Radiolabeled Monoclonal Antibodies
Targeting radiation to leukemic cells with radiolabeled monoclonal antibodies that bind to cell surface molecules could increase the radiation dose at sites of tumor and spare normal organs such as the liver and lung. An iodine-131–labeled anti-CD45 antibody has been used for this purpose in conjunction with conventional MAC in younger patients, or with RIC in patients over the age of 55. These studies have demonstrated that it is feasible to deliver >25 Gy to the BM while limiting liver and lung exposure, and achieve long-term survival in patients with high-risk AML.26,27 A similar approach using an yttrium-90–labeled anti-CD20 monoclonal antibody combined with RIC for patients with refractory CD20+ B-cell malignancies has shown promise in early phase studies.28,29 The use of pretargeting and new radioisotopes with more favorable tissue penetration might improve therapeutic efficacy and reduce toxicity.30
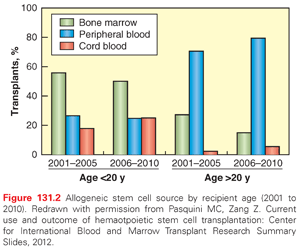
Donor/recipient genetic nonidentity at loci within the major histocompatibility complex (MHC) on chromosome 6p that encode specific HLA molecules is the most important risk factor for severe GVHD.4 Thus, hematopoietic cell grafts from donors who are identical with the recipient at the critical HLA loci are the preferred source of hematopoietic stem cells for allogeneic HCT. Although BM continues to be the most common graft source obtained from donors <20 years old in the United States, hematopoietic cells collected from the peripheral blood (PB) by apheresis after stimulation with filgrastim (granulocyte colony-stimulating factor) to mobilize CD34+ stem cells account for a majority of grafts harvested from donors >20 years of age (Fig. 131.2). For the large number of patients for whom a suitably HLA-matched donor cannot be identified, grafts derived from umbilical cord blood (UCB) or harvested from BM or PB of HLA-mismatched or HLA-haploidentical donors can provide a source of hematopoietic stem cells for allogeneic HCT.
Human Leukocyte Antigen–Matched Donors
The preferred donor for any patient who requires allogeneic HCT continues to be a full sibling who is genetically identical with the recipient for specific exons within the HLA-A, -B, -C, and -DRB1 genes. BM or PB grafts from HLA-identical siblings have been associated with the lowest risks for severe acute and chronic GHVD, and with superior posttransplant hematologic and immunologic reconstitution. Filgrastim-stimulated PB grafts in general contain a several-fold higher number of CD34+ hematopoietic progenitor cells than BM, but also contain a ≥10-fold higher number of T-lymphocytes, which are the central mediators of GVHD. Several large, randomized trials of transplantation with BM or PB grafts from HLA-identical sibling donors have shown that PB provides superior hematopoietic engraftment, but at the cost of a higher risk for both acute and chronic GVHD.31–35 Some of these studies have shown a decreased rate of relapse and superior survival in recipients of PB as compared with BM grafts, primarily in the subset of patients with high-risk malignancies.31,34 Donors of BM and PB generally report similar levels of discomfort associated with the stem cell donation, but PB donors report faster resolution of symptoms.36 Limited data also suggest that the economic cost of BM donation exceeds that of PB donation.37 Despite the higher risk of GVHD associated with PB grafts, the superior engraftment and lower relapse risk and the strong preference of donors for PB as opposed to BM donation have made PB grafts the preferred and, in fact, the default source of stem cells for patients with HLA-identical sibling donors. BM remains the preferred stem cell source for patients with nonmalignant disorders such as aplastic anemia, in whom there is no need for a GVL effect.
Unrelated Donors
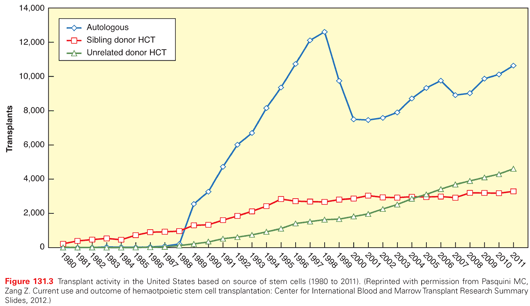
Most patients who are candidates for allogeneic HCT do not have an HLA-identical sibling donor available; for many such patients, suitably HLA-matched volunteer unrelated donors can be identified in worldwide registries that now include more than 22 million individuals (www.bmdw.org; accessed January 5, 2014). The annual number of transplants performed in the United States with grafts from volunteer unrelated donors has exceeded the number performed with grafts from HLA-identical sibling donors since 2005 (Fig. 131.3). Improved understanding of the role of the MHC in allogeneic HCT has driven steady evolution of the laboratory procedures used to establish that patients and prospective donors are suitably “matched.” The National Marrow Donor Program currently recommends matching recipients and prospective donors at the DNA sequence level in exons 2 and 3 of the HLA-A, -B, and -C genes, and exon 2 of the HLA-DRB1 gene.38 In practice, recipients and prospective donors are also “typed” and matched for exon 2 of the HLA-DQB1 gene, primarily to ensure optimal donor/recipient matching at HLA-DRB1. Retrospective analysis of 3,857 unrelated donor transplants between 1998 and 2003 revealed that donor/recipient matching at HLA-A, -B, -C, and -DRB1 (termed 8/8 matches) was associated with superior survival, and that a single mismatch at any one of these loci (7/8 matches) was associated with higher mortality.39 Accumulating evidence suggests that donor/recipient matching at an additional HLA locus, HLA-DPB1, also influences the outcome of unrelated donor HCT.40 Although donor/recipient matching at HLA-DQB1 is typically achieved with current selection algorithms for unrelated donors, no data demonstrate that HLA-DQB1 matching is associated with a survival benefit.
