostatic carcinoma. J Urol 1988;140(4):775-777.
15. Seidenfeld J, Samson DJ, Hasselblad V, et al. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Ann Intern Med 2000;132(7):566-577.
16. Klotz L, Boccon-Gibod L, Shore ND, et al. The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallelgroup phase III study in patients with prostate cancer. BJU Int 2008;102(11):1531-1538.
17. Neri RO. Antiandrogens. Adv Sex Horm Res 1976;2:233-262.
18. Schellhammer P, Sharifi R, Block N, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Casodex Combination Study Group. Urology 1995;45(5):745-752.
19. Prostate Cancer Trialists’ Collaborative Group. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Lancet 2000;355(9214):1491-1498.
20. Sanford EJ, Paulson DF, Rohner TJ Jr, et al. The effects of castration on adrenal testosterone secretion in men with prostatic carcinoma. J Urol 1977;118(6):1019-1021.
21. Geller J, de la Vega DJ, Albert JD, et al. Tissue dihydrotestosterone levels and clinical response to hormonal therapy in patients with advanced prostate cancer. J Clin Endocrinol Metab 1984;58(1):36-40.
22. Geller J, Albert JD, Nachtsheim DA, et al. Comparison of prostatic cancer tissue dihydrotestosterone levels at the time of relapse following orchiectomy or estrogen therapy. J Urol 1984;132(4):693-696.
23. Trachtenberg J. Optimal testosterone concentration for the treatment of prostatic cancer. J Urol 1985;133(5):888-890.
24. Kyprianou N, Isaacs JT. Quantal relationship between prostatic dihydrotestosterone and prostatic cell content: critical threshold concept. Prostate 1987;11(1):41-50.
25. Kyprianou N, Isaacs JT. Biological significance of measurable androgen levels in the rat ventral prostate following castration. Prostate 1987;10(4):313-324.
26. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68(11):4447-4454.
27. Labrie F, Dupont A, Belanger A, et al. New hormonal therapy in prostate cancer: combined use of a pure antiandrogen and an LHRH agonist. Horm Res 1983;18(1-3):18-27.
28. Labrie F, Belanger A, Simard J, et al. Combination therapy for prostate cancer. Endocrine and biologic basis of its choice as new standard first-line therapy. Cancer 1993;71(3 Suppl):1059-1067.
29. Dijkman GA, Janknegt RA, De Reijke TM, et al. Long-term efficacy and safety of nilutamide plus castration in advanced prostate cancer, and the significance of early prostate specific antigen normalization. International Anandron Study Group. J Urol 1997;158(1):160-163.
30. Boccardo F, Pace M, Rubagotti A, et al. Goserelin acetate with or without flutamide in the treatment of patients with locally advanced or metastatic prostate cancer. The Italian Prostatic Cancer Project (PONCAP) Study Group. Eur J Cancer 1993;29A(8):1088-1093.
31. Bono AV, DiSilverio F, Robustelli della Cuna G, et al. Complete androgen blockade versus chemical castration in advanced prostatic cancer: analysis of an Italian multicentre study. Italian Leuprorelin Group. Urol Int 1998;60(Suppl 1):18-24.
32. Crawford ED, Kasimis BS, Gandara D, et al. A randomized, controlled trial of leuprolide and anandron (LA) vs leuprolide and placebo (LP) for advanced prostate cancer. Proc Am Soc Clin Oncol 1990;9:A523.
33. de Voogt HJ, Studer U, Schroder FH, et al. Maximum androgen blockade using LHRH agonist buserelin in combination with short-term (two weeks) or long-term (continuous) cyproterone acetate is not superior to standard androgen deprivation in the treatment of advanced prostate cancer. Final analysis of EORTC GU Group Trial 30843. European Organization for Research and Treatment of Cancer (EROTC) Genito-Urinary Tract Cancer Cooperative Group. Eur Urol 1998;33(2):152-158.
34. Di Silverio F, Serio M, D’Eramo G, et al. Zoladex vs. Zoladex plus cyproterone acetate in the treatment of advanced prostatic cancer: a multicenter Italian study. Eur Urol 1990;18(Suppl 3):54-61.
35. Eisenberger MA, Blumenstein BA, Crawford ED, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med 1998;339(15):1036-1042.
36. Fourcade RO, Colombel P, Mangin M. Zoladex plus flutamide versus Zoladex plus placebo in advanced prostatic carcinoma: extended followup of the French multicentre study. In: Murphy G, Khoury S, Chatelain C, et al., eds. Proceedings of the 3rd International Symposium on Recent Advances in Urological Cancer: Diagnosis and Treatment, June 1992. Paris, France: Scientific Communication International Ltd., 1993:102.
