Agents targeting microtubules and mitotic processes
Eric K. Rowinsky, MD
Overview
The treatment of many diseases owes much to the importance of medicines derived from natural sources, and the treatment of malignant disease is no exception. Billions of years of evolutionary pressure have resulted in the natural selection of plants, fungi, and microorganisms capable of producing potent and specific toxins. After several plant-derived compounds and other natural products, many of which targeted the mitotic processes, demonstrated prominent anticancer activity in the 1950s and 1960s, the microtubule was recognized as a subcellular target of major strategic importance.
The first widely used class of antimicrotubule agents, the plant-derived vinca alkaloids, had been the mainstay of both palliative and curative regimens for treating malignancies for several decades. The addition of the plant-derived taxanes, which possess a unique mechanism of action and anticancer spectra, to our therapeutic arsenal several decades later resulted in renewed interest in the microtubule and mitotic processes as targets for which to develop cancer therapeutics, as well as in the identification of other natural products to treat cancers. More recently, several plant- and marine-derived compounds as well as synthetic agents with yet even more distinctive disruptive actions on microtubules and other mitotic constituents have been identified. These include the analogs of the epothilones and halichondrin B, which were isolated from soil-dwelling myxobacterium and marine sponges, respectively. They also include potent antimicrotubule agents, such as analogs of maytansine and dolastatin, which are components of antibody-drug conjugates. This chapter focuses on the microtubule as a target for therapeutic development and antimicrotubule agents that comprise our therapeutic armamentarium.
Introduction
The treatment of many diseases owes much to the importance of medicines that have been derived from natural sources, and the treatment of malignant disease is no exception. In essence, billions of years of evolutionary pressure have resulted in the natural selection of plants, fungi, and microorganisms that are capable of producing highly potent and specific toxins. After several plant-derived compounds and other natural products, many of which were noted to suspend cell division in mitosis by affecting the mitotic spindle, demonstrated prominent anticancer activity in patients with advanced malignancies in the 1950s and 1960s, the microtubule was recognized as a subcellular target of major strategic importance.
The first widely used class of antimicrotubule agents, the plant-derived vinca alkaloids, had been the mainstay of both palliative and curative regimens for treating malignancies for several decades. The addition of the plant-derived taxanes, which possess a unique mechanism of action and anticancer spectra, to our therapeutic arsenal several decades later resulted in renewed interest in the microtubule and mitotic processes as targets for which to develop cancer therapeutics, as well as in the identification of other natural products to treat cancers. More recently, several plant- and marine-derived compounds as well as synthetic agents with yet even more distinctive disruptive actions on microtubules and other mitotic constituents (e.g., mitotic kinesins and kinases) have been identified and added to our therapeutic arsenal. These include the analogs of the epothilones (e.g., ixabepilone) and halichondrin B (e.g., eribulin), which were isolated from soil-dwelling myxobacterium and marine sponges, respectively. They also include the highly potent antimicrotubule agents, such as analogs of maytansine and dolastatin, which are components of antibody-drug conjugates (ADCs) that utilize monoclonal antibodies (mAbs) to deliver them selectively cancer cells that express the mAb targets. This chapter focuses on the microtubule as a target for therapeutic development and antimicrotubule agents that comprise our therapeutic armamentarium, particularly the vinca alkaloids and taxanes, as well as several classes of promising antimicrotubule agents undergoing clinical evaluation.
Microtubules as strategic targets against cancer
Microtubules are highly regulated and integral components of the cellular cytoskeleton that can be disrupted by various natural products (e.g., vinca alkaloids, taxanes, epothilones, halichondrins, others), as well as an increasingly number of synthetic compounds.1–6 Microtubules, together with microfilaments and intermediate filaments, form the cytoskeleton and maintain cell structure. Although the most important functions of microtubules in proliferative cells are through their actions as components of the cytoskeleton and mitotic spindle apparatus, which pulls apart chromosomes and is vital to cell division, they are involved in many other critical functions throughout the cell cycle, including intracellular transport of vesicles and organelles, trafficking of proteins including many oncoproteins, locomotion, adhesion, and anchorage of subcellular organelles and receptors.1–8 The cytoskeleton, particularly microtubules, also influences gene expression and signal transduction, and vice versa; however, the mechanisms involved in these interactions are not well understood. The specific expression of transcription factors in concert with drug-mediated depolymerization of microtubules has been well described and such has provided information on the differential expression of specific genes.9 Lastly, the state of the cytoskeleton mediates cellular response via the action of various growth factors.9 Antimicrotubule agents disrupt these functions.1–9
Microtubule structure and dynamics
Microtubules are dimeric structures made up of two globular subunits, α-tubulin and β-tubulin monomers, each of which consists of approximately 450 amino acids with a molecular weight of 50 kD.10 The α/β-tubulin dimers assemble into microtubules by forming linear protofilaments. Typically, microtubules are formed by the parallel association of 13 protofilaments, although microtubules composed of fewer or more protofilaments have been observed in vitro. Microtubules have distinct polarity, which is conferred by the unique alignment of the protofilaments. The protofilaments form a cylindrical wall around a hollow core; tubulin polymerizes end to end, with the α-tubulin subunit of one dimer in contact with the β-tubulin subunit of the next, as shown in Figure 1. Therefore, one end of a protofilament will have the α-tubulin subunits exposed, while the other end will have the β-tubulin subunits exposed. These ends are designated the minus and plus ends, respectively. The protofilaments align parallel to one another with the same polarity, so, in a microtubule, there is one end, the plus end, with only β-tubulin subunits exposed, while the other end, the minus end, has only α-tubulin subunits exposed. At the plus end, there is guanosine triphosphate (GTP) binding, rapid assembly, and net elongation, whereas assembly is slow and net shortening occurs at the minus end. The unique functions of microtubules are related to their polymerization dynamics, involving a dynamic equilibrium between an intracellular pool of α/β-tubulin dimers and microtubule polymers, and simultaneous release of the α/β-tubulin dimers into the soluble tubulin pool. Tubulin polymerization occurs by a nucleation–elongation mechanism, in which the slow formation of a short microtubule “nucleus” is followed by rapid elongation of the microtubule at its ends by the reversible, noncovalent addition of α/β-tubulin dimers. The dynamic equilibrium between free α/β-tubulin dimers and the microtubule occurs simultaneously at both ends of the microtubule.1–7 Each tubulin molecule is associated with two molecules of GTP. The nucleoside bound to α-tubulin, at the N site, is nonexchangeable, whereas the one bound to β-tubulin, at the E site, can be exchanged with free guanosine diphosphate (GDP). The assembly process utilizes energy provided by the hydrolysis of GTP.
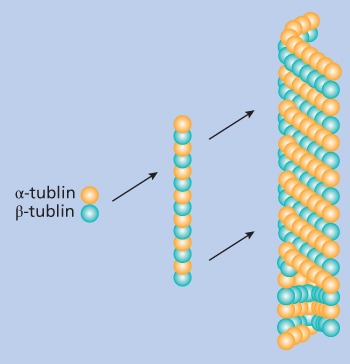
Figure 1 Schematic model of the α/β-tubulin dimer with the monomers represented by shades of gray and white. The dimers associate linearly to form protofilaments that then in turn associate laterally to form the hollow cylindrical wall of the microtubule. Protofilaments can twist slowly around the microtubule axis, although these shown here are in parallel as in microtubules containing 13 protofilaments. Throughout most of the microtubule, lateral contacts involve α–α and β–β monomer interactions. Monomers of each type thus are in contact along a shallow spiral path around the microtubule.
Tubulin binds GTP with high affinity, and as tubulin-GTP is added to the ends of growing microtubules, GTP is gradually hydrolyzed to GDP and Pi.11 The Pi ultimately dissociates, leaving a microtubule core that consists of tubulin bound to GDP. The GDP nucleotide remains nonexchangeable until the tubulin subunit dissociates from the microtubule. Although tubulin polymerization and dissociation, and consequently microtubule elongation and shortening, occur simultaneously at each end of the microtubule, the net changes in length at the more kinetically dynamic plus end are much larger over time than those at the minus end. If the polymerization reaction is followed in vitro, an initial lag phase is noted, after which microtubules form rapidly until a plateau phase is reached. In the intact cell, the minus ends of the microtubule are anchored at each of the two microtubule-organizing centers (MTOCs), whereas the plus end is free in the cytoplasm radiating toward the cell periphery. The centrosome, the principal MTOC of most cells, is the nidus for mitotic spindles; however, the Golgi apparatus can also serve as a platform for the nucleation of microtubules.12 Because nucleation from the centrosome is inherently symmetrical, Golgi-associated microtubule nucleation permits formation of an asymmetrical microtubule network. In interphase, the MTOC organizes the microtubules that provides polarity to the cytoplasm and is involved in the trafficking of proteins, including several oncoproteins.8
Treadmilling and dynamic instability
Two principal processes govern microtubule dynamics in live cells.1–15 The first, known as treadmilling, is the net growth at one end of the microtubule and the net shortening at the opposite end.8
Mitotic spindle formation and chromosomal segregation during the anaphase stage of mitosis, among other microtubule processes, occur by treadmilling. The second dynamic process, termed dynamic instability, occurs when the plus ends of microtubules switch spontaneously between states of slow sustained growth and rapid shortening.11, 13–15 Dynamic instability is dependent on a cycle of GTP hydrolysis and exchange. Mechanistically, the GTP cap model, in which a cap on the ends of the microtubule, consisting of GTP or GDP with associated nonexchangeable Pi, stabilizes the microtubule, thus allowing growth at the plus end, likely explains the process of dynamic instability. In essence, the switch between growth and shortening depends on the end conformation of the microtubule where growing ends are stabilized by a layer of GTP-tubulin subunits (GTP cap), whereas shortening ends have lost their GTP, allowing terminal GDP-tubulin dimers to dissociate from the microtubule lattice.16
Microtubules and mitosis
At the onset of mitosis, the interphase array of microtubules, which are attached to the MTOC, disassemble and are replaced by a new population of spindle microtubules that are much more dynamic.2, 13, 17, 18 Contained within the MTOC is another type of tubulin, γ-tubulin, which associates with other proteins to form a lock washer-like structure called the γ-tubulin ring complex. This complex acts as a template for α/β-tubulin dimers to begin polymerization; it caps the negative end, while microtubule growth occurs at the free positive end.19 A recently identified protein complex called augmin is critical for MTOC-dependent, spindle-based microtubule generation as it interacts with the γ-tubulin ring complex to increase microtubule density around the mitotic spindle origin.20 As the spindle-shaped array of newly assembled microtubules is organized, the nuclear envelope breaks down and releases the newly condensed chromosomes, and the centrosomes, which were duplicated before mitosis, separate into the poles of the forming mitotic spindle. The rate of dynamic instability is accelerated during mitosis, resulting in the formation and attachment of the mitotic spindles to the chromosomes. In most cells, mitosis progresses rapidly, and the highly dynamic microtubules that comprise the mitotic spindle render them sensitive to antimicrotubule agents that disrupt polymerization dynamics.3, 13
Dynamic instability and treadmilling are vital to the assembly and function of the mitotic spindle, and the high dynamaticity of the microtubules that comprise the mitotic spindle is required for the precise alignment of the chromosomes and their attachment to the mitotic spindle during metaphase, as well as chromosome separation during anaphase. Dynamic instability and treadmilling enable the microtubules of the mitotic spindle to make vast growing and shortening excursions, often termed search and capture, essentially probing the cytoplasm, until their positive ends become attached to a chromosome at its kinetochore. If even a single chromosome is unable to achieve a bipolar attachment to the spindle, perhaps because of drug-induced suppression of microtubule dynamics, the cell will not traverse beyond a prometaphase/metaphase-like state and instead will undergo apoptosis due to a complex series of processes involving the spindle assembly checkpoint.21, 22 Although mitotic spindles can assemble even in the presence of low concentrations of antimicrotubule agents, mitosis cannot progress beyond the mitotic checkpoint at the metaphase/anaphase transition.13, 17, 22 Such perturbations in mitotic spindle dynamics may delay cell-cycle progression at critical mitotic checkpoints, ultimately triggering apoptosis through a complicated interplay of checkpoint and mitotic kinases.7, 13, 17, 22 In the unperturbed normal state, oscillations of the duplicated chromosomes, dynamic instability, and treadmilling, in which there is addition of tubulin to the spindle at the kinetochore and loss of tubulin at the spindle poles, exert considerable tension on the chromosomes in metaphase and facilitate progression to anaphase. In anaphase, microtubules attached to the chromosomes undergo shortening, while another subpopulation of microtubules, called interpolar microtubules, lengthen, resulting in polar movement of the chromosomes. Suppression of spindle-microtubule treadmilling and dynamic instability by antimicrotubule agents reduce spindle tension and impede progression from metaphase to anaphase, thereby triggering cell death.3–5, 13, 17, 18, 23, 24 A similar process of positive-end capping activity for interphase microtubules, which is mediated by formins, the adenomatous polyposis coli protein, and EB1, has also been described.