The strong preference for utilization of PB grafts in transplants between HLA-identical sibling pairs has profoundly influenced clinical practice in the unrelated donor setting, and PB grafts are currently used in over three-fourths of unrelated donor transplants. Unrelated PB grafts, like PB grafts from sibling donors, are associated with superior hematopoietic engraftment, and unrelated donors who donate PB grafts experience faster resolution of donation-related discomfort than do BM donors.36 A recent multicenter international randomized trial demonstrated no significant difference in 2-year survival between the recipients of unrelated BM and PB grafts; however, trends toward more graft failure in the BM recipients and more chronic GVHD in the PB recipients were observed.41
Although the outcome of allogeneic HCT from unrelated donors was for many years inferior to that of transplants between HLA-identical siblings, advances in donor selection and in transplant practice—particularly conditioning regimens and supportive care—are steadily eliminating this critical differential. Retrospective analyses of HCT between HLA-matched related and 8/8 locus-matched unrelated donor/recipient pairs have shown comparable overall survival between the two groups, albeit with significantly more acute and chronic GVHD after unrelated donor transplant.42,43 The equivalent outcome of transplantation from HLA-matched related versus unrelated donors is all the more impressive given that patients who are referred for unrelated donor HCT have, on average, more advanced and higher risk disease.
Umbilical Cord Blood
The development and applications of UCB transplantation have been recently reviewed,44 and this stem cell source provides an increasingly used alternative for patients that do not have a readily available, suitably HLA-matched family or unrelated donor. There are now >131 voluntary banks in 35 countries that house >500,000 UCB units available for searches to identify a suitable UCB unit for patients who need an allogeneic HCT.45 The advantages of UCB include rapid availability, little or no risk to the mother or newborn infant, and a low risk of transmitting viruses. There is also a decreased requirement for stringent HLA matching, and UCB transplants that match at only four of six HLA class I and II molecules lead to acceptable rates of GVHD. The disadvantages of UCB include delayed engraftment, especially in patients that receive a nucleated cell dose of <2.5 × 107/kg or a CD34+ cell dose of <1.2 × 105/kg, and delayed immune reconstitution, which contributes to higher transplant-related mortality.46 The initial applications of UCB transplant focused on pediatric patients, who because of their size could receive a higher dose of CD34+ cells/kg of body weight. Data from registry studies demonstrate that the overall survival of patients receiving single UCB transplant compares favorably to HLA-matched unrelated donor BM or PB transplants.47,48
To overcome the low CD34+ cell dose in a single UCB unit, two UCB units that were matched for at least four of six HLA alleles with the recipient have been administered. Such double UCB transplant improved the time to engraftment in adult recipients,49,50 even though chimerism analysis revealed that one UCB unit often rapidly dominated engraftment.51 A comparison of outcomes of double UCB transplant to those using HLA-matched related or unrelated donors has suggested a lower risk of relapse but higher transplant-related mortality with UCB grafts, and a similar overall survival.50 UCB transplant has also been successfully employed with RIC, providing an option for older patients that could not tolerate MAC and do not have a suitably matched related or unrelated donor.52,53
Innovations that may improve the outcome of UCB transplant are the subject of intense research. A promising approach to shorten the duration of neutropenia is to infuse an uncultured UCB unit along with in vitro expanded progenitor cells derived either by culturing UCB CD34+ cells with the Notch ligand delta 1, or with mesenchymal progenitor cells.54,55 These approaches currently require specialized cell culture facilities, but in the future cryopreserved banks of third-party “off-the-shelf” progenitors could be derived and used for this purpose. Strategies to improving homing of the infused stem cells to hematopoietic niches are also being explored to improve engraftment.56 Immune reconstitution might be improved by the transfer of virus-specific cells or lymphoid progenitors derived de novo from UCB T cells,57,58 and GVHD reduced by infusing additional CD4+ regulatory T cells at the time of transplant.59,60
