Alkylating agents and platinum antitumor compounds
Zahid H. Siddik, PhD
Overview
Alkylating agents and platinum-based compounds are highly potent antitumor drugs used in the treatment of a variety of cancers. These drugs target DNA (deoxyribonucleic acid) but require activation by spontaneous or metabolic transformation to induce formation of DNA monofunctional adducts and interstrand and intrastrand crosslinks. As a result of this damage, the DNA unwinds and/or bends, and such distortions are then recognized by specialized DNA damage recognition proteins to activate checkpoints, which arrest the cell cycle to allow cells time to repair the damage and survive. If the DNA damage is extensive and repair cannot be completed, then cells activate p53-dependent or independent apoptosis (programmed cell death) to affect antitumor drug response. As activated species from alkylating agents and platinum compounds are not tumor-selective, they will also interact with DNA and other endogenous macromolecules in normal cells to induce severe side effects; at times the toxicity can be irreversible and cumulative and, thereby, presents a dose-limiting barrier. Another limitation is that genetic changes in tumors that are either intrinsic or acquired can inhibit apoptosis and induce resistance to alkylating and platinating drugs. Resistance mechanisms may be observed in the form of reduced drug accumulation, increased drug inactivation, increased DNA repair, failure of DNA damage recognition system to recognize the damage, and aberrant apoptotic signal transduction pathways. Rational strategies to circumvent resistance mechanisms are, therefore, needed desperately to enhance patient care.
Introduction
Hallmarks of cancer include sustained proliferative signaling and replicative immortality as biological features that contribute to uncontrolled increase in tumor mass.1, 2 These characteristics are consistent with absence of feedback or inhibitory control on DNA (deoxyribonucleic acid) replication and the cell cycle that is commonly found in tumor cells. It is not surprising, therefore, that therapeutics targeting or covalently interacting with DNA to inhibit DNA replication and, thereby, cellular proliferation, have been the mainstay for clinically managing patients with cancer. Such therapeutics are broadly known as DNA alkylating agents and include platinum-based antitumor compounds. These agents form monofunctional and/or bifunctional adducts, with bifunctional adducts existing in several forms, including interstrand and intrastrand DNA crosslinks (Figure 1).
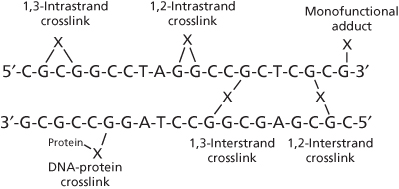
Figure 1 Monofunctional DNA adducts and interstrand and intrastrand crosslinks induced by DNA damaging agents (X = alkyl or platinum species).
The foundation for alkylating agents as therapeutics dates back to the 1914–1918 Great War when poison gases, such as chlorine or the fatally vesicant sulfur mustard gas, were used indiscriminately against enemy soldiers in chemical warfare. The sulfur mustard (Figure 2) was first used by the Germany military in 1917 against British, Canadian, and French soldiers near the town of Ypres, Belgium. During autopsy of dead soldiers, it was noted that mustard gas was highly myelosuppressive and produced lymphoid aplasia.3 Thus, the notion that such agents could combat uncontrolled growth of cancers was conceptualized, but the high toxicity and reactivity of mustard gas precluded its clinical application. Efforts in early 1940s at Yale University by Goodman and Gilman4, 5 eventually resulted in the less-reactive and less-toxic nitrogen mustard, mechlorethamine or mustine (Figure 2), as the first agent to be tested in antitumor clinical trials in lymphoma patients, and its demonstrated activity firmly laid the foundation for modern cancer chemotherapy. Its activity also spawned the development of more effective and less-toxic alkylating agents, and some of these remain part of the present-day clinical anticancer armamentarium. Apart from the nitrogen mustards, several structural classes of alkylating agents are also of considerable interest, including the aziridines, hexitol epoxides, alkyl sulfonates, nitrosoureas, and the triazines/hydrazines, and these, together with the platinum-containing antitumor drugs, form the basis of this article.
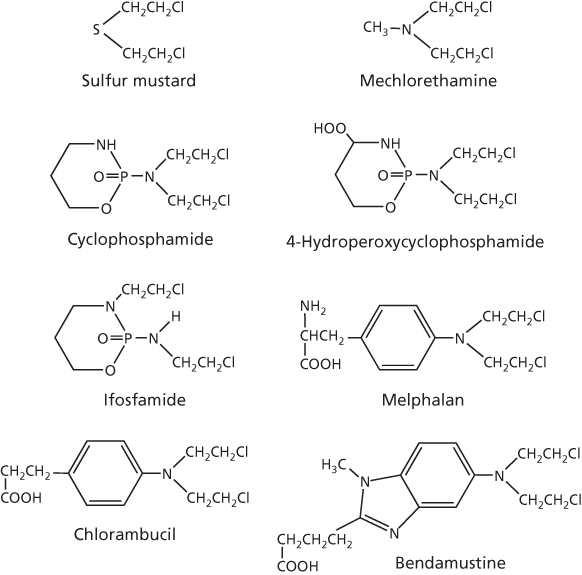
Figure 2 Structures of sulfur and nitrogen mustards.
General mechanisms of cytotoxicity
Alkylating agents and platinum antitumor compounds can transform to highly reactive species that indiscriminately form strong chemical bonds with endogenous macromolecules having electron-rich (nucleophilic) centers, such as nitrogen and sulfur atoms. The macromolecules can be peptides, proteins, or nucleic acids, but the interaction with DNA is the basis for their potent cytotoxic effects. Interactions with DNA can be monofunctional, when only a single alkylating or platinating reaction is permissible, or bifunctional, when covalent bonds are formed at nucleophilic centers of two different bases on DNA to induce crosslinks or adducts. Moreover, such crosslinks, formed predominantly through the N7 position of guanine, can be interstrand (between opposite DNA strands; induced preferentially by alkylating agents) or intrastrand (on the same DNA strand; induced preferentially by platinum drugs) (Figure 1).