Regulating microtubule dynamics and functional diversity
The net direction of the dynamic equilibrium between the intracellular pool of tubulin and the microtubule polymer is influenced by several factors that regulate the critical concentration of tubulin required for polymerization, including the GTP/GDP ratio, the ionic microenvironment, cell-cycle modulators, and various microtubule-associated proteins (MAPs) and regulatory proteins.1, 4, 5, 25, 26 MAPs are partly responsible for the functional diversity of microtubules in different tissues and are directly and indirectly being targeted for therapeutic development against cancer.26 The major classes of MAPs include those that favor microtubule polymerization such as the tau proteins (molecular weights, 40–60 kD) and the high molecular weight (200–300 kD) proteins MAP1, MAPc (an adenosine triphosphatase [ATPase]), MAP2, MAP4, and XMAP215.4, 25–27 Other regulatory proteins include stathmin, XKCM1, XKIF2, and katanin, all of which favor depolymerization.27 MAPs generally possess two binding domains, one of which binds to microtubules and facilitates the initial nucleation step of tubulin polymerization and a second that links microtubules to other cellular constituents. Still, other MAPs, such as the dyneins (GTPases) and kinesins (ATPases), function as microtubule motor proteins, transmitting chemical energy to mechanical force and moving various solutes and subcellular organelles along the microtubule.1, 4, 5, 25–27 Motor proteins play key roles in mitosis, premeiotic events, and organelle transport and are being evaluated as targets for anticancer therapeutic development (see section titled “Targeting Mitotic Motor Proteins and Kinases”).28, 29
In addition to MAPs, the functional diversity of microtubules in various tissues is, in part, conferred by differences in tubulin isotypes and posttranslational modifications. There are at least six isotypes of α-tubulin and β-tubulin each in human cells, which are distinguished by different C-terminal amino acid sequences and encoded by a large multigene family that has been highly conserved throughout evolution.4, 30–33 Analysis of tubulin isotype expression in various tissues has demonstrated a complex, albeit highly conserved, pattern of isotype distribution, suggesting functional specificity.1, 4, 5, 25–27 Both forms of tubulin also undergo various posttranslational modifications, including phosphorylation, detyrosylation, polyglycylation, delta 2 tubulin formation, acetylation, and polyglutamylation, by microtubule-bound enzymes.34, 35 Posttranslational modifications confer stability to the microtubule polymer, as well as structural and functional diversity.34, 35 Most modifications are rapidly reversed by soluble enzymes when microtubule depolymerization occurs. Posttranslational modifications generally occur on the C-terminal region of α-tubulin. This region, which is rich in negatively charged glutamate, forms relatively unstructured tails that project out from the microtubule and contact motor proteins. Therefore, posttranslational modifications appear to regulate the interactions of motor proteins with microtubules.
Vinca alkaloids: Introduction and indications
The vinca alkaloids are naturally occurring or semisynthetic nitrogenous bases extracted from the pink periwinkle plant Catharanthus roseus G. Don. The early medicinal uses of this plant led to screening these compounds for their hypoglycemic activity, which was ultimately of minor importance compared to their cytotoxic effects. Many vinca alkaloids have been extensively evaluated, but only vincristine (VCR), vinblastine (VBL), and vinorelbine (VRL) are approved for use in the United States and elsewhere, whereas vinflunine (VFL) and vindesine (VDS) are approved for use in some regions. VCR sulfate liposomal injection is also available for use in the United States and elsewhere.
The vinca alkaloids have dimeric structures composed of two basic multiringed units (Figure 2), an indole nucleus (catharanthine), and a dihydroindole nucleus (vindoline), joined together with other complex systems.36–38 Structurally, VCR and VBL are identical except for a single substitution on the vindoline nucleus, where VCR and VBL possess formyl and methyl groups, respectively. Although this minor difference does not fundamentally alter the mechanisms and tubulin-binding properties of these agents, the anticancer and toxicologic profiles of VCR and VBL differ significantly.4, 36–41
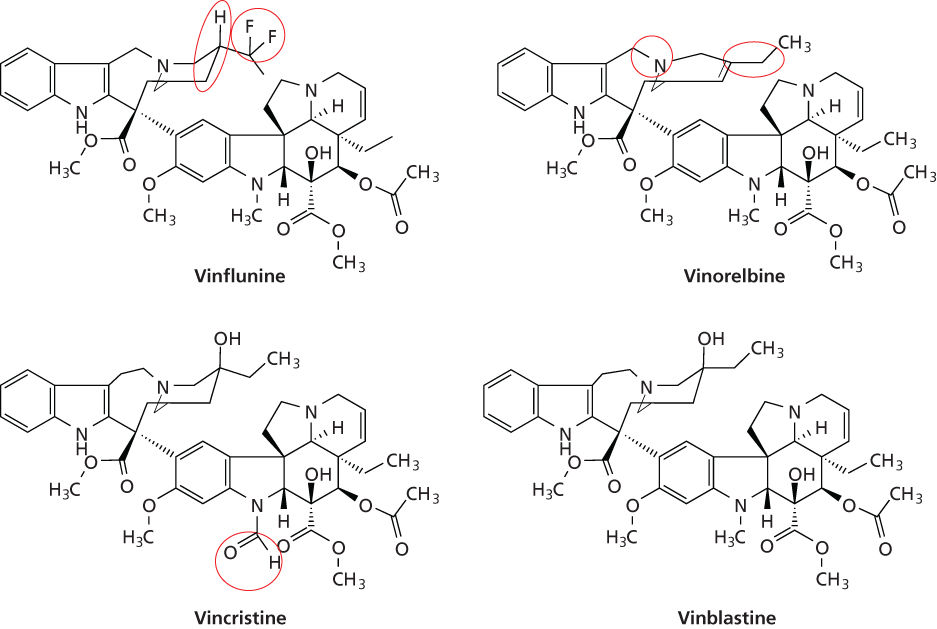
Figure 2 Chemical structures of the vinca alkaloids. Structural modifications with vinblastine as the reference are circled.
VCR is used more commonly to treat pediatric malignancies, which is due, in part, to a generally greater level of intrinsic sensitivity of pediatric malignancies to VCR and better tolerance of therapeutic VCR doses in children. In both children and adults, however, VCR is an essential component of the chemotherapy regimens used to treat acute lymphocytic leukemia (ALL), lymphoid blast crisis of chronic myeloid leukemia (CML), and both Hodgkin and non-Hodgkin lymphoma. VCR also plays a role in some multimodality therapies of Wilms’ tumor, Ewing’s sarcoma, neuroblastoma, and rhabdomyosarcoma and is occasionally used in treating multiple myeloma, particularly as a component of the VAD (VCR, doxorubicin, and doxorubicin) regimen, recurrent small cell lung cancer, and sarcoma of both soft tissue and bone. The agent has also been used as a component of a combination regimen, known as PCV (procarbazine, lomustine, and VCR), which is used in early and advanced settings of anaplastic oligoastrocytoma and oligodendroglioma.42 More for a historical sense, indicating a wide range of utility, VCR and VCR-laden platelet transfusions have been effective in refractory autoimmune thrombocytopenia; VCR has been effective in treating both nonmalignant and immunologically mediated disorders such as autoimmune hemolytic anemia, hemolytic uremic syndrome, thrombotic thrombocytopenia purpura, and steroid-dependent nephritic syndrome.38–40, 43
VBL has been a mainstay component of chemotherapy for germ cell malignancies and some types of advanced lymphoma.38 For many years, a regimen termed PVB, consisting of cisplatin, VBL, and bleomycin, was a standard therapy for germ cell carcinoma; however, VBL has largely been replaced by etoposide in this setting because of the more favorable toxicity profile of the cisplatin–etoposide regimen. For Hodgkin’s lymphoma, VBL is often used in combination with doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD).38 This regimen has been either administered alone, alternated with nitrogen mustard, VCR, procarbazine, and prednisone (MOPP), which is noncross-resistant to ABVD, or used as a hybrid (MOPP/ABV) that includes both VCR and VBL, which has also been extensively studied. Anticancer activity has been observed with VBL as a single agent or in combination with other anticancer drugs in patients with advanced and/or relapsed carcinoma of the breast, bladder, and lung, as well as Kaposi’s sarcoma, choriocarcinoma, terminal phase of CML, cutaneous T-cell lymphoma, and histiocytosis X.38 Infusions of VBL or VBL-laden platelets have been effective in some cases of refractory autoimmune thrombocytopenia because of its avidity to platelets.38, 43 VBL has also been used alone or in combination with other agents to treat Kaposi’s sarcoma and bladder, breast, and some types of malignancies of the central nervous system.38, 44–48
The semisynthetic VBL derivative vinorelbine (5′-norhydro-VBL; VRL), which is structurally modified on its catharanthine nucleus, resulting in much greater lipophilicity as compared with the other vinca alkaloids, is approved in the United States for treating nonsmall cell lung cancer as either a single agent or in combination with cisplatin and has been registered elsewhere to treat patients with advanced breast cancer. The agent has also demonstrated clinical activity in advanced ovarian carcinoma and lymphoma, but a unique role in treatment of these cancers has not been defined. VRL has also been shown to confer favorable therapeutic indices to patients with advanced breast and lung cancers who are elderly and/or have diminished performance abilities.4, 38, 44–48
Several other vinca alkaloids or reformulated versions of older vinca alkaloids are available for treating relevant malignancies worldwide. One widely studied vinca alkaloid, vindesine (desacetyl-VBL caroxyamide), a semisynthetic derivative and human metabolite of VBL, was introduced in the 1970s and several regions outside the United States. In some reports, response rates in nonsmall cell lung cancer with combinations of VDS and cisplatin or mitomycin appear to be superior to those achieved with standard combinations or with either agent alone.38, 40, 41 In addition, anticancer activity has been seen in ALL, blast crisis of CML, malignant melanoma, various pediatric solid neoplasm, and advanced renal, breast, and esophageal carcinoma. In addition, VFL, which is a bifluorinated vinca alkaloid, has shown notable clinical activity in bladder, breast, lung, and other cancers.4, 38, 44–52 In Europe, VFL is approved as a treatment option for patients with advanced urothelial cancer who have failed a prior platinum-containing regimen.52 Lastly, a sphingomyelin- and cholesterol-based liposomal nanoparticle formulation of VCR (vinCRIStine sulfate LIPOSOME injection (VSLI)) is registered in the United States for the treatment of adult patients with Philadelphia chromosome-negative ALL whose disease has progressed following at least two prior therapies.53, 54 Although the liposomal formulation permits delivery of higher VCR doses, has a longer circulatory time, and delivers greater amounts of active VCR to target tissues compared to standard VCR, the overall clinical benefits of the new formulation of VCR over standard VCR are not known.