Mismatched and Haploidentical Donors
HLA-mismatched or related haploidentical donors also provide a transplant option for the 40% to 50% of patients who do not have a readily available HLA-matched family or unrelated stem cell donor. Transplantation from donors mismatched at two or three HLA antigens was complicated in early studies by an unacceptably high incidence of graft rejection and severe GVHD resulting from potent bidirectional alloreactive T-cell responses.61–63 The use of intense MAC, depletion of host and donor T cells, and infusion of high numbers of donor CD34+ T cells was necessary to overcome both the graft rejection and GVHD barriers, but resulted in a prolonged immunodeficiency and high risk of opportunistic infections, including invasive mold infection.64 Relapse rates in AML were low, particularly in recipients in which donor-versus-recipient natural killer (NK) cell alloreactivity was present, due to the absence of MHC class I killer cell immunoglobulin-like receptor (KIR) inhibitory ligands in the recipient compared with the donor.65
An alternative to T-cell depletion, and one that is used with RIC to prevent GVHD and graft rejection after haploidentical HCT, is the administration of high-dose Cy posttransplant. This approach has the advantage of selectively eliminating alloreactive T cells that become activated in the recipient, resulting in preservation of immune reconstitution to pathogens, thus avoiding the severe opportunistic infections that are observed after T-cell–depleted haploidentical HCT.66–68 A recent Blood and Marrow Transplant Clinical Trials Network study of two parallel phase 2 trials of RIC and haploidentical donor BM transplant with high-dose Cy posttransplant or double UCB transplant demonstrated similar overall and progression-free survival at 1 year (62% and 48% haploidentical HCT; 54% and 46% double UCB transplant), and similar incidence of grade II to IV acute GVHD (32% haploidentical HCT; 40% double UCB transplant).69 The 1-year cumulative incidences of nonrelapse mortality and relapse, respectively, were 7% and 45% after haploidentical HCT, and 24% and 31% after double UCB transplant. These results illustrate the vast improvements in outcomes after haploidentical HCT and suggest that randomized comparisons of UCB, matched unrelated donor, and haploidentical related donor HCT will be needed to determine if any of these procedures is superior.
IMMUNOBIOLOGY OF ALLOGENEIC HEMATOPOIETIC CELL TRANSPLANTATION
The outcome of allogeneic HCT is in large part determined by recognition of alloantigens expressed on recipient cells by donor lymphocytes. Genetic disparity between donor and recipient is a key to the therapeutic efficacy of allogeneic HCT, but also the root cause of GVHD, which is its primary limitation. GVL and GVHD are mediated primarily by lymphocytes contained in or derived from the donor hematopoietic cell graft. GVL activity is closely linked to the development of GVHD, but clinically significant GVHD is not required for GVL, implying that important mechanistic distinctions exist between the two phenomena.
Graft-versus-Host Disease
GVHD comprises two distinct but variably overlapping clinical syndromes—acute and chronic GVHD—that differ in their time course, clinical manifestations, and response to therapy. The pathophysiology of acute GVHD is complex and involves tissue injury caused by cytotoxic conditioning and subsequent release of inflammatory cytokines, activation of donor lymphocytes and recipient antigen-presenting cells, recognition by donor lymphocytes of alloantigens on recipient antigen-presenting cells, proliferation and differentiation of activated donor lymphocytes, trafficking of donor lymphocytes to secondary lymphoid organs, and infiltration of target organs by activated donor effector cells with subsequent target organ injury. The pathophysiology of chronic GVHD is poorly understood, but it cannot readily be attributed to the same mechanisms that mediate acute GHVD. Many of the features of chronic GVHD are phenocopies of manifestations of autoimmune diseases such as scleroderma and Sjögren syndrome, and many transplant physicians view chronic GVHD as a condition in which loss of immune tolerance is a central feature. That distinct mechanisms mediate the pathogenesis of acute and chronic GVHD is underscored by the fact that pharmacologic strategies for prevention of acute GVHD have limited efficacy for preventing the chronic form of the disease (and vice versa).
