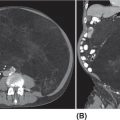
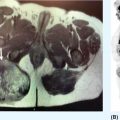
20915 Alveolar Soft Part Sarcoma Alveolar soft part sarcoma (ASPS) is a rare, translocation-driven soft tissue sarcoma (STS) classically affecting adolescents and young adults. The disease was first formally described in 1952 by Christopherson et al., who coined the term “alveolar soft part sarcoma” based on its unique histologic features, which included the presence of an organoid to pseudoalveolar pattern and large eosinophilic tumor cells. Clinically, ASPS is known for its indolent behavior, propensity to metastasize early, and inherent resistance to conventional doxorubicin-based chemotherapy typically employed in the management of STS. Given the rarity of ASPS, the majority of our understanding of the disease comes from small case series, retrospective analysis, and clinical experience. This chapter reviews the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for ASPS. Alveolar Soft Part Sarcoma, ASPS, immunotherapy, angiogenesis alveolar soft part sarcoma, clinical presentation, diagnostic approach, epidemiology, molecular drivers, retrospective analysis, soft tissue sarcoma, treatment options Diagnosis, Epidemiology, Retrospective Studies, Sarcoma, Alveolar Soft Part, Therapeutics Alveolar soft-part sarcoma [is] a type of soft-part sarcoma quite unlike any well-documented tumor of which we are aware. … Even though one cannot classify a tumor, it is desirable to recognize it as something one does not fully understand, for only through such recognition can one offer opinions as to probable behavior. —William M. Christopherson, et al., 19521 INTRODUCTION Alveolar soft part sarcoma (ASPS) is a rare, translocation-driven soft tissue sarcoma (STS) classically affecting adolescents and young adults. The disease was first formally described in 1952 by Christopherson et al., who coined the term “alveolar soft part sarcoma” based on its unique histologic features, which included the presence of an organoid to pseudoalveolar pattern and large eosinophilic tumor cells.1 Further description of ASPS is credited to Dr. Pierre Masson, who first described the intracytoplasmic crystalline structures in his 1956 text, Tumeurs Humaines: Histologie, Diagnostics et Techniques.2 With advances in technology and our improved understanding of molecular genetics, we further elucidated that ASPS is defined not only by its characteristic histologic features but also by a conserved unbalanced translocation t(X;17)(p11;q25), which leads to a chimeric ASPL–transcription factor E3 (TFE3) fusion protein that leads to aberrant transcription.3,4 Clinically, ASPS is known for its indolent behavior, propensity to metastasize early, and its inherent resistance to conventional doxorubicin-based chemotherapy typically employed in the management of STS. Given the rarity of ASPS, the majority of our understanding of the disease comes from small case series, retrospective analysis, and clinical experience. The discovery of the ASPL–TFE3 fusion protein and identification of angiogenic factors as critical mediators of ASPS pathophysiology has opened new doors to the exploration of novel treatment options including the use of tyrosine kinase inhibitors (TKIs) and most recently, modern immunotherapy including immune checkpoint inhibitors (ICIs). In this chapter, we review the epidemiology, clinical presentation, diagnostic approach, molecular drivers, and treatment options for ASPS. EPIDEMIOLOGY While STSs are a rare and heterogeneous group of cancers with an estimated 13,040 new cases diagnosed in 2018 representing only 0.8% of all new cancer diagnosis, ASPS accounts for <1% of newly diagnosed STS cases.5 Based on data from the Surveillance, Epidemiology, and End Results (SEER) database, a mere 267 new cases were diagnosed in the United States between 1973 and 2014.6 Most commonly, ASPS presents in young adults between the ages of 15 and 35. Diagnosis before age 30 has been shown to have a female-to-male predominance, with reversal of this ratio in patients older than 30 at the time of diagnosis.4,7 No links to particular ethnicity or race have been reported. CLINICAL PRESENTATION AND NATURAL HISTORY ASPS frequently presents as a slow-growing mass in the deep tissues of the extremities, typically in the buttock and thighs. This is the primary disease site in approximately 60% of adult patients. Alternatively, unusual primary disease sites have been described, including the gastrointestinal tract, 210female genital tract, mediastinum, breast, urinary bladder, bone, and lung. Children with ASPS commonly present with tumors in the orbits or head and neck region. Given the indolent nature of the tumor, patients often do not develop functional impairment and describe a history of a slow-growing mass over years. Most commonly, patients present once the mass has become symptomatic or with signs and symptoms of metastatic