Mechanism of action
The vinca alkaloids principally induce cytotoxicity by disrupting microtubule function, particularly that of microtubules that comprise the mitotic spindle apparatus, leading to metaphase arrest and cell death.2–7, 36–38, 55–59 However, they are capable of many other biochemical activities that may or may not be related to their effects on microtubules, including inhibiting synthesis of proteins and nucleic acids, elevating oxidized glutathione, altering lipid metabolism and membrane lipids, elevating cyclic adenosine monophosphate (cAMP), and inhibiting calcium–calmodulin-regulated cAMP phosphodiesterase.38 Many effects that do not involve microtubule disruption occur only after treatment with superphysicological concentrations that are not readily attained in vivo, whereas nanomolar concentrations, which are readily achievable in following drug administration in clinical practice, induce typical antimicrotubule effects. The vinca alkaloids also disrupt the structural integrity of platelets and other cells, which are rich in tubulin. In addition to mitotic disruption, the vinca alkaloids and other antimicrotubule agents perturb cells in nonmitotic cell-cycle phases, which is not surprising because microtubules are involved in many nonmitotic functions.38, 57–59 The vinca alkaloids bind rapidly and reversibly to sites on tubulin (called the vinca domain), which are distinct from those of the taxanes, colchicine, podophyllotoxin, and GTP but similar to those of maytansine and several other plant alkaloids.
The vinca alkaloids appear to bind to two binding sites with different affinities.2, 4, 6, 7, 10, 37, 38, 51, 55–57, 60, 65 Binding of the vinca alkaloids to high-affinity sites (Kd, 1–2 µmol), which are at the ends of microtubules, is responsible for the potent suppression of tubulin exchange at low concentrations (<1 µmol). At low vinca alkaloid concentrations, processes dependent on microtubule dynamics (treadmilling and dynamic instability) are disrupted, but microtubule mass is not affected. The overall result is a potent mitotic block at the metaphase/anaphase boundary. Binding of vinca alkaloids to low-affinity sites (Kd, 0.25–3.0 mmol) along the sides of microtubules appears to result in reduced microtubule mass due to tubulin depolymerization and the splaying of microtubules into spiral aggregates or spiral protofilaments that form paracrystals, ultimately leading to microtubule disintegration. These effects occur at high drug concentrations (>1–2 µmol) by a self-propagated mechanism, initially involving drug binding to a limited number of sites, which progressively weakens the lateral interactions between the protofilaments, thereby exposing new sites and augmentation of the process.
The vinca alkaloids induce a potent block in mitosis at the metaphase/anaphase transition.2, 4, 66 Following nuclear envelop breakdown, the vinca alkaloids block mitotic spindle formation and reduce the tension at the kinetochores of the chromosomes. Although chromosomes may condense, they remain scattered in the cells. The chromosomes separate along their lengths but still remain attached at their centromeres.2, 4, 9, 13, 18, 24, 58, 59, 67 Mitotic progress is delayed in a metaphase-like state, with chromosomes “stuck” at the spindle poles, unable to move to the spindle equator. The cell-cycle signal to the anaphase-promoting complex, which is required for the cell to transition from metaphase to anaphase, is blocked, and apoptotic cell death ensues. Cell-cycle progression in the absence of anaphase or cytokinesis may occur, resulting in chromatin decondensation and formation of multilobed nuclei.2, 4, 13, 24 Disruption of dynamics without depolymerization of the microtubules that constitute the mitotic spindle ultimately induces apoptosis that involves expression of proapoptotic genes and both activation and inactivation of proapoptotic and antiapoptotic proteins, respectively (see section titled “Vinca Alkaloids, Mechanism of Action” and “Vinca Alkaloids, Mechanisms of Resistance” and “Taxanes, Mechanisms of Action, and Resistance”).2, 4, 9, 13, 18, 21, 23, 51, 67–72 Induction of apoptosis, however, is not dependent on the presence of an intact TP53 checkpoint. The loss of p21, a protein that controls entry into mitosis at the G2/M checkpoint, sensitizes tumor cells to both vinca alkaloids and taxanes, possibly by hastening entry of drug-damaged cells into mitosis.23, 68–70
The relationships between microtubule depolymerization caused by the vinca alkaloids and their resultant effects on cell proliferation, mitotic arrest, and mitotic spindle disruption have been characterized in a series of studies whose results indicate that cell growth inhibition is directly related to metaphase arrest.18 The inhibition of proliferation and blockage of cells in metaphase at the lowest effective drug concentration occur with little or no microtubule depolymerization or disorganization of the mitotic spindle apparatus. With increasing drug concentrations, the organization of microtubules and chromosomes in arrested mitotic spindles breaks down in a manner similar to most antimicrotubule drugs, regardless of the underlying mechanism.2–4, 13, 14
In addition to their direct cytotoxic effects on tumor cells, the vinca alkaloids as well as the maytansinoids, taxanes, and various other antimicrotubule agents confer anticancer effects by inhibiting processes associated with malignant angiogenesis with surprising potency.73 These antiangiogenic effects are most likely caused by drug-induced microtubule perturbations in vascular endothelial cells; however, the relative contribution of these effects to the clinical anticancer activity is unclear.
The reasons for the disparate sensitivity of various tissues and tumors to the vinca alkaloids are multifactorial. One possible explanation is the differential sensitivity of tissues with varying tubulin isotype composition, which may affect intracellular drug accumulation and drug binding to tubulin.4, 33, 74, 75 In addition, differences in the type and concentration of MAPs and posttranslational tubulin modifications, which may influence drug interactions with tubulin, as well as variability in cellular permeability and retention, may affect the formation and stability of complexes formed between the vinca alkaloids and tubulin. Palmitoylation, a posttranslational tubulin modification, is directly inhibited by the vinca alkaloids, and depalmitoylation of tubulin may not only be a mechanism of action, but may also relate to drug sensitivity.76 Other putative factors include differences in tubulin isotypes (see sections titled “Vinca Alkaloids, Mechanisms of Resistance” and “Taxanes, Mechanisms of Resistance”), GTP content, rates of GTP hydrolysis, and cellular permeability.74, 77–81
Mechanisms of resistance
In most preclinical models, resistance to the vinca alkaloids develops rapidly and is associated with decreased drug accumulation and retention. At least two types of mechanisms of resistance have been characterized. The first typifies the “classic” pleiotropic or multidrug-resistant (MDR) phenotype that can be either primary or acquired. Although many proteins that mediate MDR have been characterized, the most well known are the ATP-binding cassette (ABC) transporters that belong to the largest known transporter gene family and translocate a wide range of substrates across cellular compartments.82, 83 These intracellular and extracellular membrane-spanning proteins, which transport both endobiotics and xenobiotics, confer resistance to the vinca alkaloids and other structurally bulky natural products. The best characterized ABC transporters are the permeability glycoproteins (Pgp) or the MDR-encoded gene product MDR1 (ABC subfamily B1) and the multidrug resistance protein (MRP; ABC subfamily C2).82
MDR1 is a 170-kDa Pgp energy-dependent transmembrane pump that regulates the efflux of a wide range of bulky hydrophobic substances.82 The protein is constitutively overexpressed in various normal tissues, which include renal tubule epithelium, colonic mucosa, and adrenal medulla, as well as tumors derived from tissues that constitutively overexpress Pgp. Pgp, an ATPase, functions to bind and extrude the vinca alkaloids from the tumor cell in a process that requires energy in the form of ATP. The MDR phenotype also confers varying degrees of cross-resistance to other structurally bulky natural products such as the taxanes, anthracyclines, epipodophyllotoxins, and colchicine. The specific Pgp associated with vinca alkaloid resistance shows slight antigenic and amino acid sequence differences and a different amino acid map after digestion than does Pgp from cells selected for resistance to colchicines or taxanes.84–87 In fact, two forms of the protein have been demonstrated to be produced by a single clone of VCR-resistant cells, and these forms undergo posttranslational N-glycosylation and phosphorylation, which lead to further structural diversity and may explain the greater degree of resistance for the specific agent used compared with the resistance of other drugs conferred by MDR and may also account for the variable patterns of resistance among MDR cells. The composition of membrane gangliosides in cancer cells resistant to the vinca alkaloids has also been shown to differ from that of wild-type cells. The clinical ramifications of these resistance mechanisms are not known. In one study in childhood ALL, however, VCR resistance measured in vitro did not correlate with Pgp overexpression.88
MRP1, a 190-kD membrane-spanning protein that shares 15% amino acid homology with MDR1, also confers resistance to the vinca alkaloids in vitro.39, 82, 83, 89–92 MRP1 expression is found in many tumors and has been associated with the MDR phenotype in cancers of the lung, colon, breast, bladder, and prostate, as well as leukemia. MRP1 transports glutathione conjugates of alkylating agents and several xenobiotics such as etoposide and doxorubicin but confers resistance only to the latter agents. MRP1 confers resistance to the vinca alkaloids and methotrexate.39, 82, 83, 89–92 Although other ABC transporters have been identified and several confer cellular resistance to the vinca alkaloids in vitro, their roles in conferring inherent or acquired drug resistance in the clinic are less clear than those of MDR1 and MRP1.
Drug resistance conferred by MDR1 and MRP in vitro is reversible after treatment with agents that have distinctly different structural and functional characteristics, such as the calcium channel blockers, calmodulin inhibitors, detergents, progestational and antiestrogenic agents, antibiotics, antihypertensives, antiarrhythmics, antimalarials, and immunosuppressives, which has been a source of clinical interest.39, 82, 93, 94 Those reversal agents bind directly to Pgp, thereby blocking the efflux of the cytotoxic drugs and increasing intracellular drug concentrations. However, clinical studies of resistance modulation have been confounded by the fact that MDR modulators, particularly MDR1 reversal agents, also enhance drug uptake in normal cells, decrease biliary elimination and drug clearance, and lead to enhanced toxicity.39, 82, 93, 94 Overall, clinical strategies aimed at reversing resistance to the vinca alkaloids with pharmacologic modulators of both MDR1 and MRP have been disappointing, most likely owing to the fact that the MDR1 expression is associated with overexpression of a large number of resistance proteins.82
Structural and functional alterations in α- and β-tubulins due to either genetic mutations, posttranslational modifications, or differential expression of tubulin isotypes, particularly the ßIII-tubulin isotype, have been identified in tumor cells with resistance to the vinca alkaloids and taxanes in both preclinical and clinical studies.95–106 These studies suggest that the ßIII-tubulin isotype could be both a prognostic and a predictive determinant of clinical benefit. Expression of ßII-tubulin, which appears to depend on TP53 suppressor function, may also relate to vinca alkaloid resistance.107
Increased expression of MAPs, particularly MAP4, has al so been associated with vinca alkaloid resistance.75 This alteration, as well as alterations in α- and β-tubulin, promotes resistance to agents that inhibit microtubule assembly by enhancing microtubule stability, possibly by promoting longitudinal inter- and intradimer interactions and/or lateral interactions between protofilaments. Furthermore, these “hyperstable” microtubules are collaterally sensitive to the taxanes (see sections titled “Taxanes, Mechanisms of Resistance”). Although such tubulin modifications have been demonstrated repeatedly in tumor cells that are continuously exposed to the vinca alkaloids in vitro, the relevance of “hyperstable tubulin” caused by alterations in tubulins or MAPs is not known.