The central role of donor T-lymphocytes in mediating acute GVHD is suggested by the complementary observations that depletion of T-lymphocytes from the donor graft can prevent clinically significant GVHD,7 whereas supplementation of conventional grafts with additional donor T cells markedly exacerbates it.70 The occurrence of GHVD in recipients of grafts from donors that are genotypically identical throughout the entire MHC implicates donor/recipient disparity at polymorphic genetic loci outside the MHC in causing GVHD. Such loci encode short peptides (8 to 12 residues in length) termed “minor histocompatibility antigens” that are presented on the cell surface by HLA molecules encoded in the MHC. Minor histocompatibility (H) antigens are created by the extensive sequence and structural variation present in the human genome, and all donor/recipient transplant pairs, despite selection for identity at the MHC, will be mismatched for many minor H antigens.71
The etiology of chronic GVHD is distinguished from acute GVHD by a significant role for B-lymphocytes and dysregulated B-cell homeostasis. Abnormal levels of naïve B cells are observed in chronic GVHD72 and are associated with elevated levels of B-cell activating factor,73 which can promote the survival and activation of autoreactive B cells.74 Autoantibodies specific for nuclear, mitochondrial, parietal cell, smooth muscle, and parotid gland antigens have been detected in patients with severe symptoms of chronic GVHD.75,76 Autoantibodies against platelet-derived growth factor have been detected in patients with chronic GVHD with prominent sclerosis and fasciitis.77
Graft versus Leukemia
The elimination of malignant cells in recipients of allogeneic HCT is intimately associated with GVHD, but GVL activity is observed in the absence of GVHD, suggesting important distinctions between the two. It is clear that the GVL effect is not a single homogeneous phenomenon, and that the mechanisms that mediate GVL are determined by the genetic disparity between the donor and recipient, the source, composition, and processing of the hematopoietic cell graft from the donor and the recipient tumor type. Donor T-lymphocytes and NK cells are the primary GVL effector cells. Donor T cells specific for minor H antigens expressed on recipient malignant cells are particularly important in patients who receive T-replete grafts from HLA-identical donors. Minor H antigen-specific T cells exhibit potent antileukemic activity both in vitro78 and in vivo,79,80 and specifically eliminate the leukemic stem cells that establish human leukemia in immune-deficient mice.79,80 Minor antigen-specific T cells with reactivity against recipient leukemic cells have frequently been detected in the blood of HCT recipients demonstrating regression of leukemia following donor lymphocyte infusion.81 It is thought that donor T cells recognizing minor H antigens that are selectively or exclusively expressed on recipient hematopoietic cells—including leukemic cells—may mediate GVL activity without contributing to GVHD.
Donor T cells recognizing nonpolymorphic antigens encoded by genes that are over- or aberrantly expressed in myeloid and lymphoid leukemic cells can contribute to GVL activity. CD8+ T cells recognizing peptides encoded by genes overexpressed in myeloid leukemic cells such as myeloblastin and neutrophil elastase,82 by the WT1 oncogene that is expressed in a wide range of leukemic cells,83 or from the cancer-testis gene NY-ESO-1 that is often expressed in AML and multiple myeloma84 have been detected in the blood of HCT recipients. Donor T-cell responses against tumor-specific antigens can also develop in allogeneic HCT recipients and contribute to GVL activity.85 It is likely that donor T-cell responses against this class of recipient antigens are enabled and facilitated by the immunogenic milieu created by responses against recipient minor H antigens, as they have not in general been detected in recipients of syngeneic transplants.
Alloreactive donor NK cells can be major mediators of GVL in recipients of grafts from multiple HLA-mismatched and haploidentical donors, in whom extensive in vitro or in vivo T-cell depletion of the graft is required to prevent lethal GVHD.65 The recognition of recipient target cells by cytotoxic donor NK cells in allogeneic HCT is in large part determined by donor and recipient genotypes at the KIR locus on chromosome 19q, as well as at the HLA-A, -B, and -C loci on chromosome 6p. KIR genes encode transmembrane receptors that are expressed selectively or exclusively in NK cells and subsets of αβ or γδ T cells; bind to specific subsets of HLA-A, -B, and -C alleles in a manner that is influenced by HLA-bound peptide; and participate in specific recognition of target cells by NK and T cells. In the HLA-haploidentical or multiple HLA-mismatched setting, cytotoxic donor NK cells can eliminate recipient leukemic cells that express molecules for which they are specific. The interaction of donor NK and recipient leukemic cells is in part determined by the gene content and haplotype of the donor at the KIR locus, but the nature of the interactions have yet to be completely defined. Several models of NK alloreactivity have been proposed, but it is not yet clear which of these models has the greatest accuracy.