disease. Gastrointestinal bleeding, vaginal bleeding, and proptosis have been reported as the presenting symptoms in patients with unusual disease sites. On physical exam, the clinician may appreciate a pulsatile mass and bruit on auscultation, as these tend to be highly vascular tumors (Figure 15.1A).4,8 A recent case series of 69 patients diagnosed with ASPS retrospectively reviewed from four major institutions reported that the median age at diagnosis was 17 years and 64% of patients were female.8 The primary sites of disease included limbs (58%), trunk/retroperitoneum/pelvis (16%), and less commonly, tongue/pharynx (9%) and orbits (3%). All the 26 patients tested for ASPL–TFE3 translocation were positive. Thirty-one patients presented with localized disease with Intergroup Rhabdomysarcoma Study (IRS) staging I, II, and III being 28%, 10%, and 7%, respectively. Thirty-eight (55%) of the patients had stage IV disease at the time of diagnosis. The sites of metastatic disease included lung (97%), bone (26%), and brain (4%). In the 31 patients presenting with localized disease, the 5-year event-free survival (EFS) rate was 80% and overall survival (OS) rate was 87%; alternatively, in those presenting with stage IV disease, the EFS and OS rates were 7% and 61%, respectively.8 In another relatively large series of 70 patients over 39 years from MD Anderson Cancer Center (MDACC), the population was slightly older with a median age of 26 years.9 Overall, 51% of patients in this series were female; however, in patients over the age of 30, 58% of the patients were male. Metastatic disease at diagnosis was detected in 65% of patients.9 At a median follow-up of 9 years, two of 22 patients who initially presented with localized disease developed local recurrence and three of 22 patients developed metastatic disease to the lungs. Five-year disease-free survival (DFS) rate of patients with localized disease was 71% and OS rate was 88%. In contrast, the median survival of patients with metastatic disease was 40 months, with a 5-year survival rate of 20%. Importantly, this series revealed an even higher proportion of patients with brain metastases. In contrast to other subtypes of STS, where brain involvement is rare, nine of 48 patients (19%) who presented with metastatic ASPS were noted to have brain lesions, which were always found in the presence of metastases to other sites as well. No patients with isolated brain lesions as the sole site of metastasis were identified, suggesting that routine intracranial imaging is unlikely to benefit patients without evidence of other metastatic disease. Despite the historically dire prognosis of metastatic disease, it is critical to recognize that not all patients will experience rapid disease progression. Like other indolent STSs, such as epithelioid hemangioendothelioma, some patients may have relatively low-burden metastatic disease in the lungs that can remain stable and controlled without progression for months to years. In the MDACC series, two patients were noted to be alive with disease for 11 and 28 years, respectively, after the diagnosis of metastatic ASPS. Because of this observation, many providers will opt for close monitoring for newly diagnosed metastatic patients to identify patients with indolent biologic behavior and to reserve treatment for radiographic or symptomatic disease progression. DIAGNOSTIC APPROACH Given the rarity of this tumor, high clinical suspicion is required for accurate diagnosis. Like any soft tissue mass, early evaluation with gadolinium-enhanced MRI is critical to identify features suggestive of a malignant process. Referral to a multidisciplinary sarcoma center for initial management, including biopsy, pathologic review, and surgical resection for localized disease, is paramount for the best outcomes.10 Initial staging should include a high-quality contrast-enhanced CT scan of the lungs, as well as abdomen, pelvis, and bone scan. PET/CT scan may be useful to identify occult lesions particularly in advanced disease, but early lung metastases are often subcentimeter in size and below the threshold of PET detection. Additionally, PET avidity is variable, particularly given the indolent nature of some ASPS. MRI of the brain should be performed at diagnosis and periodically in patients with metastatic disease in other sites. Radiographic Features ASPS tumors appear highly vascular on angiography and MRI with evidence of torturous, dilated veins (Figure 15.1B and C). On MRI, it is typical to see signs of prominent venous vasculature and high signal intensity on T1- and T2-weighted images. Cui et al. performed a retrospective analysis of 211MRI features in a case series of 12 patients with ASPS confirmed by pathology. They concluded that low signals or radiating flow voids accompanied by high signals of slow blood flow or blood sinuses in the center part of the tumor have high significance for the diagnosis of ASPS.11 