Although the chance interaction between alkylating agents and DNA can be influenced by a number of factors, such as formation, detoxification, and nuclear access of the active alkylating moiety, the actual interaction with DNA is site specific. The N7 site of guanine, for instance, is electron rich and facilitates preferential covalent interaction with the positively charged alkylating species. The interaction is further facilitated by the nucleotide sequence, that is, interstrand GG crosslinks with nitrogen mustards are favored when guanine is in the 5′-GC-3′ or 5′-GXC-3′ sequence (where X = any nucleotide) in both DNA strands.6, 7 On the other hand, N2-guanine in the 5′-CG-3′ sequence (cytosine preceded by guanine) is preferred for crosslinking by mitomycin C owing to its orientation in the minor groove of DNA.8 Alkylation at O6-guanine and N3-cytosine can also occur.
The critical nature of N7-diguanyl crosslinks in the cytotoxicity of alkylating agents was established in early 1960s.6, 9 Although interstrand crosslinks may be envisioned as preventing separation of DNA strands during replication of the genome to induce cytotoxicity, this is by no means certain; it is clear that monofunctional, interstrand, and intrastrand adducts all have cellular effects, including inhibition of DNA synthesis, cell cycle arrest and apoptosis; however, the bifunctional drugs have greater potency than monofunctional agents. These cellular effects are the net result of specific distortions and unwinding in DNA induced by the alkylating or platinating drug and resultant coordinated assembly of distinct proteins at the DNA damage site to activate a number of signal transduction pathways. The final cellular effect is dependent on the concentration of the drug and, thereby, the extent of DNA damage; low level of DNA damage induces cell cycle arrest to permit DNA repair and cell survival, whereas extensive DNA damage overwhelms the capacity of the repair machinery and activates apoptosis. These events occur in both tumor and normal cells, and the small difference in relative tolerance to DNA damage between normal and tumor cells makes side effects of therapy inevitable that prevent administration of larger doses.
Alkylating agents
Bifunctional alkylating agents
Nitrogen mustards
Soon after the initial report of clinical activity of mechlorethamine in 1946,4, 5 many nitrogen mustards were evaluated for their antitumor potential through structure–activity relationships. Of the handful that advanced to clinical trials, a few were approved by the FDA and are still in use today. In addition to mechlorethamine, these include cyclophosphamide, ifosfamide, melphalan, chlorambucil, and bendamustine, which are shown in Figure 2. Also shown in this figure is 4-hydroperoxycyclophosphamide, which is the prodrug form of active 4-hydroxycyclophosphamide.
The sequence of events and specificity of the reaction between nitrogen mustards and DNA have been well described by Colvin et al.10 The bis-chloroethyl group of these agents is critically important for the predominant alkylation of N7-guanine in DNA, although alkylation can occur but infrequently at O6-guanine and at N3- and N7-adenine. The alkylation is sequence specific and requires guanine in the 5′-GXC-3′ sequence (where X = any nucleotide). For interaction, the chloroethyl moiety cyclizes to form the highly reactive aziridinium ion intermediate that reacts readily with electron-rich sites, as shown with N7-guanine in Figure 3. It is possible that the aziridinium ion may first rearrange to the reactive carbonium ion intermediate for the alkylation reaction, but this remains uncertain. As there are two chloroethyl groups present, this results in sequential bifunctional alkylation to crosslink DNA. As indicated above, interstrand crosslinks are preferred by nitrogen mustards, with 1,3-crosslinks being the favored interaction. However, DNA–DNA intrastrand, DNA–protein, and DNA–glutathione crosslinks are also formed, but lack cytotoxic potential. Interstrand DNA crosslinks are the critical lesions, and according to Roberts et al.,11 bifunctional interstrand DNA crosslinks are about 100-fold more effective at killing cells than monofunctional DNA adducts.
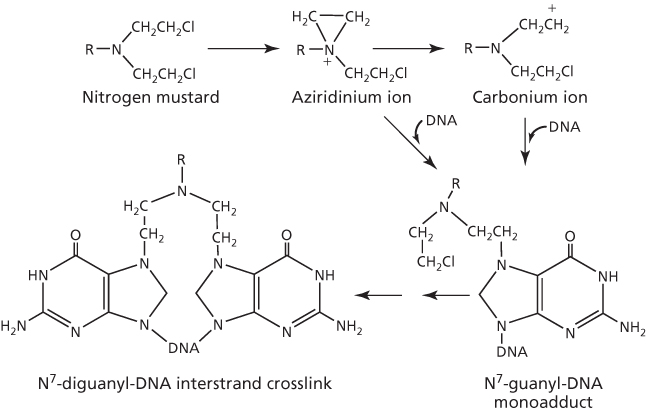
Figure 3 DNA alkylation by nitrogen mustard to form interstrand crosslink.
All nitrogen mustards are neutral and must be activated to interact with DNA target sites. Mechlorethamine, for instance, is activated spontaneously at physiological pH, but the rapid rate of activation is a major cause of much of the unwanted side effects. Chemical modification of the molecule, however, has played a crucial role not only to stabilize nitrogen mustard congeners, but also to influence their physicochemical properties that regulate transport, distribution, and reactivity of the molecule in order to enhance clinical utility.12 Cyclophosphamide, which has an oxazaphosphorine ring, was selected from over a 1000 oxazaphosphorine candidates, and its chemical inertness and dependence on metabolic activation were considered as desirable characteristics.13, 14 The primary site for this activation is the liver, where the P450-mediated mixed function oxidase reaction initiates a complex series of chemical transformation events15–18 that is depicted in part in Figure 4. The 4-hydroxycyclophosphamide is the most significant metabolite, which is relatively nonpolar and can readily distribute throughout the body, including tumor cells. This metabolite can be formed directly from the prodrug 4-hydroperoxycyclophosphamide (Figure 1), which has demonstrated utility in purging tumor cells in bone marrow aspirates of patients undergoing autologous bone marrow transplant.19 At physiologic pH, a small amount of the 4-hydroxycyclophosphamide exists in equilibrium with aldophosphamide, which is relatively unstable and spontaneously decomposes to form the highly reactive phosphoramide mustard and then nornitrogen mustard.