37. Iversen P, Rasmussen F, Klarskov P, et al. Longterm results of Danish Prostatic Cancer Group trial 86. Goserelin acetate plus flutamide versus orchiectomy in advanced prostate cancer. Cancer 15 1993;72(12 Suppl):3851-3854.
38. Jorgensen T, Tveter KJ, Jorgensen LH. Total androgen suppression: experience from the Scandinavian Prostatic Cancer Group Study No. 2. Eur Urol 1993;24(4):466-470.
39. Robinson MR, Smith PH, Richards B, et al. The final analysis of the EORTC Genito-Urinary Tract Cancer Co-Operative Group phase III clinical trial (protocol 30805) comparing orchidectomy, orchidectomy plus cyproterone acetate and low dose stilboestrol in the management of metastatic carcinoma of the prostate. Eur Urol 1995;28(4):273-283.
40. Thorpe SC, Azmatullah S, Fellows GJ, et al. A prospective, randomised study to compare goserelin acetate (Zoladex) versus cyproterone acetate (Cyprostat) versus a combination of the two in the treatment of metastatic prostatic carcinoma. Eur Urol 1996;29(1): 47-54.
41. Tyrrell CJ, Altwein JE, Klippel F, et al. Multicenter randomized trial comparing Zoladex with Zoladex plus flutamide in the treatment of advanced prostate cancer. Survival update. International Prostate Cancer Study Group. Cancer 1993;72(12 Suppl):3878-3879.
42. Zalcberg JR, Raghaven D, Marshall V, et al. Bilateral orchidectomy and flutamide versus orchidectomy alone in newly diagnosed patients with metastatic carcinoma of the prostate—an Australian multicentre trial. Br J Urol 1996;77(6):865-869.
43. Moinpour CM, Savage MJ, Troxel A, et al. Quality of life in advanced prostate cancer: results of a randomized therapeutic trial. J Natl Cancer Inst 1998;90(20):1537-1544.
44. Samson DJ, Seidenfeld J, Schmitt B, et al. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer 2002;95(2):361-376.
45. Schmitt B, Wilt TJ, Schellhammer PF, et al. Combined androgen blockade with nonsteroidal antiandrogens for advanced prostate cancer: a systematic review. Urology Apr 2001;57(4):727-732.
46. Bertagna C, De Gery A, Hucher M, et al. Efficacy of the combination of nilutamide plus orchidectomy in patients with metastatic prostatic cancer. A meta-analysis of seven randomized double-blind trials (1056 patients). Br J Urol 1994;73(4):396-402.
47. Denis L, Murphy GP. Overview of phase III trials on combined androgen treatment in patients with metastatic prostate cancer. Cancer 1993;72(12 Suppl):3888-3895.
48. Blumenstein BA. Some statistical considerations for the interpretation of trials of combined androgen therapy. Cancer 1993;72(12 Suppl):3834-3840.
49. Bales GT, Chodak GW. A controlled trial of bicalutamide versus castration in patients with advanced prostate cancer. Urology 1996;47(1A Suppl):38-43; discussion 48-53.
50. Chodak G, Sharifi R, Kasimis B, et al. Singleagent therapy with bicalutamide: a comparison with medical or surgical castration in the treatment of advanced prostate carcinoma. Urology 1995;46(6):849-855.
51. Iversen P, Tveter K, Varenhorst E. Randomised study of Casodex 50 MG monotherapy vs orchidectomy in the treatment of metastatic prostate cancer. The Scandinavian Casodex Cooperative Group. Scand J Urol Nephrol 1996;30(2):93-98.
52. Kaisary AV, Tyrrell CJ, Beacock C, et al. A randomised comparison of monotherapy with Casodex 50 mg daily and castration in the treatment of metastatic prostate carcinoma. Casodex Study Group. Eur Urol 1995;28(3):215-222.
53. Boccardo F, Barichello M, Battaglia M, et al. Bicalutamide monotherapy versus flutamide plus goserelin in prostate cancer: updated results of a multicentric trial. Eur Urol 2002;42(5):481-490.
54. Tyrrell CJ, Kaisary AV, Iversen P, et al. A randomised comparison of ‘Casodex’ (bicalutamide) 150 mg monotherapy versus castration in the treatment of metastatic and locally advanced prostate cancer. Eur Urol 1998;33(5):447-456.
55. Kolvenbag GJ, Iversen P, Newling DW. Antiandrogen monotherapy: a new form of treatment for patients with prostate cancer. Urology 2001;58(2 Suppl 1):16-23.
56. Fourcade RO, Chatelain C, Poterre M. An open multicentre study to compare the effect and safety of Casodex (bicalutamide) 150 mg monotherapy with castration plus nilutamide in metastatic prostate cancer [Abstract]. Eur Urol 1998;33(Suppl 1):88.