Ultrasound can also reveal vascular flow through ASPS lesions, which can sometimes be mistaken for large hemangiomas. In metastatic disease, virtually all patients will have innumerable lung metastases, and remarkably most patients can tolerate substantial burden of disease with mild clinical symptoms such as cough or decreased exercise tolerance (Figure 15.1D).12 FIGURE 15.1 Clinical, radiographic, and pathologic features of ASPS. (A) Clinical image of large soft tissue mass of the left upper arm. (B) Angiogram of right lower extremity showing marked vascularity of the ASPS tumor with large tortuous arteries and veins and arteriovenous shunting. (C) MRI showing sagittal view of large lower extremity tumor. (D) CT of the chest showing substantial burden of metastatic disease common in ASPS. (E) Biopsy stained with hematoxylin and eosin, low-power view, showing the organoid and pseudoalveolar appearance of the tumor with central clearing and thin-walled vascular spaces. ASPS, alveolar soft part sarcoma. Source: Figure 15.1B reproduced with permission from Temple HT, Scully SP, O’Keefe RJ, et al. Clinical presentation of alveolar soft-part sarcoma. Clin Orthop Relat Res. 1994:300: Figure 5, p. 216. PMID:8131338. 212Histopathologic Features ASPS has a distinctive appearance microscopically and interestingly varies little from case to case; however, the differentiation and cell origin remain ambiguous. Uniform organoid nests of polygonal tumor cells, separated by fibrovascular septa, and delicate capillary-sized vascular channels define the histologic appearance (Figure 15.1E). The “alveolar” component of the name is due to the pseudoalveolar pattern, which appears to be due to necrosis of centrally located cells in the nests. Intravascular extension is present at the periphery in nearly all cases and may contribute to the high rate of metastatic disease. The cells often contain an abundant eosinophilic cytoplasm with a central nucleus and prominent nucleolus and intracytoplasmic crystals.3 The role of immunohistochemistry has historically been limited for the diagnosis for ASPS. Typically, these tumors are negative for epithelial markers including cytokeratins and epithelial membrane antigen, negative for neuroendocrine markers such as chromogranin A and synaptophysin, and negative for melanocytic markers including HMB-45 and Melan-A. Nonspecific markers including desmin and antibodies to actins in pan, smooth, and skeletal muscle have been reported to be positive in nearly 50% of patients. This has been an area of interest, given the controversy over cell of origin and possible myogenous differentiation.3 MOLECULAR AND BIOLOGIC DRIVERS ASPS is defined by a tumor-specific translocation, t(X;17)(p11;q25).13 This translocation results in a fusion of the transcription factor TFE3 located on Xp11.22 with a novel gene at 17q25 named ASPL, also known as ASPSCR1. Detection of this translocation can be achieved through polymerase chain reaction (PCR) or identification of TFE3 rearrangements by fluorescence in situ hybridization (FISH). Historically, a small subset of ASPS has been thought to be fusion negative; however, with modern whole-genome sequencing, the fusion is generally detected in these patients, likely with an alternative breakpoint that is missed by FISH probes. The recognition of this translocation provides an important diagnostic tool in differentiating ASPS from other tumor types based on histology alone, including renal cell carcinoma, hepatocellular carcinoma, paraganglioma, granular cell tumor, melanoma, and adrenal cortical carcinoma. It is important to note that some renal cell carcinomas have also been shown to be driven by related TFE3 translocations.14 The ASPS1–TFE3 fusion protein acts through altered regulation of activity of TFE3, a member of the microphthalmia transcription factor (MITF) transcription factor family, which drives ASPS biology through aberrant transcription and upregulation of several key pathways. First, a TFE3-binding site is located in the promoter of the proto-oncogene MET, which encodes the tyrosine kinase receptor c-MET, and multiple studies have identified increased functional protein production of c-MET and its ligand hepatocyte growth factor (HGF) in the setting of ASPS1–TFE3 activity.15–17 Downstream effects include upregulation of MAPK (mitogen-activated protein kinases)/ERK (extracellular signal-regulated kinases) and AKT (protein kinase B) pathways, key oncogenic regulators in other types of cancer. Additionally, numerous proangiogenic factors including vascular endothelial growth factor (VEGF), midkine, angiogenin, hypoxia-inducible factor 1 alpha (HIF-1alpha), and transforming growth factor beta (TGF-beta) have been shown to be highly expressed in ASPS, with subsequent chromatin immunoprecipitation analysis confirming direct binding of the fusion protein to the promoters of some targets such as angiopoietin-like 2 protein.18 