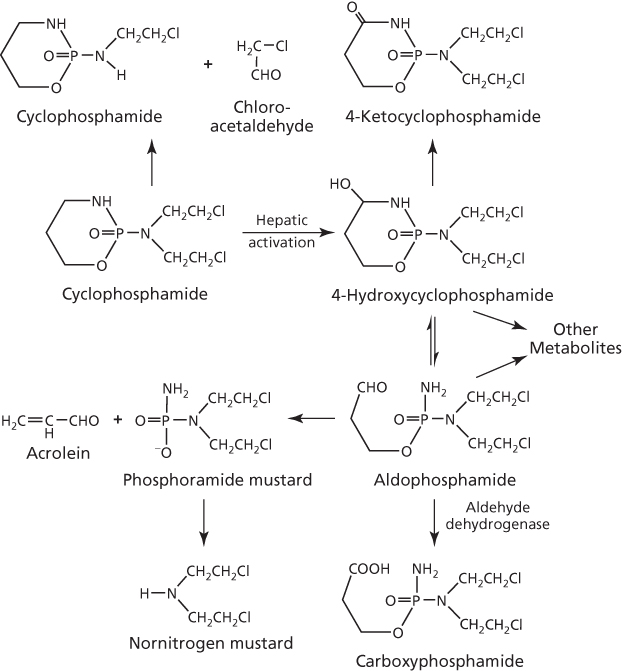
Figure 4 Metabolic activation and inactivation of cyclophosphamide.
An important by-product of the metabolism of cyclophosphamide, resulting from the conversion of aldophosphamide to phosphoramide mustard (Figure 4), is acrolein,18 which induces hemorrhagic cystitis as a serious side effect,20 but whether it has antitumor activity is of interest. Although a study with the model compound didechloro-4-hydroperoxycyclophosphamide indicated that the spontaneously released acrolein lacked cytotoxicity,21 other studies have demonstrated that acrolein indeed has antitumor and carcinogenic activities but is inactivated by O6-alkylguanine-alkyltransferase.22–24 Aldehyde dehydrogenase can also play a significant role in moderating activity by converting aldophosphamide to the inactive carboxyphosphamide metabolite, which accounts for about 80% of the cyclophosphamide dose excreted in the urine. Not surprisingly, this enzyme can confer resistance to cyclophosphamide in tumor cells when ectopically overexpressed.25 Thus, high levels of aldehyde dehydrogenase in the liver, early hematopoietic cells, and intestinal stem and mucosal absorptive cells are consistent with reduced hepatic, gastrointestinal (GI), and hematopoietic toxicities associated with cyclophosphamide as compared to other alkylating agents. Urinary excretion of metabolites can be compromised at high doses of cyclophosphamide by an antidiuretic effect owing to sodium loss and water reabsorption by renal tubules, with resultant edema.26 The electrolyte imbalance may also contribute to the reported dose-limiting and fatal cardiotoxicity of high-dose chemotherapy.27 This limitation has been recognized and doses are now monitored clinically to prevent this lesion from becoming serious.
Ifosfamide (Figure 2) is a structural isomer of cyclophosphamide that is important in the treatment of testicular cancer and sarcomas. Like cyclophosphamide, it also requires activation by hepatic mixed function oxidase,13 but the low rate of metabolic activation by 4-hydroxylation and conversion to the corresponding active isophosphoramide mustard decreases its potency approximately fourfold.28, 29 On the other hand, increase in the dose to compensate for the lower potency leads to a greater cumulative production of acrolein, which increases the incidence of urothelial toxicity that requires concurrent administration of the thiol-based mesna to inactivate the toxic by-product. Renal and bladder toxicity, as well as neurotoxicity, of ifosfamide have also been ascribed to the formation of chloroacetaldehyde,30 the product of oxidation of the chloroethyl side chain (Figure 4) that is generated in greater quantities with ifosfamide than with cyclophosphamide (∼50% vs ∼10% of the dose). The primary metabolite of ifosfamide, aldoifosfamide, is also a substrate for aldehyde dehydrogenase, and this is consistent with low toxicity of the drug to the bone marrow and the GI tract due, as mentioned previously with cyclophosphamide, to the presence of high levels of the enzyme in these organs.
Melphalan, chlorambucil, and bendamustine do not have the oxazaphosphorine ring of cyclophosphamide and ifosfamide in their structure, and so do not require metabolic activation, although metabolism of these agents does occur in the liver, but only for its disposition. These agents instead have an unsaturated electron-withdrawing aromatic ring (Figure 2) that moderates activation of the bis-chloroethyl group and formation of the aziridinium ion in order to minimize side effects. Owing to its phenylalanine-based structure, melphalan can be transported actively into cells and across the blood–brain barrier.31–33 These nitrogen mustards have found important applications against several cancers: melphalan against multiple myeloma, breast cancer, and ovarian carcinoma34; chlorambucil against ovarian cancer, chronic lymphocytic leukemia (CLL), and Hodgkin’s and non-Hodgkin’s lymphoma35–37; and bendamustine against CLL and lymphomas.38 Owing to its improved tolerance, bendamustine is reported to be more cost-effective as a therapeutic drug than the structurally similar chlorambucil.39
Aziridines
Also known as ethyleneimines, the aziridines alkylate through the three-membered heterocyclic aziridine ring. Aziridine family of drugs includes thiotepa (N,N′,N″-triethylenethiophosphoramide), mitomycin C, and triethylenemelamine, which are shown in Figure 5. Also shown is hexamethylmelamine, which is similar to triethylenemelamine, but lacks the classical aziridine ring. Of these, thiotepa and mitomycin C are of clinical interest for a number of diseases, including refractory osteosarcoma and bladder cancer, respectively.40, 41 High-dose thiotepa has also been used in drug combination regimens with42 or without43 autologous stem cell support for breast cancer. Hexamethylmelamine, on the other hand, has found application in salvage therapy for recurrent ovarian cancer.44
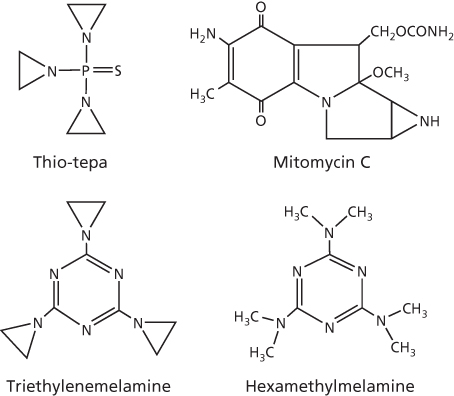
Figure 5 Chemical structures of aziridines and hexamethylmelamine.