Accumulating evidence, much of it indirect, also suggests that donor NK cells may make a significant contribution to the elimination of myeloid leukemias after allogeneic HCT between fully HLA-matched donor/recipient pairs (reviewed in Warren and Deeg86). The most compelling data have been provided by several large retrospective studies of MHC and KIR genotype and posttransplant relapse, and have shown significant associations between the presence in the donor of a specific type of haplotype at the KIR locus, or one or more components thereof, and decreased rate of relapse in recipients undergoing unrelated HCT for AML and other myeloid neoplasms.87,88
COMPLICATIONS OF ALLOGENEIC HEMATOPOIETIC CELL TRANSPLANTATION AND THEIR MANAGEMENT
Acute and Chronic Graft-versus-Host Disease
The syndrome of acute GVHD is characterized by fever, skin rash, gastrointestinal symptoms referable to involvement of the upper or lower tract, and cholestatic liver dysfunction, often accompanied by elevations in hepatic transaminases. It typically occurs within the first 60 days after HCT, but can occur later, particularly in recipients of RIC. The most important risk factors for the development of acute GVHD are donor/recipient HLA mismatch, donor and recipient age, a filgrastim-stimulated PB graft rather than a BM graft, myeloablative conditioning with a TBI-containing regimen, and donor/recipient cytomegalovirus (CMV) seropositivity. The skin rash of GVHD is most commonly an erythematous, variably pruritic, macular or maculopapular eruption that can involve any part of the body surface. Blistering and desquamation is seen in severe cases (Fig. 131.4A,B). The symptoms of gut GVHD range widely in severity and can include anorexia, nausea, vomiting, abdominal cramping, and diarrhea, the latter of which can be voluminous (measurable in liters per day) and bloody. Endoscopic evaluation of gut GVHD reveals mucosal edema and erythema in mild cases, frankly hemorrhagic epithelium in more severe cases, and pseudomembranes and extensive denudation in life-threatening disease. The pathologic hallmarks of mild to moderate skin and gut GVHD are apoptotic epithelial cells and an infiltrate of inflammatory cells, composed chiefly of lymphocytes and neutrophils, the latter primarily in the gut. Hepatic GVHD is most specifically characterized by damage to bile ducts with bile stasis, often accompanied by inflammatory cell infiltration. Recent studies have identified plasma biomarkers for skin89 and lower gastrointestinal and liver90 GVHD, and a plasma biomarker associated with therapy-resistant, life-threatening GVHD.91
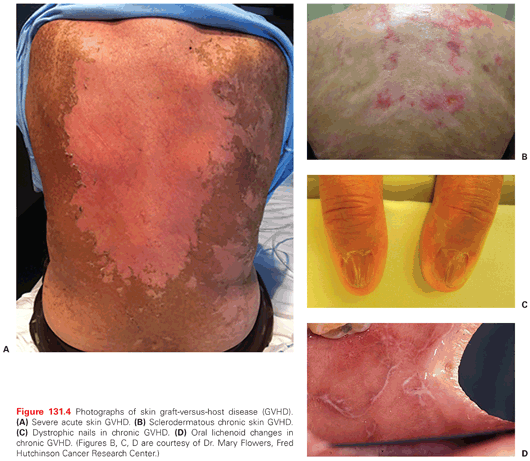
Chronic GHVD has protean manifestations that can appear in a broader range of target organs than acute GHVD, including the eyes, mouth, skin and nails, gut, liver, lungs, joints, subcutaneous tissue, and genital tract, particularly in women. Symptoms and signs of chronic GVHD include skin tightness and thickening, hypo- or hyperpigmentation (Fig. 131.4C,D), skin erosion or ulcers, pruritus, brittle and/or ridged nails, dry eyes and dry mouth, hair thinning or loss, edema, fatigue, weight loss, anorexia, dysphagia or odynophagia, joint stiffness or contractures, chronic cough, dyspnea, vaginal dryness, and dyspareunia, among others. Laboratory signs include eosinophilia, thrombocytopenia, and hypo- or, less commonly, hypergammaglobulinemia. Biopsy of involved sites reveals a spectrum of characteristic changes; fibrosis is a common feature. Evaluation of advanced lung involvement often reveals bronchiolitis obliterans. Immune deficiency is a prominent feature of chronic GVHD, and manifests as increased susceptibility to bacterial, fungal, and viral pathogens.
Pharmacologic Prophylaxis/Therapy
The incidence and severity of acute GVHD can be reduced with pharmacologic prophylaxis. Calcineurin inhibitors such as cyclosporine and tacrolimus combined with methotrexate have been the mainstay of prophylaxis in MAC transplants for several decades. Salts of mycophenolic acid have successfully replaced methotrexate in prophylaxis regimens in some RIC regimens. Two- and three-drug regimens combining tacrolimus, or tacrolimus and mycophenolic acid, with sirolimus have been extensively evaluated in clinical trials in both the MAC and RIC settings, with promising results.92 Novel approaches to preventing GVHD based on donor and/or recipient statin treatment93 or blockade of lymphocyte chemotaxis94 also appear to have significant potential. None of the pharmacologic strategies for preventing acute GVHD have yet been shown to be effective at preventing chronic GVHD.
Synthetic corticosteroids such at prednisone or methylprednisolone at 0.5 to 2 mg/kg per day are the cornerstone of therapy for acute and chronic GVHD that requires systemic therapy. Topical tacrolimus or halogenated steroids, and the combination of an oral photosensitizing psoralen with ultraviolet A irradiation can provide useful, steroid-sparing adjuvant therapy for skin GVHD. Similarly, orally administered, poorly absorbed synthetic steroids such as beclomethasone95 and budesonide are effective agents for mild to moderate gut GVHD, and facilitate a reduction in the intensity of systemic therapy. The treatment of moderate to severe (grade 3 to 4) acute GVHD that is refractory to treatment with 2 mg/kg per day prednisone equivalent continues to be an enormous clinical challenge. Anti–T-cell antibody preparations such as antithymocyte globulin of equine or lapine origin, and monoclonal anti–T-cell antibodies such as daclizumab are commonly used in this setting, but there is no agent that has been shown to be effective in a randomized, double-blind, placebo-controlled fashion.96
Alternative Approaches to Prevention of Graft-versus-Host Disease
Several approaches to GVHD prevention have been investigated as adjuncts or alternatives to the traditional combination of a calcineurin inhibitor and methotrexate. Depletion of donor T cells in vitro, using one of several different methodologies, or in vivo, typically using polyclonal or monoclonal anti–T-cell antibodies, is effective prophylaxis for both acute and chronic GVHD, but at the cost of more frequent graft failure, slower immune reconstitution, increased opportunistic infections, and higher rates of relapse in some but not all malignancies.