However, the majority of upregulated angiogenesis genes have not been shown to be direct transcriptional targets. Emerging evidence suggests that ASPS biology, including the dependence on angiogenesis, may be highly impacted by the tumor, stromal, and immune microenvironmental factors. In a mouse model of ASPS, with conditional and inducible expression of the ASPSCR1–TFE3 fusion gene transcript, lactate critically affected tumor cell metabolism, induced HIF-1alpha stabilization, and enhanced proliferation and angiogenesis.19 Additionally, a different ex vivo mouse model of ASPS demonstrated that the tumor-associated vasculature permitted intravasation of tumor cells surrounded by nonmalignant 213hemangiopericytes, a phenomenon observed in human ASPS as well.20 The authors postulated that hemangiopericytic encapsulation might protect tumor cells from attack by the immune system and facilitate metastatic spread. The natural history of ASPS, with a prolonged indolent state followed by rapid tumor growth and metastasis, suggests that the human immune system may be capable of suppressing the growth of ASPS at least initially. The cancer immunoediting hypothesis describes an initial period of equilibrium, where the immune system is capable of suppressing tumor cell growth and proliferation; but with acquisition of additional immune evasion mechanisms, immunoedited resistant clones gradually take over and escape, leading to clinically apparent tumors.21 A few case reports have described spontaneous regression of ASPS metastatic lesions, particularly after resection of the primary tumor.22 Patients undergoing metastasectomy in the setting of limited disease generally show improved OS, which can also be seen in other types of STS. Features consistent with an immunosuppressive microenvironment in ASPS tumors include robust stroma and angiogenic networks, expression of genes like TGF-β, a potential immunosuppressive cytokine, as well as reports of expression of the immune checkpoint protein programmed-death ligand 1 (PD-L1). Another potential mechanism in which ASPL–TFE3 may play a role in tumorigenesis and promote an immunosuppressive microenvironment was investigated by Ishiguro et al., who demonstrated that in vitro models with ectopic expression of ASPS1–TFE3 resulted in cell cycle arrest and increased levels of protein and mRNA of p21 (p21WAF1/CIP1). When ASPL–TFE3 was expressed in human bone marrow mesenchymal stem cells, they observed upregulation of p21 and induction of senescence-associated galactosidase activity. When p21 was suppressed, they observed significant decrease in the induction of ASPL–TFE3-mediated cellular senescence. Furthermore, the expression of ASPL–TFE3 was shown to induce proinflammatory cytokines including interleukin (IL)-1, IL-6, and IL-8 with senescence-associated secretory phenotype (SAPS), which further promote an immunosuppressive tumor microenvironment.23 Finally, faulty metabolism within tumor sites, including lactate dependence, has been recently shown to greatly impact the activity and functionality of tumor-infiltrating lymphocytes, which could be a critical factor in ASPS.24 Overall, further exploration and characterization of the immune microenvironment in ASPS are critical, particularly given the early activity observed with immunotherapy. TREATMENT CONSIDERATIONS Management of the Primary Tumor The definitive treatment for ASPS with localized disease is complete surgical resection upfront. In a recent SEER analysis of 251 patients with ASPS from 1973 to 2012, 67% of patients had locoregional disease at presentation and 43% had distant metastasis. The 5-year OS rate for patients was 56%; but for those with localized disease, it was 82% and for those with distant metastasis at presentation, it was only 27%. The analysis identified that older age at diagnosis, tumor size >10 cm, distant metastasis, and truncal primary site were independent risk factors predicting worse OS. For the patients with localized disease, the analysis found that patients treated with surgery plus radiation had improved OS in comparison to surgery alone (p = .014); however, even for patients with distant metastasis, surgery of the primary site significantly improved OS (p < .001).25 The surgical margins postoperatively and/or the presence of metastatic disease have largely guided the clinician’s decision to treat with chemotherapy and/or radiation in the adjuvant setting. It is largely accepted that ASPS is resistant to typical sarcoma doxorubicin-based chemotherapy regimens. Based on one European review of the literature of 68 patients who received upfront chemotherapy with anthracycline- or ifosfamide-containing regimens, only 4% achieved complete remission (CR), 3% had partial response (PR), 41% had stable disease, and 51% had progression of disease.26 In another prospective European study in the pediatric population, 22 patients with a diagnosis of ASPS were