The aziridine ring in the chemical structure of thiotepa is not charged, which makes it less reactive with electron-rich centers in RNA (ribonucleic acid), DNA, protein, and thiols (such as glutathione) than the structurally similar aziridinium ring formed with nitrogen mustards. In DNA, thiotepa alkylates sites in all four nucleotides, particularly at O2-cytosine, N1-thymine, N1-, N2-, and N7-adenine, and N1-, N7-, and O6-guanine.45 However, N7-guanine remains the preferential target, as with other alkylating agents, and results in guanine–guanine (GG) and adenine–guanine (AG) interstrand crosslinks, primarily 1,2-crosslinks.46 These crosslinks are formed sequentially; the first reaction at N7-guanine leads to the monofunctional adduct, and the second reaction with a nearby guanine results in the crosslink, as shown by pathway A in Figure 6. Crosslinking may also proceed via metabolism of thiotepa to tepa (sulfur replaced by oxygen), followed by opening of the aziridine ring and conversion to the chloroethyl-containing crosslinking species, as found in nitrogen mustards.47 Alternatively, the aziridine ring is cleaved to mediate alkylation of N7-guanine and forms a monofunctional DNA adduct (Figure 6, pathway B), which induces DNA single-strand breaks and then cell death.48, 49
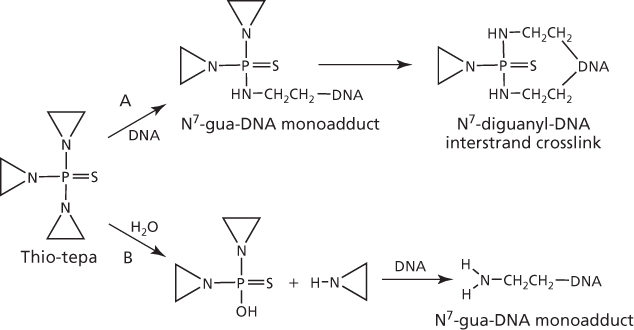
Figure 6 Metabolism and interaction of thiotepa with DNA to form interstrand crosslinks (pathway A) or monofunctional adducts (pathway B).
Unlike thiotepa, interaction of mitomycin C with DNA requires metabolic reduction to leucoaziridinomitosene for the formation of the monofunctional adduct.50 Spontaneous intramolecular rearrangement and elimination of the carbamate group at C10 of mitomycin C results in a second alkylation reaction with the opposite strand of DNA to induce cytotoxic 1,2-GG interstrand crosslink lesions (Figure 7). Interestingly, alkylation at the N2-guanine site of DNA is preferred by mitomycin C, particularly if guanine is present in the 5′-purine-CG-pyridine-3′ sequence in both strands of the duplex.8
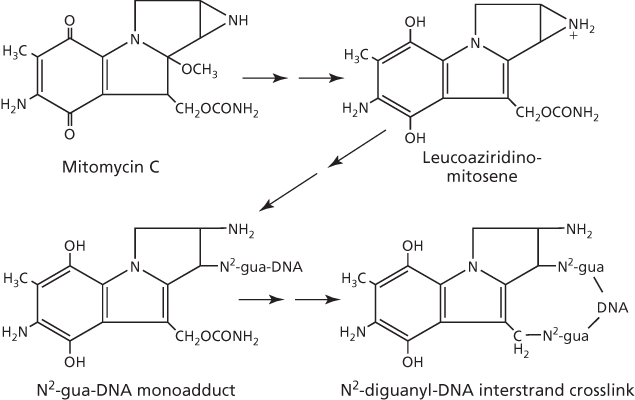
Figure 7 DNA interstrand crosslinks induced by mitomycin C.
On the basis of the presence of the aziridine ring in the triethylenemelamine structure, alkylation by this agent likely occurs in the same manner as that described for thiotepa. However, with hexamethylmelamine, metabolic activation in the liver is required.51, 52 The metabolism generates a carbinolamine intermediate, N-methylol-pentamethylmelamine, which rearranges to the reactive iminium ion as the species responsible for alkylation of guanine in DNA (Figure 8). As hexamethylmelamine metabolism can also result in the dicarbinolamine product N,N′-dimethylol-tetramethylmelamine,53 bifunctional interstrand DNA crosslink is the likely outcome, particularly as the tricarbinolamine-based prodrug of hexamethylmelamine, trimelamol, readily forms such crosslinks.54 It is noteworthy that trimelamol has been investigated in clinical trials,55 but the development did not continue, owing likely to limited clinical activity56 and/or drug formulation/stability issues.57
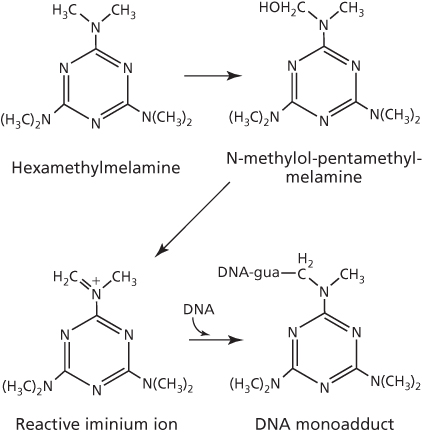
Figure 8 Activation of hexamethylmelamine to form DNA adducts.