Variability in cellular permeation and retention may also influence the formation and stability of vinca alkaloid-tubulin complexes.51, 79, 108–114 The vinca alkaloids are rapidly taken up into cells and then accumulate intracellularly, with intracellular to extracellular concentration ratios as high as 5- to 500-fold, depending on the cell type.80, 81 In murine leukemia cells, the intracellular concentrations of VCR are 5- to 20-fold higher than the extracellular concentrations, and ratios ranging from 150- to 500-fold have been reported with other vinca alkaloids.81, 108, 115 There are also marked differences in cellular uptake and retention between the vinca alkaloids, the latter of which may relate to potency and the duration of drug action. For example, VRL is more rapidly taken up and metabolized than other vinca alkaloids in isolated human hepatocytes,116 and the greater potency of VCR during exposures of short duration compared with VBL was related to the greater cellular retention of VCR in another model system. A key determinant of drug accumulation and retention is lipophilicity. The differences in the catharanthine ring of VRL render it a more lipophilic agent and increase its retention in tissues, which may explain why it is more effective at disrupting the microtubules of the mitotic spindles compared with axonal microtubules.114 The differential effects of the vinca alkaloids on axonal microtubules due to variable cellular retention and lipophilicity may explain why VRL treatment results in less neurotoxicity than other vinca alkaloids. The mechanisms responsible for the intracellular accumulation of the vinca alkaloids and other agents that bind to tubulin are not fully known, but may relate to their tubulin-binding properties and several other factors. Differential drug uptake among different tumor types may also relate to tubulin isotype expression, differential uptake and efflux, and intracellular reservoirs for drug accumulation. Lastly, pharmacokinetic differences related to the formulation vehicle appear to influence tissue uptake. For VSLI, liposomal entrapment appears to be associated with increased circulating time and tumor permeability in preclinical studies.53 Increased tumor uptake of VCR following administration of VSLI may be due, in part, to the enhanced permeability and retention effect conferred by the liposomal formulation.
Pharmacology
Cellular pharmacology
Temperature-independent nonsaturable mechanisms, analogous to simple diffusion, are responsible for most transport of the vinca alkaloids, and temperature-dependent saturable processes are less important.117 Although drug concentration and duration of treatment are determinants of drug accumulation and cytotoxicity, the duration of drug exposure above a critical threshold concentration is perhaps the most important pharmacologic determinant of cytotoxicity.80 Cytotoxicity is directly related to the extracellular concentration of drug when the duration of treatment is kept constant; for prolonged exposure to VCR, the concentration yielding 50% inhibition lies in the range of 1–5 nmol/L.118
Clinical pharmacology
The clinical pharmacology of the vinca alkaloids, which were largely studied decades ago, largely reflects the fact that sensitive, specific, and reliable analytic assays capable of measuring the minute plasma concentrations resulting from the administration of milligram quantity doses were not available. Early results may have been confounded by the use of radiolabeled drugs and assay methods. The vinca alkaloids, particularly VCR and VBL, undergo spontaneous degradation, forming degradative products that can be separated using high-pressure liquid chromatography (HPLC). Radiolabeled compounds coupled to HPLC improved separation of the various chemical moieties. Radioimmunoassay (RIA) and enzyme-linked immunosorbent assay (ELISA) methods can measure drug concentrations in the picomolar range; however, these methods cannot distinguish between the parent compounds and related derivatives and, therefore, may not provide sufficient quantitative information about degradation products and metabolites. To meet the challenge, more refined RIA and ELISA methods using mAbs with considerably greater sensitivity and specificity have been developed. Furthermore, recent technical advances in extraction and detection methodologies have increased the sensitivity and specificity of chromatographic methods, particularly tandem mass spectroscopy in conjunction with HPLC. Table 1 summarizes the pharmacokinetic characteristics of the vinca alkaloids, which generally exhibit triphasic clearance in human plasma following intravenous administration as a brief infusion. Except for VSLI, the pharmacokinetic behavior of these agents is typified by large volumes of distribution (Vd), high early clearance rates, long terminal half-lives, extensive hepatic metabolism, and biliary/fecal elimination. The relatively short α and ß phases, which are likely a result of rapid distribution and uptake and binding to tubulin in peripheral tissues and formed blood elements, are similar for all vinca alkaloids. However, the terminal (γ) phase, which is relatively long because of the avid tissue sequestration and slow release of drug, has been reported to vary by approximately fourfold.
Table 1 Vinca alkaloids: comparative pharmacokinetic and toxicologic characteristics
| Vincristine | Vinblastine | Vindesine | Vinorelbine | Vinflunine | Vincristine sulfate | |
| liposome injection | ||||||
| Standard adult dose range (mg/m2) | 1–2 (weekly) | 6–8 (weekly) | 3–4 (weekly) | 15–30 (weekly) | 320 (every 3 weeks) | 2.25 (weekly) |
| Pharmacokinetic behavior | Triphasic | Triphasic | Triphasic | Triphasic | Multiphasic | Biphasic |
| Plasma half-lives | ||||||
| α(min) | <5 | <5 | <5 | <5 | — | — |
| β(min) | 50–155 | 53–99 | 55–99 | 49–168 | — | — |
| γ(h) | 23–85 | 20–64 | 20–24 | 18–49 | 40 | 7.66a |
| Clearance (L/h/kg) | 0.16 | 0.74 | 0.25 | 0.4–1.29 | 0.64 | 0.35 (L/h) |
| Primary route | Hepatic metabolism and biliary elimination | Hepatic metabolism and biliary elimination | Hepatic metabolism and biliary elimination | Hepatic metabolism and biliary elimination | Hepatic metabolism and biliary elimination (≈66%); renal (≈33%) | Hepatic metabolism and biliary elimination |
| Principal toxicity | Neurotoxicity | Neutropenia | Neutropenia | Neutropenia | Neutropenia | Neutropenia |
| Other common toxicities | Constipation, SIADH | Alopecia neurotoxicity, mucositis | Alopecia, neurotoxicity | Neurotoxicity vomiting, constipation, mucositis | Neurotoxicity, myalgia, fatigue, anorexia, constipation, nausea/vomiting, anemia, injection site, thrombocytopenia, hyponatremia | Neurotoxicity, thrombocytopenia, anemia |
a Represents noncompartmental half-live value.
SIADH, syndrome of inappropriate antidiuretic hormone secretion.
Vincristine (VCR)
VCR is rapidly distributed to from the circulatory to the peripheral compartment following administration. There is extensive binding to both plasma proteins and formed blood elements, particularly platelets, which contain large quantities of tubulin. This served as the rationale for using VCR-laden platelets to treat disorders of platelet consumption such as refractory thrombocytopenia.38, 43 In contrast, penetration of VCR and all other vinca alkaloids across the blood–brain barrier is poor, probably because of the molecule’s large size and high substrate affinity for ABC transporter pumps that maintain the integrity of the blood–brain barrier. Following administration of conventional doses (1.4 mg/m2), given as a brief intravenous infusion, peak plasma levels approach 0.4 µmol.38, 117, 119–124 Total VCR clearance is slow, which reflects avid tissue binding and slow release, and terminal half-life (t1/2) values in the range of 23–85 h have been reported.38, 117, 119–124 VCR is metabolized and excreted primarily by the hepatobiliary system. Seventy-two hours after the administration of radiolabeled VCR, approximately 12% and 70% of the radioactivity are recovered in the urine (50% of which consists of metabolites) and feces (40% of which consists of metabolites), respectively.38, 117, 119–126 As many as 11 metabolites have been detected in humans and other species.113 The nature of the VCR metabolites identified to date, such as 4-deacetyl-VCR and N-deformyl-VCR, which have been isolated from human bile, and 4′-deoxy-3′-hydroxyVCR and 3′, 4′-epoxy-VCR N-oxide following incubation of VCR with bile, indicates that the agent is principally metabolized by hepatic cytochrome P450 CYP3A.38, 117, 119–127 There has been conflicting, albeit sparse, evidence that peak plasma concentration and systemic exposure are the principal pharmacologic determinants of neurotoxicity.127
Vinblastine (VBL)
The pharmacologic behavior of VBL is similar to that of VCR. Following rapid intravenous injection of VBL at standard doses, peak plasma drug concentrations are approximately 0.4 µmol/L.38, 117, 128 Tissue distribution is also extensive. Like VCR, binding of VBL to plasma proteins and formed elements of blood is considerable. Furthermore, distribution is rapid with t1/2 values of approximately 4 min and 1.6 h for α and ß phases, respectively. Terminal half-life values ranging from 20 to 24 h have been reported. Like VCR, VBL disposition is principally through the hepatobiliary system. The principal mode of VBL disposition is hepatic metabolism and biliary excretion. Fecal excretion of the parent compound is low, indicating that metabolism is substantial. The cytochrome P450 CYP3A isoform appears to be principally responsible for its biotransformation.116 At least one metabolite, 4-deacetyl-VBL, or VDS, which appears to be as active as the parent compound, has been identified in humans, and small quantities have been detected in both urine and feces.
Vinorelbine (VRL)
The pharmacologic behavior of VRL is similar to those of the other vinca alkaloids, and plasma concentrations decline in a biexponential or triexponential manner. Immediately after intravenous administration, plasma concentrations decline rapidly followed by much slower elimination phases (t1/2, 18–49 h).38, 46, 47, 129, 130 Plasma protein binding has been reported to range from 80% to 91%, with binding primarily to α1-acid glycoprotein, albumin, and lipoproteins. VRL is widely distributed with extensive sequestration in virtually all tissues, except the brain and testes. The drug is also extensively bound to platelets. The wide distribution of VRL may reflect the agent’s lipophilicity, which is among the highest of the vinca alkaloids. Tissue to plasma ratios range from 20 to 80, although VRL concentrations in human lung have been reported to be 300-fold greater than plasma concentrations and 3.4–13.8-fold higher than lung concentrations achieved with VDS and VCR, respectively. Hepatic metabolism and biliary excretion of metabolites and VRL are the principal modes of drug disposition.38, 46, 47, 129–132 Approximately 33–80% of the administered dose of VRL is excreted into the feces, whereas urinary excretion represents only 16–30% of total drug disposition, the majority of which is metabolized. Cytochrome P450 isoform CYP3A is principally involved in biotransformation in humans.46–48, 132–137 The principal metabolites appear to be 4-O-deacetyl-VRL, 3,6-epoxy-VRL, and several hydroxy-VRL isomers. Most metabolites are inactive. Although 4-O-deacetyl-VRL may be as active as VRL, this finding is of minor importance since concentrations are minute.