evaluated. The patients were risk stratified postoperatively based on the completeness of the resection, IRS staging, and the size and grade of the tumor. Chemotherapy with doxorubicin and ifosfamide was offered to patients in IRS group III (local tumor that has not metastasized, but cannot be completely resected) and radiation was performed in patients in IRS groups II and III (II: resected with positive margins) and/or with tumor grades 2 and 3. The majority of patients (20) had localized disease and 19 had upfront surgical resection. Of the four patients who received conventional chemotherapy, none had a response. Three of 20 patients with localized disease later developed metastasis; all patients were alive at median follow-up of 61.7 months from diagnosis and the 5-year EFS rate was 94.7% and the OS rate was 100%. Of the three patients who developed metastasis, two had IRS I and small tumors <5 cm at the time of diagnosis and were treated with resection alone, developing 214metastatic relapse at 10 years and 4 months, respectively. The third patient was IRS III and was initially treated with two cycles of doxorubicin and ifosfamide followed by surgery and radiation; the patient developed metastatic relapse after 85.8 months.27 Overall, primary ASPS lesions should undergo complete resection when possible without significant morbidity, even in the setting of low-burden metastatic disease, and remains the best strategy for cure. Unfortunately, metastasis is common even in tumors <5 cm and recurrence can occur after a much longer disease-free interval than in other STSs, even a decade beyond the primary tumor diagnosis.28,29 Therefore, these patients should be followed closely with serial imaging of the initial primary site with MRI or ultrasound and long-term chest imaging with CT scans while weighing the risks of radiation exposure from imaging versus early detection of metastatic disease. At the time of development of metastatic disease, like in other STSs, selected patients may benefit from metastasectomy, although the overall effect that surgical resection has on survival in these patients is unclear.30 Management of Metastatic Disease Anti-VEGF Receptor TKIs As discussed in the section “Molecular and Biologic Drivers,” upregulation of angiogenesis-related transcripts has been identified in ASPS, prompting clinical investigation of antiangiogenic TKIs. Although none of these drugs are approved by the U.S. Food and Drug Administration (FDA) specifically for this indication, overall, anti-VEGF receptor TKIs have proved the most effective therapies for ASPS to date. Most of the data support the activity of cediranib, based on two completed clinical trials in the United States and Europe. The international CASPS trial compared cediranib versus placebo in patients with metastatic ASPS in a Phase 2, 2:1 double-blind randomized trial. Patients were unblinded at 24 weeks or sooner if the disease had progressed, and those in the placebo arm were allowed to crossover to cediranib if they had progressed. The primary endpoint was a percentage change in the sum of target marker lesions, with progression-free survival (PFS) and RECIST (Response Evaluation Criteria in Solid Tumors) response rate being the secondary endpoints. Forty-four evaluable patients were recruited between 2011 and 2016 from 12 centers in the United Kingdom, Australia, and Spain. Median change in target lesions was −8% in cediranib versus 13% in placebo (p = .0013). Best response at 24 weeks was RECIST partial response seen in six of 28 patients on cediranib versus zero of 16 receiving placebo. Median PFS on cediranib was 10.8 months versus 3.7 months in the placebo arm. CAPS is the largest randomized trial to date and confirms the activity by reduction in tumor burden and improvement in PFS.31 Kummar et al. also observed substantial single-agent activity of cediranib in treatment of metastatic ASPS in a Phase 2 trial of cediranib 30 mg daily. They enrolled 46 patients and 43 were evaluable at the time of analysis. The primary endpoint of objective response rate (ORR) was 35%, with 15 of 43 patients achieving a partial response. Twenty-six patients (60%) had stable disease at 24 weeks. The disease control rate (partial response or stable disease) was 84% at 24 weeks. These results led to an ongoing open-label, multicenter, randomized Phase 2 trial for patients with metastatic ASPS comparing cediranib with another VEGF receptor inhibitor, sunitinib (ClinicalTrials.gov Identifier: NCT01391962).32 Anlotinib is a similar pan-VEGF receptor TKI with remarkable activity in ASPS. In a report of the Phase 2B trial conducted in China, out of 56 patients with ASPS, the median PFS was 18.23 months with anlotinib versus 3 months with placebo.33 An ongoing Phase III trial in the United States includes an ASPS arm with open-label dosing of anlotinib, with intent for registration (ClinicalTrials.gov Identifier: NCT03016819). Activities