T-cell depletion has been examined in randomized studies of allogeneic HCT between 8/8 or 7/8 HLA locus-matched donor/recipient pairs. A multicenter trial in which 405 patients with hematologic malignancies were randomized to receive in vitro T-depleted BM grafts from unrelated donors and posttransplant cyclosporine A, or T-replete marrow grafts with cyclosporine A and standard-dose methotrexate demonstrated a lower rate of grade 3 to 4 acute GVHD in the T-cell depletion arm (18% versus 37%) but no significant difference in 3-year disease-free survival.97 A subsequent randomized, multicenter trial of standard cyclosporine and methotrexate GVHD prophylaxis with or without in vivo antithymocyte globulin treatment in 202 patients receiving unrelated donor HCT similarly showed a lower cumulative incidence of grade 3 to 4 acute GVHD (11.7% versus 24.5%) and of clinical extensive chronic GVHD (12.2% versus 42.6%) in the antithymocyte globulin group, but no significant difference in overall survival between the two groups.98
A novel approach to the prevention of acute and chronic GVHD based on administration of high-dose intravenous Cy within 3 to 4 days of infusion of the hematopoietic cell graft (high-dose Cy posttransplant) has shown promise in the prevention of both acute and chronic GHVD. Long studied in preclinical models of solid organ and hematopoietic cell transplantation,68 this approach was first tested in the clinic for GVHD prophylaxis in haploidentical HCT.66 These studies demonstrated that high-dose Cy posttransplant, combined with a calcineurin inhibitor, was comparable if not superior to T-cell depletion for haploidentical transplantation. High-dose Cy posttransplant alone, without additional prophylactic immunosuppression, has subsequently been studied in the setting of HLA-matched HCT from related and unrelated donors, and in both single-center99 and multicenter studies, and has proved to be strikingly effective in the prevention of chronic GVHD, without compromising GVL activity.
Infectious Complications
Infections, both early after allogeneic HCT during the period of severe pancytopenia and later as a result of persistent immunodeficiency and/or immunosuppressive drug therapy, remain a significant cause of morbidity and mortality. Several strategies for reducing infectious complications are routinely employed, including handwashing policies for patients, families, and health-care workers; prophylactic antibiotics; screening for respiratory viruses; monitoring and preemptive therapy for CMV infection; and prompt empiric therapy for infections in at-risk patients. The types of pathogens to which the patient is most susceptible varies over time after HCT: The risk of bacterial infections is highest during the early neutropenic phase after HCT, and again later in patients with chronic GVHD. Viral infections occur most often in the first 3 months after HCT when cellular and humoral immunity are impaired by immunosuppressive drug therapy.
Bacterial Infections
Factors that predispose allogeneic HCT recipients to bacterial infections are disruption of oral and gastrointestinal mucosal barriers, neutropenia (absolute neutrophil count <500 cells/μL), indwelling central catheters, glucocorticoid therapy, and alterations in the microbiota from prior antibiotic therapy.100,101 The degree of mucosal damage and the duration of neutropenia vary depending on the intensity of the conditioning regimen, and RIC is associated with fewer early infections.
Up to 60% of HCT recipients develop a bloodstream infection with a gram-positive or gram-negative bacteria, and bacterial infections are the most common cause of infection-related mortality after allogeneic HCT.102–104 Antibiotic prophylaxis and prompt evaluation and empiric therapy of patients with neutropenic fever are critical to reduce the risk of life-threatening infections. Prophylaxis with a broad-spectrum antibiotic such as levofloxacin or ceftazidime is typically initiated at the onset of neutropenia and continued until neutrophil recovery.105,106 Levofloxacin has broad activity against gram-positive and gram-negative pathogens, an excellent safety profile, and can be administered orally in once daily dosing. Patients that develop a fever should be evaluated by physical exam for potential portals of entry, blood and urine cultures, and a chest X-ray or other imaging studies informed by the clinical presentation. Empiric antibiotic therapy should be initiated promptly and modified depending on the results of cultures. The choice of antibiotics for empiric therapy of febrile neutropenia depends on clinical presentation, results of prior surveillance cultures, local susceptibility patterns, toxicity of individual agents, and allergic history. Typically, a third- or fourth-generation cephalosporin, carbapenem alone or in combination with an aminoglycoside, to provide coverage for pseudomonas and other gram-negative pathogens, are considered in seriously ill patients with neutropenic fever, with adjustment of the antibiotic regimen as culture data becomes available. Nocardia, Legionella, and Mycobacterium infections are uncommon but should be considered in patients with pulmonary infiltrates. The potential for antibiotic-resistant bacteria, such as methicillin-resistant Staphylococcus and vancomycin-resistant Enterococcus, to cause serious infections in HCT recipients who have often been heavily exposed previously to antibiotics should be carefully evaluated.