The total body clearance of VRL (1.2 L/h/kg) and mean t1/2 values of approximately 26 h were found to be the same in elderly and younger patients with normal hepatic function in one study. Clearance has been found to be adversely affected in patients who have liver metastases that involve more than 75% of the organ; clearance can be predicted in such patients by the monoethylglycinexylidide clearance test, which assesses CYP3A4 function.137 Dexamethasone has also been used as a probe of CYP3A metabolism in the assessment of VRL pharmacokinetics.131 Although VRL clearance is not accurately predicted by bilirubin concentrations in serum, markedly elevated levels have been associated with significant reductions in clearance in the few patients studied. VRL is active when given orally. In animals, 100% of radioactivity is absorbed after the ingestion of tritium-labeled VRL. The bioavailability of the parent compound in human studies is 43% and 27% for the powder-filled and liquid-filled capsules, respectively; the bioavailability of the gel-filled capsule was negligibly affected by food.129, 138, 139 Cmax values are achieved within 1–2 h after ingestion, and interindividual variability is moderate.
Vinflunine (VFL)
The pharmacokinetics of VFL have been demonstrated to be linear at clinically relevant doses (30 mg/m2 to 400 mg/m2).49, 50 VFL undergoes multiexponential decay following intravenous administration, with a rapid initial distribution phase. Similar to other vinca alkaloids, the Vd of its terminal phase is large (2.42 L or 35 L/kg), indicating wide distribution to a large tubulin-rich tissue compartment.49, 50 Total clearance is large (43.9 L/h, 0.640 L/h/kg), reflecting its initial rapid distribution and binding to tubulin-rich tissues. Its terminal t1/2 is approximately 40 h, reflecting its slow clearance from the peripheral compartment. VFL is moderately bound to human plasma proteins (67.2%). Protein binding is nonsaturable in the range of clinically achievable VFL concentrations and mainly involves high-density lipoproteins and serum albumin; binding to α1-acid glycoprotein and to platelets is negligible (<5%). Two-thirds of VFL is disposed of via hepatic metabolism and biliary excretion, whereas renal excretion accounts for one-third of administered drug.140, 141 Except for the principal metabolite, 4-O-deacetyl-VFL (DVFL) that is formed via multiple esterases, hepatic metabolism is mediated through CYP3A4. DVFL has comparable activity to the parent compound and a terminal t1/2 of about 120 days. VFL has not been demonstrated to inhibit nor induce CYP3A4 nor other CYP metabolism enzymes in in vitro studies; however, coadministration of ketoconazole resulted in 30% and 50% increases in exposures to VFL and DVFL, respectively. Despite VFL’s extensive liver metabolism and excretion, the pharmacokinetics of both VFL and DVFL were similar among 25 patients with varying degrees of liver dysfunction in one study. Nevertheless, dose adjustments for hepatic dysfunction are recommended (see section titled “Administration, Dose, and Schedule”).142 Since VFL clearance has been shown to be significantly decreased in patients ≥80 years old as compared to a control group of younger patients <70 years, dose reductions are recommended for elderly patients (≥75 years (see section titled “Administration, Dose and Schedule”).
Vincristine sulfate liposome injection (VSLI)
The pharmacokinetics of VCR following administration of VSLI are typified by much slower clearance rates from the central compartment compared to VCR administered in a conventional nonliposomal formulation.53 The lipid composition and the approximate 100 ηm mean particle size of the VSLI liposome contribute to low protein binding that result in a longer circulation time for the nanoparticle.53 In vitro protein binding assays demonstrated negligible levels of human plasma proteins absorbed to VSLI. Approximately 18–39% of encapsulated VCR was released at 24 h in vitro using human plasma. These characteristics are likely responsible for preferential accumulation of VSLI accumulation in tumors and tissues of the macrophage phagocytic system (e.g., lymph nodes, bone marrow, spleen, and liver), possibly due to the larger microvascular fenestrations in those tissues, as well as enhanced permeability and retention.143 In studies of mice with implanted human tumor xenografts, significantly faster accumulation of drug occurred in tumors than in nontumor tissues after a single dose of VSLI.144 The interstitial amounts of drug were approximately 70-fold higher in tumor tissues compared to nontumor tissues at 1 h and remained higher at 48 h. Combined, these distribution data are consistent with VSLI exiting the systemic circulation, accumulating at the site of tumors where they act as a reservoir for the release of localized VCR to enhance the anticancer activity.
In human subjects receiving treated with VSLI 2.25 mg/m2, mean plasma clearance was determined to be 345 mL/h.53 Peak plasma concentration (Cmax) values averaged 1220 ng/mL, whereas values for the elimination t1/2 averaged 7.66 h, and mean values for total and steady-state Vd were 3.57 and 2.91 L, respectively. Following intravenous administration of VSLI, fecal and urinary excretion over 96 h accounted for 69% and <8% of administered drug.
Drug interactions
The vinca alkaloids, particularly VCR and VBL, enhance methotrexate accumulation in tumor cells in vitro, an effect mediated by a vinca alkaloid-induced blockade of drug efflux; however, the minimal VCR concentrations required to achieve this effect (0.1 µmol/L) are attained transiently in vivo.145–147 The vinca alkaloids also inhibit the cellular influx and cytotoxicity of the epipodophyllotoxins in vitro, but the clinical ramifications of this effect are unknown.148 L-Asparaginase may reduce the hepatic clearance of the vinca alkaloids, particularly VCR, which may result in increased toxicity. To minimize the possibility of this interaction, it is recommended that VCR be given 12–24 h before L-asparaginase. A pharmacokinetic interaction between VFL and pegylated liposomal doxorubicin was observed, resulting in a 15–30% apparent increase and a two to threefold decrease in VFL and doxorubicin exposure, respectively, whereas for doxorubicinol, the concentrations of the metabolite were not affected. According to an in vitro study, such changes could be related to adsorption of VFL on the liposomes and a modified blood distribution of both compounds. Therefore, caution should be exercised when this type of combination is used. A possible interaction between VFL and the taxanes, paclitaxel and docetaxel, which are CYP3A substrates, has been suggested based on the results of an in vitro study.
Treatment with the vinca alkaloids has precipitated seizures associated with subtherapeutic plasma phenytoin concentrations, most likely due to the induction of CYP3A-mediated clearance of phenytoin.149–152 Reduced plasma phenytoin levels have been noted from 1 to 10 days after treatment with both VCR and VBL. Administration of the any of the vinca alkaloids with erythromycin, itraconazole, ketoconazole, ritonavir, ketoconazole, grapefruit juice, and other pharmacologic inhibitors of CYP3A may lead to severe toxicity due to reduced hepatic metabolism.41, 152, 153 Drugs that induce CYP3A4, such as rifampin, and Hypericum perforatum (St. John’s wort) should be avoided since they may accelerate the clearance of vinca alkaloids and increase drug exposure.50 Concomitantly administered drugs, such as pentobarbital and H2-histamine antagonists, may also influence VCR clearance by modulating hepatic cytochrome P450 metabolic processes.152, 154 It is important to be cognizant that concurrent use of the 3′-azido-3′-deoxythymidine (AZT) and the vinca alkaloids, such as in patients with Kaposi’s sarcoma and acquired immune deficiency syndrome, may result in reduced metabolism of AZT since the vinca alkaloids inhibit glucuronidation of AZT to its 5′-O-glucuronide metabolite.155 Based on a report of a constellation of severe toxicities, including syndrome of inappropriate antidiuretic hormone secretion (SIADH), bilateral cranial nerve palsies, peripheral neuropathy, cranial nerve palsies, heart failure, and cardiovascular effects following VCR treatment in children with ALL who had been receiving treatment with nifedipine and itraconazole, it is possible that these medications may enhance the neurologic and cardiovascular effects of the vinca alkaloids.154 Lastly, the significant interindividual and intraindividual variability of VCR pharmacokinetics in children has been attributed to the variable induction of P450 metabolism due to concurrent use of P450-inducing corticosteroids.150, 151
Mitomycin C combined with the vinca alkaloids has been associated with pulmonary toxicity (see sections titled “Toxicity, Miscellaneous”).
Toxicity
The principal toxicities of the vinca alkaloids differ despite their structural and pharmacologic similarities. Neurotoxicity is the predominant toxicity of VCR, whereas myelosuppression predominates with VBL, VDS, VRL, VFL, and VSLI. However, peripheral neurotoxicity is noted after cumulative treatment with VBL, VDS, VRL, VFL, and VSL and in patients who are inherently susceptible.
Neurotoxicity
Although the vinca alkaloids are similar from a structural standpoint, their toxicologic profiles differ significantly. All of the vinca alkaloids induce a characteristic peripheral neurotoxicity, but VCR is most potent in this regard. The neurotoxicity is principally characterized by a peripheral, symmetric mixed sensory motor and autonomic polyneuropathy.37, 38, 41, 48, 156–160 The primary pathologic effects are axonal degeneration and decreased axonal transport, most likely caused by a drug-induced perturbation of microtubule function. At onset, only symmetric sensory impairment and paresthesia in a length-dependent manner, initially in the distal extremities, are often encountered. Neuritic pain and loss of deep tendon reflexes may develop with continued treatment, which may be followed by foot drop, wrist drop, motor dysfunction, ataxia, and paralysis. Back, bone, and limb pains may occur. Electrophysiologic studies typically reveal normal nerve conduction velocities; however, diminished amplitude of sensory and motor nerve action potentials and prolonged distal latencies, resembling axonal degeneration, may be noted.156–160 Rarely, cranial nerves are affected, resulting in hoarseness, diplopia, jaw pain, facial palsies, and laryngeal paralysis.38, 161, 162 The uptake of VCR into the brain is low, and the central nervous system effects, such as confusion, mental status changes, depression, hallucinations, agitation, insomnia, seizures, coma, SIADH, and visual disturbances, may occur in rare situations.38, 48, 163–165 Auditory effects, possibly secondary to disruption of the medial olivocochlear bundle, have also been reported. Acute, severe autonomic neurotoxicity is uncommon but may arise as a consequence of high-dose therapy (>2 mg/m2) or in patients with diminished drug clearance because of altered hepatic function.38, 158–160, 164–169 Toxic manifestations of autonomic neurotoxicity include constipation, abdominal cramps, paralytic ileus, urinary retention, orthostatic hypotension, and hypertension.38, 156–159, 166–169 Acute neurotoxic manifestations resembling Guillain–Barré syndrome have also been reported.160 In adults treated with VCR, the neurotoxic effects may begin with cumulative doses as little as 5–6 mg, and manifestations may be cumulative doses of 15–20 mg. Neurotoxic manifestations are generally cumulative and resolve slowly after treatment, often requiring many years. Neurotoxicity occasionally worsens for a short time following treatment.170 Although it has been remarked that children are less susceptible to neurotoxicity than adults and that the elderly are particularly prone, these apparent age-related differences may, in fact, be caused by previously inaccurate dose calculation using body weight in children and adults and using body surface area in infants. In infants, VCR doses are calculated now according to body weight, which may be more accurate from a pharmacologic standpoint because of ubiquitous tissue distribution. Patients with antecedent neurologic disorders, such as Charcot–Marie–Tooth disease, hereditary and sensory neuropathy type 1, Guillain–Barré syndrome, and childhood poliomyelitis, are highly predisposed to neurotoxicity.171–173 VCR treatment in patients with hepatic dysfunction or obstructive liver disease is associated with an increased risk of developing neuropathy because of impaired drug metabolism and delayed biliary excretion.174, 175 An inherited polymorphism in the promoter region of CEP72, which encodes a centrosomal protein involved in microtubule formation, was associated with increased risk and severity of VCR-related peripheral neuropathy.176 If replicated in additional populations, this finding may provide a basis for safer dosing of this widely prescribed anticancer agent.