Hexitol epoxides
The di-epoxide 1,2:5,6-dianhydrogalactitol (DAG) and its prodrug 1,2-dibromodulcitol (DBD), like the aziridines, also alkylate via a strained tricyclic ring (Figure 9). Chemical interaction between DAG and DNA results in alkylation at N7-guanine,58 a site commonly preferred by several other alkylating agents. In cell-free, cellular, and in vivo rodent systems, N7-monoguanyl and N7-diguanyl derivatives are detected as two major products that represent monofunctional and interstrand crosslinked DNA adducts, respectively.58–61
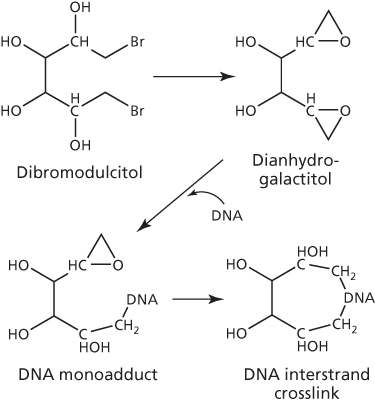
Figure 9 Conversion of dibromodulcitol to dianhydrogalactitol to form intertrand crosslinks.
Both DBD and DAG have shown antitumor properties that were similar to alkylating agents in some experimental antitumor models, but different in others. Lack of cross-resistance with DAG, for instance, was observed in a nitrosourea (BCNU)-resistant L1210 leukemia model.62 Of the two hexitols, DAG has greater chemical stability and better preclinical antitumor activity profile.63, 64 Nevertheless, both DBD and DAG entered clinical trials in 1970s and demonstrated some activity in GI and lung cancer.65, 66 Interestingly, these hexitols cross the blood–brain barrier in patients,67–69 and follow-up clinical trials found a 44% response rate with DAG in brain cancer.66 DAG was recently granted orphan designation in the United States and Europe for brain cancer, and clinical trials are currently underway in the United States (ClinicalTrials.gov Identifier: NCT01478178). In China, however, it has been marketed for the treatment of chronic myelogenous leukemia (CML) and lung cancer for over 20 years.
Alkyl sulfonates
Busulfan and the analog hepsulfam are the best known alkyl sulfonates, which are symmetrical and have either two sulfonates (busulfan) or sulfamates (hepsulfam) separated by a linear alkyl chain. Busulfan was approved in 1999 as a standard of care for CML, but its clinical application subsequently diminished by first the introduction of the less-toxic hydroxyurea and then by the targeted therapeutic Gleevec. However, busulfan is still used in the allogeneic stem cell or bone marrow transplantation setting.70, 71 Although hepsulfam was more potent than busulfan in human tumor model systems,72, 73 it has not been effective in clinical trials.
The sulfonate or the sulfamate is the critical group responsible for the alkylation reaction at N7-guanine to form GG interstrand crosslink,74, 75 as shown with busulfan in Figure 10. However, this crosslink formation by busulfan has been disputed76 and the alternative GA intrastrand crosslink has been proposed.77 Significantly, where any crosslinking has been reported, this has been correlated to cytotoxicity.
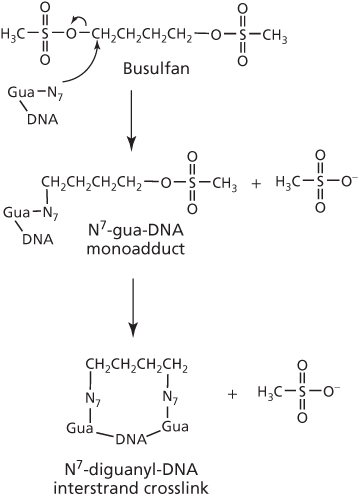
Figure 10 Busulfan-induced DNA interstrand crosslinks.
Nitrosoureas
Much of the structure–activity understanding, and the basis for clinical development of 2-chloroethylnitrosoureas (CENUs) as derivatives of urea, came from the work of Montgomery and his colleagues.78–80 With the ability to cross the blood–brain barrier, four analogs entered the clinic,81, 82 and these are shown in Figure 11. BCNU (Carmustine) and CCNU (Lomustine) are effective against several types of cancers, including glioma, glioblastoma multiforme, GI tumors, breast, and multiple myeloma, with streptozotocin (Zanosar) effective against cancers of the pancreatic islet cell, owing primarily to its uptake via the glucose transport protein GLUT2 that is present at relatively high levels in these cells.83 Absence of any advantage over other nitrosoureas has halted the use of methyl-CCNU (Semustine). The limitation in clinical utility of nitrosoureas is ascribed to their severe side effects, including hematopoietic and renal toxicities, with streptozotocin in particular demonstrating substantial toxicity to β cells of the normal pancreatic islets owing to the GLUT2 transporter.84 The development of novel nitrosoureas, therefore, continues, and a third generation analog, fotemustine (Muphoran) (Figure 11), has shown activity in brain cancer and metastatic melanoma, with or without brain involvement, and has received approval in Australia, Brazil, China, and Europe, but not as yet in the United States.85, 86
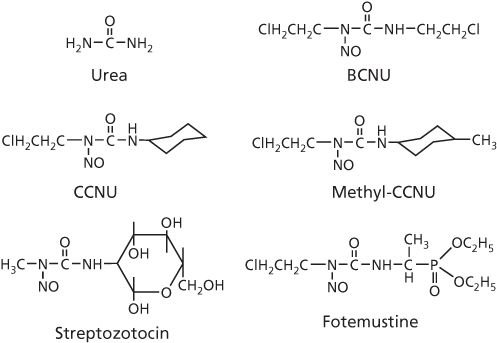
Figure 11 Chemical structures of urea and selected chloroethylnitrosourea derivatives.