Late bacterial infections can occur after recovery from neutropenia, particularly in patients with chronic GVHD who have low serum immunoglobulin levels. Such patients are at risk from serious sinopulmonary infections and systemic infections with encapsulated organisms, and should be treated with daily prophylaxis with penicillin, trimethoprim-sulfamethoxazole, or a suitable alternative.107
Clostridium difficile
There is a high incidence of Clostridium difficile infection in allogeneic HCT recipients because of frequent hospitalization, use of broad-spectrum antibiotics, and disruption of gastrointestinal mucosal barriers.108 The incidence of C. difficile in allogeneic HCT recipients in one major US transplant center was 12.5%, and infection occurred at a median of 33 days after transplant.109 This study also identified an association between C. difficile infection and acute GVHD, suggesting the possibility that alterations in the microbiome may contribute broadly to transplant outcomes. C. difficile infection after allogeneic HCT responds to standard treatment with oral metronidazole.109 Recurrent C. difficile infections in nontransplant patients that do not respond to antibiotic therapy have been treated successfully with fecal microbiota transfer,110 and this approach has been reported to be effective in an allogeneic HCT recipient with fulminant antibiotic refractory C. difficile colitis.111
Fungal and Pneumocystis jiroveci Infection
Allogeneic HCT recipients, particularly those that require intensive immunosuppression to treat GVHD or that receive a T cell–depleted HCT, are at risk for serious fungal infections. Candida and Aspergillus are the most common fungal pathogens; however, other organisms may be observed.112 The incidence of invasive and superficial Candida infections is markedly reduced by administration of prophylactic fluconazole,113–115 and this is now the standard of care in transplant centers. Fluconazole has limited activity against Candida krusei, Aspergillus spp., and Torulopsis glabrata, and patients receiving fluconazole prophylaxis remain at risk for infections with these pathogens. Azole agents such as voriconazole and itraconazole that are active against Aspergillus are more effective than fluconazole in preventing Aspergillus infection in HCT recipients, but have a higher incidence of side effects.116–118 Patients that require glucocorticoid therapy for acute GVHD are at high risk for invasive Aspergillus and Zygomycetes infection, and posaconazole has been shown to be more effective than fluconazole in this subset of patients.119
Pneuomcystis jiroveci can cause pneumonia late after allogeneic HCT, and patients should receive prophylaxis with trimethoprim/sulfamethoxazole, or, if not tolerated, an acceptable alternative such as dapsone, atovoquone, or aerosolized pentamidine, until they are off of all immunosuppressive drug therapy.120
Herpesvirus Infections
Reactivation of latent viruses of the herpes family occurs frequently in seropositive patients after allogeneic HCT, and in the absence of therapy can cause life-threatening disease. Historically, herpes simplex virus type 1 or 2 infection occurred in approximately 80% of seropositive patients, but is now nearly universally prevented by the administration of acyclovir beginning with conditioning. Varicella zoster virus can present late after HCT, most frequently in patients with GVHD, either as reactivation in dermatomal sites or less commonly as a disseminated infection with hepatitis, pneumonitis, and/or meningoenephalitis. Prolonged administration of prophylactic acyclovir for 1 year after HCT is effective in decreasing varicella zoster virus reactivation.121
Latent CMV infection is common in the general population and can reactivate and cause serious life-threatening invasive disease both early and late after allogeneic HCT. Prevention of CMV disease is the most effective management strategy. For patients who are CMV seronegative, it is standard practice to reduce the risk of the patient acquiring the virus by administering “CMV-safe” blood products as transfusion support, either obtained from CMV-seronegative donors or leukocyte reduced by filtration.122 CMV-seropositive recipients have frequent monitoring of blood for CMV reactivation using polymerase chain reaction to detect CMV DNA, and preemptive antiviral drug therapy to prevent disease is instituted in patients with reactivation to prevent progression of infection.123,124 Ganciclovir is most commonly used for preemptive therapy; however, foscarnet is a useful agent in patients with neutropenia.122 UCB transplant recipients are at particularly high risk for serious CMV infection due to the lack of transferred memory T-cell responses to CMV in the graft, the low total T-cell dose, and the delay in immune reconstitution.125 Moreover, the delayed recovery of neutrophil counts in these patients complicates preemptive therapy with ganciclovir. Thus, some centers have implemented an intensive strategy to prevent CMV disease using ganciclovir during conditioning for UCB transplant, high-dose acyclovir prophylaxis after transplant, and preemptive ganciclovir for any positive CMV DNA polymerase chain reaction.126 Ganciclovir and foscarnet cause myelosuppression and nephrotoxicity, respectively, and there is need for a safer drug for prophylaxis of CMV. CMX001 is an orally bioavailable acyclic nucleoside phosphonate that is converted intracellularly to cidofovir, and has potent antiviral activity against both herpesviruses and adenoviruses. A multicenter randomized study demonstrated that oral administration of CMX001 was effective as prophylaxis for CMV infection in CMV-seropositive allogeneic HCT recipients without myelosuppression or nephrotoxicity,126 suggesting that less toxic approaches to controlling CMV infection after allogeneic HCT may be forthcoming.