The only known effective intervention for vinca alkaloid neurotoxicity is discontinuing treatment or reduction of the dose or frequency of drug administration.166, 176, 177 Although several antidotes, including thiamine, vitamin B12, folinic acid, pyridoxine, and neuroactive agents (e.g., sedatives, tranquilizers, anticonvulsants), have been used, these treatments have not been undoubtedly shown to be consistently effective.38, 127, 178–180 Folinic acid protects mice against otherwise lethal doses of the vinca alkaloids, and there are anecdotal reports of its successful use following VCR overdosage in man; however, prospective studies have never been performed. Suggested dosages for folinic acid for the treatment of overdosage are 15 mg every 3 h for 24 h and then every 6 h for at least 48 h.38 There have been promising results with other neuroprotective agents. In one randomized double-blind trial, coadministration of glutamic acid and VCR appeared to reduce neurotoxicity.180 An adrenocorticotropin analog (ORG 2766) also demonstrated protection against VCR-induced neuropathy in an animal model and in a double-blind, placebo-controlled pilot study in patients.38 Several other agents, including nerve growth factor, insulin growth factor-1, and amifostine, appear to alter the course of neurotoxicity in experimental models, but there is no definitive evidence that these agents may be effective in the clinic. Severe neurotoxicity is observed less frequently with VBL, VDS, VRL, and VFL, as compared with VCR.114, 181 VRL has a lower affinity for axonal microtubules than either VCR or VBL, which seems to be confirmed by clinical observations.36, 38, 48, 114, 181 Characteristic vinca alkaloid neurotoxicity occurs with VSLI, but the agent has largely been evaluated in patients who have previously received VCR, and the relative neurotoxic effects of VCR and VSLI have not been studied.
Myelosuppression
Neutropenia is the principal dose-limiting toxicity of VBL, VDS, VRL, and VFL. It is also common with VSLI. Thrombocytopenia and anemia are usually less common and less severe. The onset of neutropenia is usually 7–11 days after treatment, and recovery is generally by days 14–21. Myelosuppression is not typically cumulative. Although VCR is rarely associated with hematologic toxicity, severe myelosuppression has been observed in situations, resulting in increased drug exposure, such as following inadvertent overdose, and hepatic insufficiency.
Gastrointestinal
Gastrointestinal toxicities, aside from those caused by autonomic dysfunction, may also occur.37, 38, 41, 48, 127, 182, 183 Gastrointestinal autonomic dysfunction, as manifested by bloating, constipation, and abdominal pain, occurs most commonly with VCR, high doses of the other vinca alkaloids, or settings associated with high drug exposure (e.g., hepatic dysfunction, overdosages). An initial manifestation of autonomic dysfunction due to slow intestinal transit may be impaction of stool in the upper colon. An empty rectum may be noted on digital examination, or an abdominal radiograph may be useful in diagnosing this condition, which may be responsive to high enemas and laxatives. A routine prophylactic regimen to prevent constipation is therefore recommended for all patients receiving VCR. Severe autonomic toxicity may lead to paralytic ileus, intestinal necrosis, and even intestinal perforation. The ileus, which may mimic a “surgical abdomen,” usually resolves slowly with conservative measures after termination of treatment. Patients who receive high dosages of VCR or have hepatic dysfunction may be especially prone to develop severe gastrointestinal complications due to autonomic neurotoxicity. Although success with drugs used prophylactically to minimize toxicity, including lactulose, cerulein, metoclopramide, and a cholecystokinin analog (sincalide), has been reported anecdotally, these agents also may alter the pharmacokinetic behavior of the vinca alkaloids by affecting biliary excretion and/or enterohepatic recirculation, which may ultimately result in increased drug clearance.38
Mucositis, stomatitis, and pharyngitis occur more frequently with VBL than VCR, but not uncommon with VFL, VRL, and VDS. Nausea, vomiting, and diarrhea may also occur to a lesser extent. Asymptomatic and brief elevations in serum levels of hepatic transaminases and alkaline phosphatase have been noted. Pancreatitis has also been reported rarely with VRL.182
Miscellaneous
All vinca alkaloids should be considered potent vesicants and may cause profound tissue damage if extravasation occurs. This warning should apply to VSLI as there is no evidence that the formulation protects against tissue damage if extravasation occurs. If extravasation is suspected, treatment should be discontinued immediately and aspiration of any residual drug remaining in the tissues should be attempted.38, 184–189 In experimental studies, cold packs have been shown to increase toxicity, while heat appears to limit damage. The application of local heat immediately for 1 h four times daily for 3–5 days and the injection of hyaluronidase, 150–1500 units (15 units/mL in 6 mL of 0.9% sodium chloride solution) subcutaneously, through 6 clockwise injections in a circumferential manner using a 25-gauge needle and changing the needle with each injection) into surrounding tissues are the treatment of choice in minimizing both discomfort and latent cellulitis.187–189 The use of leucovorin, diphenhydramine, hydrocortisone, isoproterenol, sodium bicarbonate, and vitamin A cream has been ineffective in animal models.185 An immediate surgical consultation to consider early debridement is recommended. Discomfort and signs of phlebitis may also occur along the course of an injected vein, with resultant sclerosis, but phlebitis may be minimized if the vein is adequately flushed after treatment.
Mild and reversible alopecia occurs in approximately 10% and 20% of patients treated with VRL and VCR, respectively. Acute cardiac ischemia, chest pains without evidence of ischemia, fever without an obvious source, Raynaud’s phenomenon, and palmar–plantar erythrodysesthesia (hand–foot syndrome) have also been reported.190–192 Respiratory reactions, characterized by dyspnea, is noted in approximately 5% of patients, particularly when the vinca alkaloids are combined with mitomycin C.193, 194 The onset of pulmonary toxicity may be acute, with bronchospasm and dyspnea as the predominant manifestations, resembling an allergic reaction. The second type of toxicity is a subacute reversible reaction associated with cough and dyspnea and occasionally with interstitial infiltrates. It typically occurs within 1 h of treatment. Corticosteroids may be beneficial in severe cases, and several patients have been re-treated without complications. There is no clear evidence that implicates VRL as a cause of chronic pulmonary toxicity.
All vinca alkaloids have been implicated in causing SIADH, and patients receiving vigorous hydration are particularly prone to severe hyponatremia due to SIADH.36–38, 48, 127 Plasma levels of antidiuretic hormone, which generally remit within 3 days’ posttreatment, have been noted. Hyponatremia generally responds to fluid restriction. VBL may cause photosensitivity reactions, possibly due to corneal irritation, and muscular effects.195 The inadvertent intrathecal administration of the vinca alkaloids causes an ascending myeloencephalopathy that is usually fatal. Reports of immediate cerebrospinal fluid withdrawal and lavage with Ringer’s lactate solution supplemented with fresh frozen plasma (15 mL/L) at a rate of 55 mL/h for 24 h have provided encouraging results, with two affected patients surviving with paraplegia, albeit intact cerebral function.196 To prevent this error, intrathecal methotrexate and intravenous VCR setting should not be administered in close temporal proximity, and the drugs should not be delivered together to staff.197 Hepatic transaminase elevations have also been reported, particularly with VSLI.
Administration, dose, and schedule
It is recommended that the vinca alkaloids be administered by rapid intravenous injection. Inadequate flushing following treatment may increase the risk of phlebitis and injection site reactions, and, therefore, the catheter should not be removed before the vein is flushed. VCR is commonly administered to children weighing more than 10 kg as a bolus intravenous injection at a dosage of 1.5–2.0 mg/m2 weekly, although 0.05–0.65 mg/kg weekly is commonly used in smaller children. For adults, the conventional weekly dose is 1.4–2.0 mg/m2. A restriction of the absolute dose of VCR to 2.0–2.5 mg in children and 2.0 mg in adults, which is often referred to as “capping,” has been generally adopted based on early reports of gastrointestinal toxicity (autonomic neuropathy) in patients treated at higher doses. However, there is little factual evidence to support this practice, and available evidence suggests that it should be reconsidered, particularly in light of the wide interpatient variability in pharmacokinetic behavior and tolerance. There is significant interpatient variability in the clearance of VCR (up to 11-fold), and some patients are able to tolerate much higher doses with little or no toxicity. Moreover, the safety and efficacy of treatment regimens that do not employ capping at 2.0 mg have been documented in adults.198 In any case, doses should not be reduced for mild peripheral neurotoxicity, particularly if VCR is being used in a potentially curative setting. Instead, doses should be modified for manifestations of more serious neurotoxicity, including severe symptomatic sensory changes, motor and cranial nerve deficits, and ileus, until toxicity resolves. In clearly palliative situations, dose reductions, lengthening dosing intervals, or selecting an alternative agent may be justified in the event of moderate neurotoxicity. A routine regimen to prevent serious consequences of severe autonomic toxicity, particularly constipation, is also recommended.
The recommended dose of VSLI is 2.25 mg/m2 intravenously every 7 days. VSLI is prepared by mixing three components from a kit supplied by the manufacturer, which includes VCR sulfate, sodium phosphate, and sphingomyelin/cholesterol liposome injection. Following the manufacturer’s procedure, VSLI should appear as a white to off-white, translucent suspension, essentially free of visible foreign matter and aggregates, comprised of sphingomyelin/cholesterol liposomes, with an approximate liposome mean diameter of 100 nm. Greater than 95% of the drug is encapsulated in the liposome. The procedure results in a solution that is diluted with either 5% dextrose injection or 0.9% sodium chloride injection to a final volume of 100 mL that is infused intravenously over 1 h.
The most common VBL schedule in chemotherapy regimens is a rapid intravenous injection at a dose of 6 mg/m2 weekly. Approved weekly dosing recommendations are 2.5 and 3.7 mg/m2 for children and adults, respectively, followed by gradual escalation in increments of 1.8 and 1.25 mg/m2 weekly based on hematologic tolerance. It is recommended that weekly VBL doses of 18.5 mg/m2 in adults and 12.5 mg/m2 in children not be exceeded as a single agent; however, these doses are much higher than most patients can tolerate because of myelosuppression, even on less frequent administration schedules. Because the severity of leukopenia that may occur with identical VBL doses varies widely, VBL should probably not be given more frequently than once each week. Five-day continuous infusions of VBL have been used at dosages ranging from 1.5 to 2.0 mg/m2 per day, which achieves biologically relevant plasma concentrations of approximately 2.0 nmol/L, but the overall advantages of using protracted infusions for a drug that is widely distributed and avidly bound to peripheral tissues are unclear.