of multiple other anti-VEGF receptor TKIs have been reported in the literature, including a small Phase 2 trial with pazopanib, which is FDA approved for other types of STS. Out of six patients, one achieved partial response and five showed stable disease with a median follow-up of 33 months. PFS was 5.5 months and OS was not reached.34 Sunitinib has also been used regularly in the off-label setting, with one study of 15 ASPS patients reporting six patients with partial response, eight with stable disease, and one with progressive disease. The median PFS was 19 months, median OS was 56 months, and 5-year OS was 49%.35 Several other therapies targeting VEGF have been reported in case studies and small clinical trials, including dasatinib, bevacizumab, and apatinib. A Phase 2 trial of dasatinib was conducted in patients with ASPS, chondrosarcoma, chondroma, epithelioid sarcoma, or solitary fibrous tumor. Twelve of the 109 patients had ASPS. The 6-month PFS, 2-year OS, and 5-year OS rates for patients with ASPS were 62%, 50%, and 30%, respectively.36 A case report of single-agent bevacizumab, a monoclonal antibody against VEGF, showed marked improvement in symptoms and prolonged PR of lung metastasis of 16 months in an elderly patient who had previously failed sunitinib.37 A case report showed a single patient with metastatic ASPS at diagnosis to the lungs which was treated with surgical resection of the primary mass and chemotherapy with gemcitabine–docetaxel 215with progression after two cycles. The patient was subsequently placed on apatinib 500 mg/day, a VEGFR-2 inhibitor, and achieved partial response at 1 month of therapy and remained progression free till the date of publication.38 In general, these drugs are reasonably well tolerated, with common toxicities including neutropenia, thrombocytopenia, hypothyroidism, hypertension, and hand–foot syndrome.35 These numerous examples suggest that blockade of VEGF receptors exhibits promising therapeutic advancement in the treatment of metastatic ASPS. Targeting c-MET With c-MET identified as a direct target of the fusion protein, the selective MET inhibitor tivantinib was also evaluated in a clinical trial including ASPS patients. Tivantinib appears to work through a different mechanism of action to block angiogenesis by impairing MET activity and likely having downstream effects on VEGF receptor activity. A multicenter, single-arm Phase 2 clinical trial was conducted on patients with advanced MITF-associated tumors. This is a rare group of tumors, which includes ASPS, clear cell sarcoma, and translocation-associated renal cell carcinoma, in which dysregulated expression of oncogenic MITF family transcription factors is found. The MET receptor tyrosine kinase gene is transcriptionally activated by MITF proteins, including TFE3. The study enrolled 47 patients with a median age of 25 years, of whom 27 had ASPS. They received tivantinib 120 mg orally twice daily and then 360 mg twice daily as per the protocol. The primary endpoint was overall response rate and the secondary endpoints included safety and PFS. There were no responses observed; however, stable disease was seen in 28 patients (60%), and median PFS was 5.5 months in the ASPS arm.39 Of notable interest, two patients with ASPS were long-term responders to tivantinib. The first was a 15-year-old girl diagnosed with ASPS on the left thigh with widely metastatic disease in the lungs at the time of diagnosis in 2007. She was previously treated with resection of the primary mass, radiation to the surgical bed, and resection of a tumor nodule in her lung. She was started on sorafenib, but was discontinued after 4 months because of progression of disease in the lungs. She was enrolled on the clinical trial, at which time she had several pulmonary nodules, which have been stable since last reported in a case series in January 2013. The second excellent responder was a 12-year-old boy with ASPS on the left thigh again with numerous lung lesions at the time of diagnosis in 2007. He was treated with preoperative radiation followed by resection of the primary mass. He was started on imatinib 600 mg daily for 4 months, but had progression of disease in the lungs. He was enrolled on the study and two target lesions in lungs have been closely followed. He was maintained on tivantinib for 199 weeks with stable disease as of January 2013. The prolonged stability of disease observed in these patients suggests this agent may show benefit in combination with another TKI or immunotherapy in the future.40 Overall, while the use of VEGF-receptor and MET TKIs has proved ability to stabilize ASPS, with some patients remaining on these agents for years prior to progression, complete responses are rare and these therapies are not curative. Immunotherapy for ASPS In the past decade, we have seen a revolution in cancer therapy with the development of modern immunotherapy, including