CMV surveillance and preemptive antiviral drug therapy has reduced the incidence of CMV disease in the first 3 months after allogeneic HCT. However, late CMV disease still occurs in up to 15% of CMV-seropositive recipients, most commonly in the first year after transplant, and has a poor outcome.127 Patients at high risk include recipients of T cell–depleted or UCB grafts, and those with low CD4+ T cells, absent reconstitution of CMV-specific T-cell immunity and GVHD. Extended monitoring for CMV reactivation and preemptive antiviral therapy is recommended in such patients to reduce late CMV disease.124
Epstein-Barr virus (EBV) reactivation in donor B cells and progression to an aggressive lymphoproliferative disorder (LPD) may occur in recipients of T cell–depleted allogeneic HCT or in patients that receive T cell–depleting antibodies to treat GVHD. Strategies for T-cell depletion that also remove donor B cells from the graft reduce the incidence of EBV-LPD.128 The pathogenesis of LPD is a deficiency of EBV-specific T cells that are necessary to control latent EBV infection. Thus, EBV-LPD can be treated effectively or prevented by infusing donor lymphocytes that contain EBV-specific T cells or are enriched for EBV-specific T cells by in vitro culture prior to infusion.129–132 An effective alternative to adoptive T-cell therapy for patients at risk for EBV-LPD is to monitor EBV-DNA in blood and preemptively administer rituximab, an anti-CD20 monoclonal antibody that depletes B cells in vivo.133,134
Other Viral Infections
Allogeneic HCT recipients have an increased susceptibility to developing lower respiratory tract disease from seasonal respiratory viruses, and mortality from influenza, respiratory syncytial virus, parainfluenza, and human metapneumovirus infections can be significant.135,136 Systemic or high-dose oral ribavirin therapy may be beneficial in patients with serious respiratory syncytial virus or parainfluenza virus infections, although controlled efficacy trials have not been performed.137,138 Active surveillance during seasonal outbreaks can identify patients with asymptomatic or symptomatic shedding, and isolation of such patients may reduce spread to health-care workers and other patients.
Adenovirus can cause disseminated infection in allogeneic HCT recipients who received T cell–depleted grafts or develop severe GVHD requiring prolonged immunosuppression. There are no antiviral drugs with proven efficacy for adenovirus; however, this infection may be susceptible to adoptive T-cell therapy with donor-derived adenovirus-specific T cells.139,140 BK virus is a prevalent latent polyomavirus that is increasingly recognized as a cause of urinary tract infection and hemorrhagic cystitis in allogeneic HCT recipients, particularly those with GVHD.141,142 The pathogenesis of symptomatic hemorrhagic cystitis associated with BK virus infection remains to be fully elucidated, and therapy is largely symptomatic.
Organ Complications
Organ complications are associated with an adverse outcome after allogeneic HCT; however, mortality has declined in recent years due to prevention strategies, improved supportive care, and the use of RIC.143
Early and Late Pulmonary Complications
Pulmonary complications occur in up to one-third of allogeneic HCT recipients and can be divided into infectious and noninfectious etiologies.144 Pretransplant pulmonary function testing is predictive of early respiratory failure and mortality, and may influence the choice of conditioning regimen in individual patients.145,146 The incidence of pulmonary infections has been declining due to effective preventive measures. Bacterial pneumonia in the neutropenic phase can be reduced by the use of prophylactic antibiotics. Similarly, the occurrence of CMV pneumonia has been reduced by preemptive antiviral drug therapy. Invasive pulmonary Aspergillus and Zygomycetes infections remain serious complications, particularly in patients with prolonged neutropenia and/or those who require intensive immunosuppression to treat GVHD, and prophylaxis with newer azoles is indicated in such patients.146
Noninfectious pulmonary complications that occur in the first 100 days after allogeneic HCT include periengraftment respiratory distress syndrome, diffuse alveolar hemorrhage (DAH), and idiopathic pneumonia syndrome. Periengraftment respiratory distress syndrome typically develops approximately 1 to 3 weeks after HCT coincident with neutrophil recovery and is characterized by fever, hypoxemia, noncardiogenic pulmonary edema, and erythroderma. This syndrome is thought to result from release of proinflammatory cytokines and influx of neutrophils into the lung, and responds to high-dose corticosteroids.147
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