VRL is usually administered at a dosage of 30 mg/m2 on a weekly or biweekly schedule as a 6–10-min intravenous injection through a side-arm port into a running infusion (alternatively, a slow bolus injection followed by flushing the vein with 5% dextrose or 0.9% sodium chloride solutions) or as a short infusion over 20 min. It appears that more rapid infusions are associated with less local venous toxicity. Oral dosages of 80–100 mg/m2 given weekly are generally well tolerated, but an acceptable oral formulation has not been approved. Other dosing schedules that have been evaluated include chronic oral administration of low doses, intermittent high doses, and prolonged intravenous infusion schedules.
The recommended dose of VLF is 320 mg/m2 as a 20-min intravenous infusion every 3 weeks. A reduced dose of 280 mg/m2 is recommended for patients with World Health Organization or Eastern Cooperative Oncology Group performance status of 1 or 0 and a history of pelvic irradiation. In the absence of hematological toxicity in the first cycle causing treatment delay or dose reduction, the dose should be increased to 320 mg/m2 for the subsequent cycles.
Because of their remarkable vesicant properties, the vinca alkaloids should not be administered intramuscularly, subcutaneously, intravesically, or intraperitoneally. Direct intrathecal injection of VCR and other vinca alkaloids inadvertently has induced a severe myeloencephalopathy characterized by ascending motor and sensory neuropathies, encephalopathy, and death (see sections titled “Toxicity, Neurotoxicity”).
Although specific dosing guidelines for patients with hepatic dysfunction have not been thoroughly formulated, the major role of the liver in the disposition of the vinca alkaloids implies that dose modifications should be considered for patients with hepatic dysfunction, particularly hepatic excretory dysfunction. For VCR, VRL, VDS, and VBL, a 50% dose reduction is often recommended for patients with total bilirubin levels between 1.5 and 3.0 mg/dL (50% dose reduction for bilirubin levels between 2.0 and 3.0 mg/dL is recommended for VRL), as well as at least a 75% dose reduction for plasma total bilirubin levels above 3.0 mg/dL. Dosing guidelines for patients with hepatic dysfunction have not been formulated, and therefore conservative measures, perhaps similar to the aforementioned guidelines for nonencapsulated vinca alkaloids, may be prudent since hepatic metabolism and biliary excretion are the principal processes involved with the disposition of VSLI. However, the pharmacokinetics of VSLI were evaluated in patients with moderate hepatic dysfunction (Child–Pugh B) and the dose-adjusted maximum plasma concentration and exposure were comparable to those with normal hepatic function.49 The recommended doses of VFL are 250 and 200 mg/m2 given once every 3 weeks for patients with mild (Child–Pugh grade A) and moderate (Child–Pugh grade B) liver impairment, respectively, as defined further in the prescribing information.
Dose reduction for renal impairment is not indicated for any of the vinca alkaloids except for VFL in which the recommended starting doses are 280 and 250 mg/m2 every 3 weeks for patients with moderate (creatinine clearance rate between 40 and 60 mL/min) and severe (creatinine clearance rate between 20 and 40 mL/min) renal impairment, respectively.53
VFL dose reductions are also recommended for elderly patients since drug clearance relates to age. In patients whose ages are between 75 and 80 years and for patients whose age exceeds 80 years, the recommended dose is 280 and 250 mg/m2 every 3 weeks, respectively.53
Taxanes: Introduction and indications
Although the taxanes affect microtubules, their principal mechanisms of action, pharmacology, clinical indications, and toxicities substantially differ from those of the vinca alkaloids. Interest in the taxanes began in 1963, when a crude extract of the bark of the Pacific yew tree, Taxus brevifolia, demonstrated notable anticancer activity in preclinical studies. In 1971, Wall and colleagues identified paclitaxel as the active constituent of the bark extract; however, the limited supply of its primary source that was exclusively derived from the Pacific yew tree, the difficulties inherent in large-scale isolation, extraction, and preparation of bulk compound, and its poor aqueous solubility delayed its development.199 Interest was maintained after characterization of its novel mechanism of action and the availability of an adequate supply for requisite preclinical and limited clinical studies. The early search for taxanes derived from more abundant and renewable resources led to the semisynthesis of docetaxel by the addition of a side chain to 10-deacetylbaccatin III, an inactive taxane precursor found in the needles and other components of more abundant yew species.200 The supply of paclitaxel is no longer preclusive due to the development of commercially feasible semisynthetic processes. A wide variety of taxane analogs, as well as paclitaxel formulations, have been evaluated. The most recent to be broadly registered is protein-bound paclitaxel particles for injection (PBPPI), sometimes called “nab-paclitaxel” or nanoparticle albumin-bound paclitaxel, which is an albumin-bound formulation of paclitaxel.201
Figure 3 shows the structures of paclitaxel, docetaxel, and cabazitaxel, which are complex esters consisting of a 15-member taxane ring system linked to an unusual 4-member oxetane ring. The taxane rings of both paclitaxel and docetaxel, but not 10-deacetylbaccatin III, are linked to an ester side chain attached to the C-13 position of the ring, which is essential for antimicrotubule activity.202 The structures of paclitaxel and docetaxel differ in substitutions at the C-10 taxane ring position and on the ester side chain attached at C-13, which render docetaxel slightly more water soluble and potent than paclitaxel. However, the clinical ramifications of these differences are not entirely clear. Another more recent addition to the therapeutic armamentarium is cabazitaxel, a dimethoxy derivative of docetaxel that is partially synthesized into a single diastereomer from 10-deacetylbaccatin III.203 Cabazitaxel is more potent and has lower substrate affinity for Pgp and other transmembrane drug efflux pumps than docetaxel, which, in part, explain its notable activity in docetaxel-resistant cancers.204
Figure 3 Chemical structures of the taxane: paclitaxel, docetaxel, and cabazitaxel.
Clinical indications
Paclitaxel initially received regulatory approval in the United States and elsewhere for the treatment of patients with ovarian cancer after failure of first-line or subsequent chemotherapy. Its use in combination with a platinum compound as primary induction therapy in suboptimally debulked stages III or IV ovarian cancer has demonstrated a survival advantage over preexisting standard therapy in randomized phase III studies.205 In the United States, paclitaxel is also indicated for treatment of patients with metastatic breast cancer after failure of combination chemotherapy for metastatic disease or at relapse within 6 months of adjuvant chemotherapy, with prior therapy that includes an anthracycline.206 Paclitaxel is also indicated for the adjuvant treatment of node-positive breast cancer administered sequentially to standard doxorubicin-containing combination chemotherapy. In the clinical trial, there was an overall favorable effect on disease-free and overall survival in the total population of patients with both hormone receptor-positive and hormone receptor-negative tumors, but the benefit has been irrefutably demonstrated only in patients with hormone receptor-negative cancers.207–209 Compelling results have been noted following treatment of patients with both metastatic breast cancer and high-risk early-stage breast cancer with alternative taxane-containing regimens, particularly “dose-dense” and weekly regimens; however, these efficacy results should not be generalized to other indications and/or other taxanes.210, 211 Weekly administration schedules using paclitaxel and other taxanes have been associated with lower incidences of some adverse effects, particularly myelosuppression, compared to less frequent dosing schedules.
Various therapeutics received regulatory approval as components of paclitaxel-based combination regimens. The combination of gemcitabine and paclitaxel demonstrated superior survival to paclitaxel alone in the first-line treatment of metastatic breast cancer, and gemcitabine received regulatory approval for use with paclitaxel in this setting. Similarly, the combination of trastuzumab and paclitaxel is indicated in the first-line treatment of women with HER-2-amplified metastatic breast cancer.212 Paclitaxel is also indicated for adjuvant treatment of HER-2-amplified lymph node positive or negative (hormone receptor negative or with at least one high-risk feature) as part of a regimen consisting of doxorubicin, cyclophosphamide, and trastuzumab.213, 214 Paclitaxel has also received regulatory approval in the United States for second-line treatment of Kaposi’s sarcoma associated with the acquired immunodeficiency syndrome and in combination with cisplatin as primary treatment advanced stage IIIB and stage IV nonsmall cell lung cancer patients who are not candidates for radiation therapy or potentially curative surgery.215, 216 Bevacizumab, in combination with paclitaxel and carboplatin, is approved for the treatment of stage IIIB and IV nonsquamous nonsmall cell lung cancer.217
Docetaxel initially received regulatory approval in the United States and elsewhere for patients with metastatic or locally advanced breast cancer after failure of anthracycline-based chemotherapy, which was later broadened to a more general indication following chemotherapy failure.200 Subsequently, the combination of capecitabine and docetaxel demonstrated superior overall survival over docetaxel alone after failure of an anthracycline in patients with locally advanced or metastatic breast cancer, and capecitabine as a component of this doublet received regulatory approval.218 Regulatory approval has also been granted for docetaxel combined with cyclophosphamide and doxorubicin as adjuvant treatment for locally advanced breast cancer.219 In nonsmall cell lung cancer, docetaxel initially is approved for treatment of unresectable locally advanced or metastatic disease after demonstrating increased survival after failure of cisplatin-based therapy, and the combination of docetaxel and cisplatin was later granted regulatory approval as first-line treatment of such patients.220 Regulatory approval was granted for docetaxel plus prednisone in patients with hormone-refractory prostate cancer as the regimen demonstrated superior overall survival compared to mitoxantrone plus prednisone.221 Similarly, the taxanes have notable activity in the treatment of squamous cell carcinoma of the head and neck, and docetaxel combined with cisplatin and 5-fluorouracil has received regulatory approval in the neoadjuvant treatment of patients with locally advanced disease.222 Docetaxel combined with cisplatin and 5-fluorouracil is approved for patients with untreated advanced gastric and gastroesophageal junction adenocarcinoma, whereas paclitaxel combined with ramucirumab is indicated for patients with relapsed or refractory disease.223, 224
Cabazitaxel plus prednisone is indicated for treatment of patients with metastatic castrate-resistant prostate cancer previously treated with a docetaxel-containing regimen since the combination showed improved overall survival compared to mitoxantrone plus prednisone in this indication.225
PBPPI, the newest taxane to receive regulatory approval in the United States and elsewhere, is a formulation of paclitaxel in 3–4% human serum albumin, similar to the concentration of albumin in the blood.201 PBPPI was initially approved in patients with metastatic breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy.226 PBPPI plus carboplatin is indicated in patients with locally advanced or metastatic nonsmall cell lung cancer based on a randomized trial demonstrating that the objective response rates were higher with the regimen compared to paclitaxel plus carboplatin; overall survival was similar.227 Additionally, PBPPI combined with gemcitabine is indicated in the first-line treatment of metastatic adenocarcinoma of the pancreas, as the regimen demonstrated increased overall survival compared to gemcitabine alone.201
It is important to note that the anticancer spectra for paclitaxel, docetaxel, cabazitaxel, and PBPPI are identical, with modest activity noted in many other cancers including endometrial, bladder, small cell lung, and germ cell carcinoma, lymphoma, and melanoma. Differences in clinical efficacy endpoints and regulatory indications between these taxanes may reflect different regulatory strategies, study designs, and dose schedules and not necessarily the inherent superiority of any specific taxane. In contrast to the vinca alkaloids, the taxanes do not seem to have relevant activity in pediatric malignancies.