It is widely acknowledged that CENUs as bifunctional DNA interstrand crosslinkers are unstable, as exemplified by a short half-life of about 50 min for BCNU82; the several products of decomposition are shown in Figure 12. An unusual feature of some CENUs is that they can generate isocyanate, which carbomylates the ϵ-amino group of lysine in proteins, including histone and other nuclear proteins, but there is disagreement whether the carbomylated-protein contributes to cytotoxicity.79, 87, 88 A second unusual feature is that CENUs can alkylate at O6-guanine in DNA, likely through formation of the 2-chloroethyl diazene hydroxide and the analogous 2-hydroxyethyl diazene hydroxide species (Figure 12).81, 82 Rearrangement of these species to carbonium ions leads to alkylation at the O6-site of guanine to form monoadducts.89 The O6-alkylation reaction is considered important as upregulation of O6-alkylguanine-DNA alkyltransferase, which repairs the O6-adduct, increases resistance and, conversely, inhibition of this enzyme with O6-benzylguanine sensitizes tumor cells to nitrosoureas.89–91 However, adducts from N7-alkylation of guanine (Figure 12) are normally the predominant species, particularly where this purine is in the middle of a GGG sequence.92 These DNA monoadducts form rapidly (often within an hour), but then slowly convert to the cytotoxic N7-diguanyl interstrand crosslink that may take up to 8 h.82 It is noteworthy that N3-cytosine–N1-guanyl crosslinks are also formed through cyclization of the O6-chloroethyl-guanine monoadduct to the O6,N1-ethanoguanine pentacyclic intermediate, which reacts with N3-site of cytosine and becomes crosslinked to the N1-site of guanine following opening and rearrangement of the ethanoguanine ring.82, 93, 94 There is also a proposal from steric considerations that only the CG crosslink of CENUs is interstrand, with the similarly cytotoxic GG adduct being intrastrand.89
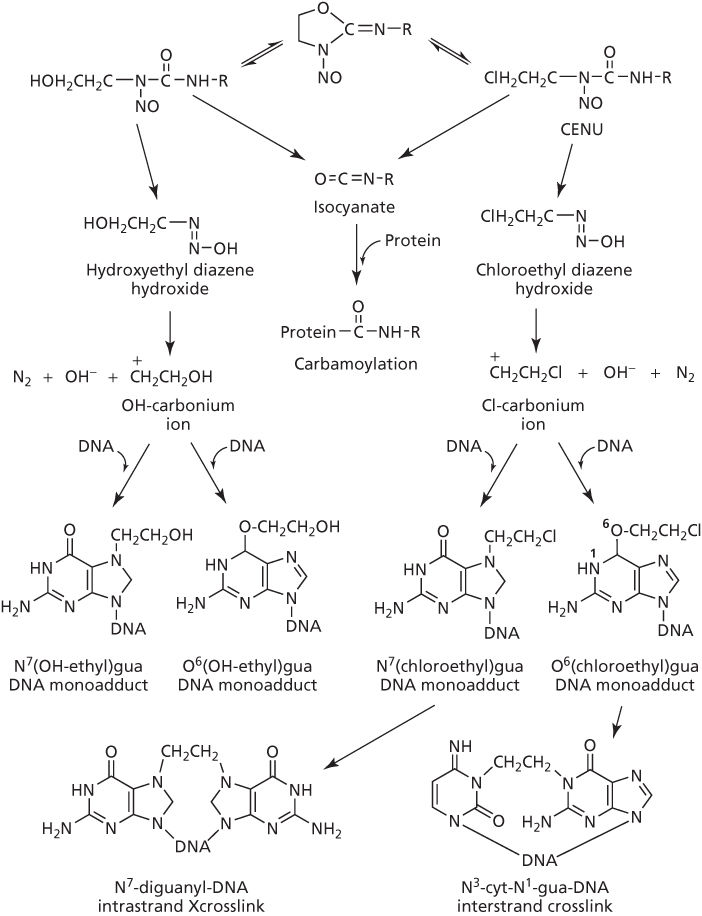
Figure 12 Activation of chloroethylnitrosourea (CENU) and reaction with DNA.
Monofunctional alkylating agents
Structures of some monoalkylators discussed below are shown in Figure 13.
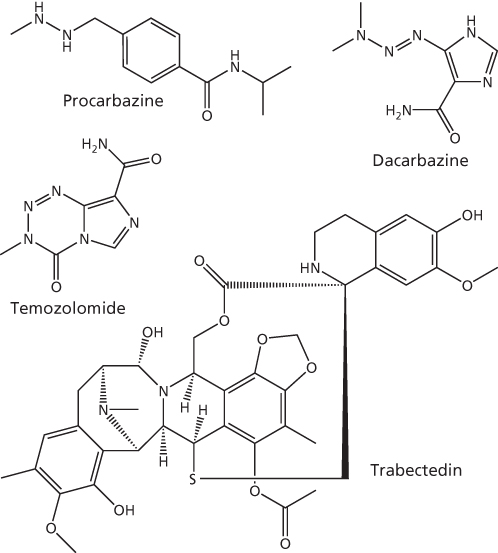
Figure 13 Structures of monofunctional alkylating agents.
Hydrazines
More than 400 hydrazine compounds have been tested preclinically, but only procarbazine has received clinical approval for the treatment of cancers, specifically Hodgkin’s lymphoma, lung cancer, and melanoma. Its activation is poorly understood, but metabolic oxidation to azoxy-procarbazine is an important step. This metabolite rearranges to a reactive methyl diazonium intermediate that forms O6- and predominantly N7-methyl-guanine adducts, which then spontaneously depurinate to induce single-strand breaks.95–97 DNA–DNA or DNA–protein crosslinks have not been reported. Another hydrazine that had been studied since the early 1970s was hydrazine sulfate, but the clinical activity was equivocal, and the compound never received approval in the United States.
Triazines
The most recognizable triazines are hexamethylmelamine (Altretamine), dacarazine (DTIC-Dome), and temozolomide (Temodar). Of these, hexamethylmelamine is likely a bifunctional alkylator and, therefore, has been grouped above with its closely resembling trimethylmelamine under “aziridines.” Dacarbazine has been used against a variety of cancers, including Hodgkin’s lymphoma, sarcoma, malignant melanoma, and pancreatic islet cell carcinoma. Temozolomide is an imidazotetrazine derivative of dacarbazine that is particularly active against astrocytoma, as well as melanoma. Triazines methylate O6- and N7-guanine,98 but, like the hydrazines, require metabolic activation for cytotoxic alkylation and DNA single-strand breaks. For dacarbazine, hepatic activation to [methyl-triazenyl]-imidazole-carboxamide (MTIC) generates the reactive DNA-methylating methyldiazonium ion.99 Temozolomide, in contrast, functions as a prodrug of MTIC and spontaneously generates the reactive species.100
Isoquinoline alkaloids
The two naturally occurring tetrahydroisoquinoline alkaloids of interest are trabectedin (ecteinascidin-743, Yondelis) and Zalypsis. Trabectedin is the first marine-derived antitumor agent to be approved in Europe, Russia, and South Korea for cancer treatment, specifically advanced soft tissue sarcoma and platinum-resistant ovarian cancer.101 Its approval in the United States, however, is pending. The drug initially binds noncovalently to TCG, CGG, AGC, or GGC sequence of one strand in the DNA minor groove, where dehydration of the carbinolamine group generates the iminium intermediate that then alkylates the unusual N2-site of guanine in the opposite strand.102 Trabectedin-induced monofunctional adducts then recruit the transcriptionally coupled nuclear excision repair (TC-NER) protein Rad13/ERCC5 to the DNA damage site and induce strand breaks and cell death. An additional unusual feature of trabectedin, not found in several bifunctional alkylating agents, is that absence of the p53 tumor suppressor activity increases antitumor activity about threefold.103 Zalypsis is essentially a synthetic derivative of trabectedin that has an identical mode of action as the parent molecule.104, 105 It is presently undergoing clinical trials.