57. Boccon-Gibod L, Fournier G, Bottet P, et al. Flutamide versus orchidectomy in the treatment of metastatic prostate carcinoma. Eur Urol 1997;32(4):391-395; discussion 395-396.
58. Pavone Macaluso M. Flutamide monotherapy versus. combined androgen blockade in advanced prostate cancer [Abstract]. Interim Report of an Italian Multicenter 1994; SIU 23rd Congress. A354.
59. Chang A, Yeap B, Davis T, et al. Double-blind, randomized study of primary hormonal treatment of stage D2 prostate carcinoma: flutamide versus diethylstilbestrol. J Clin Oncol 1996;14(8):2250-2257.
60. Pavone-Macaluso M, de Voogt HJ, Viggiano G, et al. Comparison of diethylstilbestrol, cyproterone acetate and medroxyprogesterone acetate in the treatment of advanced prostatic cancer: final analysis of a randomized phase III trial of the European Organization for Research on Treatment of Cancer Urological Group. J Urol 1986;136(3): 624-631.
61. Ostri P, Bonnesen T, Nilsson T, et al. Treatment of symptomatic metastatic prostatic cancer with cyproterone acetate versus orchiectomy: a prospective randomized trial. Urol Int 1991;46(2):167-171.
62. Tay MH, Kaufman DS, Regan MM, et al. Finasteride and bicalutamide as primary hormonal therapy in patients with advanced adenocarcinoma of the prostate. Ann Oncol 2004;15(6):974-978.
63. Beynon LL, Chisholm GD. The stable state is not an objective response in hormoneescaped carcinoma of prostate. Br J Urol 1984;56(6):702-705.
64. Isaacs JT. The timing of androgen ablation therapy and/or chemotherapy in the treatment of prostatic cancer. Prostate 1984;5(1):1-17.
65. Isaacs JT, Coffey DS. Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the Dunning R-3327-H adenocarcinoma. Cancer Res 1981;41(12 Pt 1):5070-5075.
66. Kramolowsky EV. The value of testosterone deprivation in stage D1 carcinoma of the prostate. J Urol 1988;139(6):1242-1244.
68. The Veterans Administration Co-operative Urological Research Group. Treatment and survival of patients with cancer of the prostate. Surg Gynecol Obstet 1967;124(5):1011-1017.
69. Christensen MM, Aagaard J, Madsen PO. Reasons for delay of endocrine treatment in cancer of the prostate (until symptomatic metastases occur). Prog Clin Biol Res 1990;359:7-14; discussion 15-24.
70. The Medical Research Council Prostate Cancer Working Party Investigators Group. Immediate versus deferred treatment for advanced prostatic cancer: initial results of the Medical Research Council Trial. Br J Urol 1997;79(2):235-246.
71. Wilt T, Nair B, MacDonald R, et al. Early versus deferred androgen suppression in the treatment of advanced prostate cancer (Cochrane Review). The Cochrane Library, Issue 2. Chichester, UK: John Wiley and Sons, 2004.
72.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med 2004;351(15):1488 -1490.
73.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10(1):33-39.
74.Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009;69(6):2305-2313.
75.Wang D, Montgomery RB, Schmidt LJ, et al. Reduced tumor necrosis factor receptor-associated death domain expression is associated with prostate cancer progression. Cancer Res 2009;69(24):9448-9456.
76.Mahoney EM, Harrison JH. Bilateral adrenalectomy for palliative treatment of prostatic cancer. J Urol 1972;108(6):936-938.
77.Brendler H. Adrenalectomy and hypophysectomy for prostatic cancer. Urology. Aug 1973;2(2):99-102.
78.Samojlik E, Lippman AJ, Kirschner MA, Ertel NH, Park Y, Szmal E. Medical adrenalectomy for advanced prostatic cancer: clinical and hormonal effects. Am J Clin Oncol 1988;11(5):579-585.
79.Sanford EJ, Drago JR, Rohner TJ, Jr., Santen R, Lipton A. Aminoglutethimide medical adrenalectomy for advanced prostatic carcinoma. 1976;115(2):170-174.
80.Yap TA, Carden CP, Attard G, de Bono JS. Targeting CYP17: established and novel approaches in prostate cancer. Curr Opin Pharmacol 2008;8(4):449-457.
81.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol 2004;22(6):1025-1033.
83.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006;66(5):2815-2825.
84.Locke JA, Guns ES, Lubik AA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 2008;68(15):6407-6415.
85.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324(5928):787-790.
86.Sawyers CL TC, Wongvipat J, Ouk S, Yoo D, Protter AA, Hung DT, Jung ME. Characterization of a new anti-androgen MDV-3100 effective in preclinical models of hormone refractory prostate cancer. 2007 Prostate Cancer Symposium 2007.
87.Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol 20;28(9):1489-1495.
Only gold members can continue reading. Log In or Register to continue