ICIs. Monoclonal antibodies to the immune checkpoints CTLA-4 and the programmed death 1 (PD-1)/PD-L1 axis block suppression of T-cell activation and cytotoxicity, and have shown remarkable activity in numerous chemotherapy-refractory solid and hematologic malignancies. In sarcomas, the first trials of checkpoint inhibitors have shown meaningful and durable responses in a small subset of patients, with response rates of just under 20% with anti-PD-1 antibody pembrolizumab monotherapy and combination CTLA-4/PD-1 blockade with ipilimumab and nivolumab.41,42 Unlike other cancers such as non-small cell lung cancer and urothelial cancer, in most cases, PD-1/PD-L1 expression has not been a reliable biomarker for the response to ICI in sarcomas, and additional biomarkers for patients likely to respond are still under investigation. However, early evidence of remarkable efficacy has been observed in patients with ASPS with checkpoint inhibitor therapy. In a review of 50 patients enrolled in immunotherapy trials at MDACC with 14 different subtypes of sarcoma, four patients with metastatic ASPS who had received multiple prior lines of therapy were included. Two of the ASPS patients who received anti-PD-L1 therapy had partial responses nearing complete responses lasting 8 to 12 months and the other two achieved stable disease.43 Recently presented preliminary data from a study of the PD-L1 inhibitor atezolizumab for ASPS revealed eight out of 19 patients achieving partial response (42%), nine having stable disease, and one having progressive disease. The median time to first response was five cycles, and the median time on treatment was 11 cycles.44 In an ongoing study of tremelimumab/durvalumab, three of six patients with ASPS45 had 216achieved partial response at the time of presentation. One partial response was reported in the Alliance study of ipilimumab/nivolumab in an ASPS patient receiving nivolumab monotherapy.41 Tumor angiogenesis has been shown to be a major contributor to the immunosuppressive tumor microenvironment, with cytokines such as HIF-1α and VEGF promoting accumulation of suppressive phenotypes of macrophages, dendritic cells, myeloid-derived suppressor cells, and T cells.46 In melanoma and renal cell carcinoma, combinations of VEGF receptor inhibitors with immune checkpoint blockade have led to improved immune cell infiltration and tumor responses relative to either drug alone.47,48 In an early trial of granulocyte–macrophage colony stimulating factor (GM-CSF) secreting vaccines with autologous tumor lysates in patients with ASPS and clear cell sarcoma, investigators noted postvaccine induction of serum antibodies to downstream VEGF-mediated factors including tissue-type plasminogen activator and angiopoietins-1/2. PD-1 and PD-L1 were also expressed on cytotoxic T cells and tumor cells in posttreatment tumor biopsies.49 While no responses were seen, this was further evidence of a link between tumor angiogenesis and antitumor immunity in ASPS. Given the importance of angiogenesis in the biology of ASPS, and the activity of anti-VEGF receptor TKIs in many types of STS, we conducted a Phase 2 trial of the anti-VEGF receptor TKI axitinib plus the anti-PD-1 checkpoint inhibitor pembrolizumab in patients with progressing, advanced, or metastatic ASPS and other STSs (ClinicalTrials.gov Identifier: NCT02301039). Out of 11 evaluable ASPS patients, six patients achieved partial response (54.5%, 95% confidence interval [CI]: 24.6–81.9) and two additional patients achieved stable disease for a clinical benefit rate of 72.7% (95% CI: 32.3–92.7). The median duration of response for ASPS patients was 29 weeks, with one patient maintaining partial response for 85 weeks before developing progressive disease and one patient who continues with a near-complete response for 94 weeks.50 All ASPS tumor biopsies were positive for PD-L1 expression on tumor cells with robust immune cell infiltrates, suggesting that ASPS likely displays an inflamed phenotype, which is correlated with the response to ICI in other types of cancer. However, further evaluation of escape mechanisms is critical, given that most patients still ultimately develop resistance to immunotherapy. SUMMARY ASPS is an exceedingly rare and unique subset of STS. Despite the initial indolent nature, the early tendency of ASPS to metastasize has historically made this an incurable and ultimately fatal disease in all patients, many of whom are young. With anti-VEGF receptor TKIs, outcomes have clearly improved over the past several years; however, despite prolonging the disease course, ASPS remains incurable in nearly all cases. The early activity of ICIs against PD-1/PD-L1 represents significant hope for these patients, and with better understanding of the biology and development of counteractive combinations to resistance mechanisms, cure may be achievable one day.

Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