Decomposition and metabolism
For therapeutic formulation and medical handling, alkylating agents must initially be in an inactive state to preserve maximal potency of the molecule until administered to patients. As discussed above for alkylating agents, such as temozolomide and cyclophosphamide, this potency is realized through spontaneous decomposition in the biological milieu or through metabolic activation, respectively. The resultant active species are strong electrophiles and key to inhibiting tumor growth, but are themselves also prone to decomposition by hydrolytic reactions with water or by direct inactivating interactions with the abundance of nucleophilic targets in the cell. Thiols, such as the tripeptide glutathione or cysteine-rich metallothionein protein, are major targets of interaction; in some cases, the inactivation by glutathione is facilitated by glutathione S-transferase. Consequently, depletion of glutathione by inhibiting its synthesis can enhance antitumor activity of these agents.106 In contrast, presence of glutathione is important for bifunctional activation of mitomycin C.107 Metabolism of alkylating agents in the liver and extrahepatic tissues also plays a major role in drug disposition. Apart from hyroxylation, other reactions are also important, and this includes dechlorination and denitrosation, as observed with nitrosoureas.87 As noted earlier, high levels of aldehyde dehydrogenase in the GI and hematopoietic cells can protect cells by detoxifying metabolites of cyclophosphamide. Moreover, metabolism can be influenced by other agents, such as phenobarbital,108, 109 which can increase microsomal enzymatic activity and the rate at which the active species and the inactive metabolites are generated and disposed of.
Resistance to alkylating agents
A major clinical limitation of alkylating agents is resistance of tumor cells. This resistance can be intrinsic that arises during tumor development or it can be acquired by tumor cells via drug selection pressures. In many tumors, presence of a single resistance mechanism is rare, but multiple mechanisms abound, as has been reported for melphalan110 and temozolomide.111 Thus, any effort to induce antitumor response in refractory cancers will require circumventing multiple mechanisms of resistance, and this makes the task more challenging. However, knowledge of individual mechanisms may allow identification of weakness in the signaling network within tumor cells that may be exploited for synthetic lethality approaches.112 Alternatively, identifying a resistance mechanism that, if circumvented, will provide a dominant trigger for apoptosis and over-ride remaining resistance mechanisms may have potential also.
In general terms, resistance of tumor cells can be due to (1) reduced intracellular drug concentration, (2) increased cellular inactivation, (3) enhanced DNA repair, and (4) downregulation of apoptotic signaling. Reduced uptake in resistant cells has been observed with melphalan owing to downregulation of the L-type amino acid transporter.110, 113 However, most alkylating agents rely on diffusion for cell entry and are not affected by transporters. Resistance to alkylating agents can occur from increased inactivation due to elevated levels of thiol-containing compounds or thiol-related enzymes, such as glutathione, metallothionein, γ-glutamylcysteine synthase (involved in glutathione synthesis), and glutathione S-transferase (increases drug conjugation with glutathione).114–118 Increased inactivation can also occur by elevated levels of aldehyde dehydrogenase, which readily detoxicates cyclophosphamide and related alkylating agents.119, 120
Alkylating agents target DNA, and persistence of DNA adducts correlates with cytotoxicity. In resistant tumor cells, these adducts are repaired efficiently. As discussed above, one repair mechanism involves the enzyme O6-alkylguanine-alkyltransferase, which reverses the initial alkylation of O6-guanine in DNA and prevents the formation of interstrand crosslinks. Thus, upregulation of this alkyltransferase increases resistance in cancers, as observed in human glioma cells resistant to BCNU.121 In contrast, the cytotoxic effects of O6-alkylguanine adducts (O6-methyl ≫ O6-ethyl) are facilitated by the mismatch repair (MMR) pathway122 and, conversely, resistance to the methyl-alkylator temozolomide arises when defects in the MMR pathway are present.123 Repair of crosslinks also occurs by the nucleotide excision repair (NER) and the alternative nonhomologous end-joining (NHEJ) pathways. Thus, increased repair by the NER and NHEJ system in human tumor cells induces resistance to cyclophosphamide, melphalan, and chlorambucil.124–127 In contrast, and as noted above, the activity of the monofunctional alkylating agent trabectedin is in fact enhanced by the participation of NER protein Rad13/ERCC5.102
Adduct formation and interstrand crosslinks kill cells by apoptosis, which has been demonstrated for a number of alkylating agents, including cyclophosphamide and temozolomide.128, 129 DNA damage activates a complex set of signal transduction pathways that converge on the apoptotic machinery to induce cell death. Specifically, alkylation of nucleotide bases induces unwinding and bending of DNA, and the resultant conformational changes in DNA are sensed by specialized recognition proteins that transduce DNA damage signals. This results in transcriptional activation of specific genes that finally activate caspases, which carry out the apoptotic cell death program. As a large number of proteins are involved, such as ATR, Chk1, Chk2, p53, Bak, and Bax, apoptosis can be inhibited if any critical step of the apoptotic cell death program becomes disrupted, and this leads to resistance to alkylating agents. As an example, loss of p53 function through mutation or other means leads to temozolomide resistance.111, 130 Resistance to this agent has also been noted in stem-like glioblastoma cells by overexpression of Mdm2 protein, which inhibits p53 by increasing its proteosomal degradation.131 On the other hand, overexpressed Bcl-2 protein in some cancers binds to Bax and, thereby, blocks the caspase-9/caspase-3 cascade to inhibit cyclophosphamide-induced apoptosis.128 Alternatively, resistance to cyclophosphamide, chlorambucil, and temozolomide in human B cell CLL and U87MG glioblastoma cells can occur when Bax expression is low.132, 133
Although drug resistance to alkylating agents in tumor cells can often be observed in both tissue culture and in rodent models, this is not always the case. In a classical example, EMT-6 murine mammary tumors that developed resistance to cyclophosphamide by directly injecting tumor-bearing mice with the drug did not display this resistance when cells from these tumors were exposed to this agent in vitro.134 This demonstrates that some mechanisms of resistance to alkylating agents, such as the influence of TGF-β (transforming growth factor-beta) growth factor, poor tumor perfusion, changes in intracellular pH, and the tumor microenvironment, may only operate in vivo.135–137
Clinical pharmacology
The pharmacokinetics (PK) of a drug dictates its pharmacodynamics (PD), and this PK–PD relationship is integral to understanding the basis for drug activity and toxicity and designing regimens that maximize activity against cancer and minimize side effects. The pharmacology of alkylating agents has been studied for over six decades, but it was clear as early as 1952 that an ideal alkylating agent should be administered in an inactive form and converted into an active form in patient’s tumor.12, 138 The essential knowledge on the clinical pharmacology of these agents of interest is summarized below.
Cyclophosphamide
A systemic dose of 50–75 mg/kg cyclophosphamide produces plasma levels of up to 400 µmol/L of the intact drug, which then decays with a half-life of 3–10 h.139–141 Peak plasma concentration of 50–100 µmol/L phosphoramide mustard metabolite was also observed at about 3 h. Interestingly, the AUC of this and the 4-hydroxy-cyclophosphamide metabolite were similar after intravenous and oral administration, and this indicated both routes to be therapeutically effective. However, there is considerable variation in the PK of the drug among patients, but this is likely due to a prior dose of cyclophosphamide autoinducing its own metabolism or to administration of metabolism-inducing drugs, such as phenobarbital.109, 142, 143 At conventional doses, however, variations in PK do not significantly affect its toxicity or therapeutic effects.144 One explanation for this is that the final AUC for the active metabolites is unaffected, which is supported by the demonstration that cyclophosphamide given iv over 90 min yields 4-hydroxycyclophosphamide AUC of 105–110 µmol/L that is similar to an AUC when the dose is infused over 4 days.145 Although high-dose therapy is an attractive approach in bone marrow transplantation regimens, the benefits can be limited by saturable PK.146 Urinary excretion of cyclosphosphamide, predominantly as inactive metabolites, is the main route of excretion and accounts for about 60–70% of the dose.147, 148 Renal function does not appear to correlate with the toxicity of cyclophosphamide, and this supports drug clearance of active metabolites being determined by spontaneous decomposition, and not renal excretion.
Ifosfamide
The clinical PK data on ifosfamide is limited, but appears to be similar to that of cyclophosphamide. However, levels of the active 4-hydroxy metabolite from ifosfamide are lower than from cyclophosphamide.149–151 As with cyclophosphamide, ifosfamide is orally bioavailable and can autoinduce its own metabolism.141, 152 Although such similarities may make the choice between these two agents difficult, the potential for larger interpatient variation in metabolism of the side chain and the impact of concomitantly administered drugs altering the metabolic pathway may limit the utility of ifosfamide more than that of cyclophosphamide.153–155
Melphalan
An iv dose of 0.6 mg/kg results in peak melphalan plasma levels of 4–13 µmol/L, which then decays biexponentially, with a rapid α-phase half-life (T½α) of 8 min and a relatively slower β-phase half-life (T½β) of 1.8 h.156, 157 At this dose, the mean AUC of 8 µmol h/L of melphalan was achieved and 24-h urinary excretion of melphalan was about 13% of the dose. Similar plasma PK has been observed following high-dose iv melphalan, and as spontaneous degradation of melphalan plays a major role in drug elimination, renal insufficiency was reported not to impact drug clearance.158 Another study, however, has demonstrated that renal insufficiency reduces drug clearance and increases myelosuppression, which disappears with dose reduction.159 Melphalan has good oral bioavailability, and conventional doses of 0.15–0.25 mg/kg melphalan given orally resulted in peak plasma levels of up to 0.2–0.6 µmol/L by 1–2 h, which decayed with a half-life of 0.6–3 h.160 However, absorption after oral dosing can vary, with food reducing drug absorption and high doses subject to saturable absorption kinetics.161–163
Chlorambucil
An oral dose of 0.6 mg/kg chlorambucil produced peak plasma levels of 2–6 µmol/L of the intact drug an hour later and 2–4 µmol/L of the metabolite phenylacetic acid mustard by 2–4 h.157 The β-phase half-life of the parent compound was 92 min, with that of the metabolite being 145 min. In another study, PK analysis of an oral daily dose of 0.8–0.9 mg/kg over 4 days indicated that the AUC of about 10 µmol h/L of chlorambucil on day 1 decreased 17% by day 4, with further progressive decreases noted with each 4-week treatment cycle.164 This suggests that chlorambucil, as with cyclophosphamide and ifosfamide, may also be subject to autoinduction of its metabolism and excretion, although an alternative possibility is that repeat dosing reduces absorption from the gut. A two- to fourfold inter- and intraindividual variation in the PK of oral chlorambucil has also been reported.165 Interestingly, plasma AUC of the drug or its metabolite phenylacetic acid mustard was unaffected by food intake.166
Bendamustine
A bolus iv dose of 5 mg/kg bendamustine was eliminated rapidly from the plasma, with T½α of about 10 min and T½β of 36 min, and a resultant AUC of about 28 µmol h/L.167 The oral bioavailability is good and in the range 0.25–0.94 (mean, 0.57). The drug is metabolized by hepatic mixed function oxidases to N-demethyl- and γ-hydroxy-bendamustine as major metabolites.168 Glutathione conjugates of the metabolites are also known, and an additional metabolite, dihydroxy-bendamustine, has been observed in patients plasma after an iv dose.169
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