Mechanisms of action
The binding site for paclitaxel on microtubules is different from the binding sites for exchangeable GTP, colchicine, podophyllotoxin, and the vinca alkaloids. Paclitaxel binds to the N-terminal 31 amino acids of the β-tubulin subunit of tubulin polymers; however, other sites may be involved.199, 228–234 Paclitaxel binds in a pocket that is lined by several hydrophobic residues on the luminal side of the microtubule wall, roughly in the middle of the monomer along the protofilament direction.3–7, 199, 234, 235 Docetaxel, which most likely shares the same binding site as paclitaxel, appears to have a 1.9-fold higher affinity for the site, and the tubulin assembly process induced by docetaxel proceeds with a critical protein concentration that is 2.1-fold lower than that of paclitaxel.233, 236–240 However, it is not clear whether these differences translate into increased therapeutic indices for docetaxel in the clinic since higher potency may result in more severe toxicity at identical drug concentrations. Nevertheless, the results of both preclinical and clinical studies indicate that the taxanes may not be completely cross-resistant in individual tumors, but these results may reflect differences in delivered dose and schedule. Nevertheless, the anticancer spectra of paclitaxel, docetaxel, and other taxanes are nearly identical. Although both the vinca alkaloids and taxanes produce similar disruptive effects on the mitotic spindle apparatus, their principal mechanisms of action differ. Following binding to the β-tubulin subunit, the taxanes stabilize lateral contacts between microtubule protofilaments by electrostatic interactions.7 In effect, this interaction alters the tubulin rate dissociation constants at both ends of the microtubules, reduces the critical tubulin concentration required for microtubule assembly, and promotes both nucleation and elongation of the polymerization reaction, thereby stabilizing microtubules against depolymerization and enhancing polymerization.2–7, 55, 239–245
Binding of the taxanes to polymerized tubulin strengthens the microtubule. At substoichiometric concentrations, the taxanes suppress microtubule dynamics without appreciably increasing the rate of formation of polymerized tubulin.2–7, 55, 239–245 At much higher concentrations, which are achieved in the clinic, the taxanes induce both tubulin polymerization and increased microtubule mass. The microtubules of taxane-treated cells are extraordinarily stable, resisting depolymerization by cold, calcium, GTP, and depolymerizing agents like the vinca alkaloids and colchicine. These actions result in the suppression of treadmilling and dynamic instability, which are essential for normal microtubule dynamics during both the mitotic and nonmitotic phases of the cell cycle.2, 3, 13, 17, 20 Both stoichiometric and substoichiometric binding of the taxanes inhibit the proliferation of cells, principally by inducing a sustained mitotic block at the metaphase/anaphase boundary; however, morphologic changes, such as microtubule bundle formation during the nonmitotic phases of the cell cycle, suggest that the interphase microtubules are also affected. Nevertheless, perturbations in microtubule dynamics are most relevant during mitotic spindle formation, and therefore cells are most sensitive to the taxanes in mitosis.
Perturbations of mitotic processes and the induction of mitotic arrest by the taxanes trigger apoptosis; however, the precise mechanisms by which these perturbations result in cell death are not entirely clear. Nevertheless, the disruptive effects of the taxanes on the formation of functional mitotic spindle, which is responsible for chromosome segregation, ultimately activates the mitotic checkpoint, the principal cell-cycle control mechanism in mitosis that prevents missegregation of the chromosomes. The mitotic checkpoint delays separation of the chromosomes, which enter mitosis as replicated pairs of sister chromatids, until each pair has made stable attachments to both poles of the mitotic spindle. The taxanes impede attachment of the mitotic spindle to the kinetochores, which activate a signal transduction cascade that delays mitotic progression by inhibiting the anaphase-promoting complex.22, 246–248. Paclitaxel-treated cells ultimately enter anaphase and undergo mitosis, but the presence of unattached kinetochores result in the division of chromosomes in multiple directions; chromosome segregation is random due to multipolar division followed by partial cytokinesis failure. The resultant daughter cells are aneuploid, and a portion of these cells die, presumably due to loss of one or more essential chromosomes.
Microtubule perturbations induced by the taxanes trigger a delay in the mitotic checkpoint, which ultimately results in cell death via the intrinsic pathway of apoptosis.22, 75, 244–262 This process involves various regulatory constituents such as the tumor suppressor gene TP53, inhibitors of cyclin-dependent kinases (e.g., p21/Waf-1), and regulators of other protein kinases.244, 245, 262 As a consequence, cells are arrested in G2/M, after which time they either undergo apoptosis or traverse through G2/M and ultimately divide.248, 259 The apoptotic initiating events include activation of the proapoptotic molecules Bax and Bad, as well as inactivation of the antiapoptotic regulators Bcl-2 and BclxL.248, 259–261 Various kinases have been implicated in the inactivation of Bcl-2 induced by the taxanes and other antimicrotubule agents, including Jun N-terminal kinase and its proapoptotic effectors (e.g., Bim, c-Raf, extracellular signal-regulated kinase 1 and 2, apoptosis signal-regulated kinase, cyclin-dependent kinase 1 (CDK-1), cAMP-dependent protein kinase A, and protein kinase C).69, 250, 260–264 Inactivation of Bcl-2 family members and activation of proapoptotic molecules stimulate the intrinsic pathway of apoptosis and downstream effector caspases.257, 259 The taxanes may also induce cell death independent of caspase activation.241, 265
The taxanes also perturb interphase microtubules in nonproliferating cells as manifested by the formation of microtubule bundles.248 Paclitaxel has been reported to induce transcription factors and enzymes that mediate proliferation, apoptosis, and inflammation.252, 262, 266, 267 The taxanes also enhance the effects of ionizing radiation in vitro and in vivo, most likely by inhibiting cell-cycle progression in G2/M, which is the most radiosensitive phase of the cell cycle.268–272 In addition, the taxanes inhibit angiogenesis at concentrations below those that induce cytotoxicity, but the contribution of these antiangiogenic effects to the anticancer effects of the taxanes is not entirely clear.258, 273, 274
Similar to the vinca alkaloids, the taxanes induce many other cellular effects that may or may not relate to their disruptive effects on microtubule dynamics. Although the taxanes primarily block cell-cycle traverse in the mitotic phases, the agents prevent G0– to S-phase transition.260, 275 Explanations that have been proposed to account for the nonmitotic actions of the taxanes include disruptive effects on tubulin in the cell membrane, the interphase cytoskeleton, and microtubules that are involved in growth factor signaling and vascular endothelial growth factor expression through reactive oxygen species production and/or hypoxia inducible factor-1α.38, 276, 277
The taxanes perturb nonmalignant, as well as malignant, tissues. These effects are mediated, at least, in part, by disrupting microtubule dynamics. Paclitaxel inhibits relevant morphologic and biochemical processes in human neutrophils, including chemotaxis, migration, spreading, polarization, hydrogen peroxide generation, and killing of phagocytized microorganisms.38 Paclitaxel also antagonizes the effects of microtubule-disrupting drugs on lymphocyte function and cAMP metabolism and inhibits the proliferation of stimulated human lymphocytes. Paclitaxel mimics the effects of endotoxic bacterial lipopolysaccharide on macrophages, which results in a rapid decrement in tumor necrosis factor-α (TNF-α) receptors and TNF-α release.261, 266, 267 The agent also induces expression of the gene for TNF-α, but these activities are not related to paclitaxel’s disruptive effects on microtubule assembly, which suggests that cytokine activation, in part, confers the anticancer activities of the taxanes.266 In addition, paclitaxel inhibits chorioretinal fibroblast proliferation and contractility in models of proliferative retinopathy, as well as neointimal smooth muscle cell proliferation after cardiac and peripheral vascular angioplasty.278, 279 Cardiac and femoropopliteal arterial stents coated with paclitaxel to prevent restenosis from proliferation of neointimal tissue received regulatory approval in the United States and elsewhere.280, 281 Finally, paclitaxel inhibits secretory functions in many specialized cells, such as insulin secretion in isolated rat islets of Langerhans, protein secretion in rat hepatocytes, and the nicotinic receptor-stimulated release of catecholamines from chromaffin cells of the adrenal medulla.38
Although PBPPI likely affects the microtubules’ malignant and nonmalignant tissues similar to conventional formulations of paclitaxel, the binding of paclitaxel to albumin in PBPPI increases accumulation of paclitaxel molecules in tumor tissue. Albumin has several characteristics that make it an attractive drug vehicle for cancer drugs. It is a natural carrier of endogenous hydrophobic molecules that are bound in a reversible noncovalent manner and facilitates endothelial transcytosis of protein-bound and unbound plasma constituents principally by binding to a 60-kDa glycoprotein (gp60) cell surface receptor. Following binding of albumin to gp60, the complex then interacts with caveolin-1, an intracellular protein, with subsequent formation of transcytotic vesicles (caveolae) that transport the complex to the intracellular compartment. Osteonectin, which is also known as secreted protein acidic and rich in cysteine (SPARC), also binds albumin because of a sequence homology with gp60.201, 282–284 Similar to caveolin-1, SPARC is expressed in several malignancies, including breast, lung, and prostate cancer, as well as by the stroma of these and others, which may account for the intratumoral accumulation of albumin, as well as the accumulation of albumin-bound drugs like PBPPI.201, 282–284 However, SPARC expression in tumor and/or stroma has yet to be definitely shown to predict for the anticancer activities of PBPPI.201, 282–286
PBPPI, which was developed as an alternate paclitaxel formulation to polyoxyethylated castor oil, is an albumin-bound, 130-ηm particle formulation of paclitaxel, free from any solvent. It is used as a colloidal suspension derived from the lyophilized formulation of paclitaxel and human serum albumin. Human serum albumin stabilizes the drug particle at an average size of 130 ηm to prevent capillary obstruction, and the lack of a solvent requires no particular premedication nor infusion system.201, 282–284 Further, preclinical studies conducted in athymic mice with human tumor xenografts have shown that PBPPI delivers higher paclitaxel concentrations to tumor cells, resulting in greater anticancer activity than equivalent doses of paclitaxel in polyoxythylated castor oil. SPARC–albumin interactions may also be responsible for more rapid systemic distribution and clearance of paclitaxel than poloxyethylated castor oil formulations.
Mechanisms of resistance
Two general types of mechanisms of acquired taxane resistance have been described in cells made resistant by prolonged drug treatment at low concentrations. The best characterized mechanism is the MDR phenotype, which can be mediated by several ABC multidrug transporter family members. The most important ABC transporter with respect to conferring taxane resistance is the 170-kDa Pgp efflux pump or the MDR1-encoded gene product MDR1 (ABC subfamily B1; ABCB1) and MDR2 (ABC subfamily ABCB4) (see sections titled “Vinca Alkaloids, Introduction and Indications” and “Vinca Alkaloids, Mechanisms of Resistance”).83, 86, 87, 233, 287, 288 In addition, low-level taxane resistance appears to be conferred by the bile salt export pump (BSEP) (also known as ABCC11).293, 294 Unlike the vinca alkaloids, ABCC1 (MRP1) and ABCC2 (MRP2) do not appear to be involved in transporting the taxanes.289–292 In murine systems, the particular species of Pgp found in paclitaxel-resistant cells is similar, but not identical, to that found in VBL and colchicine-resistant cells derived from the same parental line.92, 287, 288 These cells are cross-resistant with many other natural products, and resistance to both paclitaxel and docetaxel conferred by MDR can be reversed by many classes of membrane-active drugs, including the calcium channel blockers, tamoxifen, cyclosporine A, and antiarrhythmic agents.287, 291–294 Even the principal component of the formulations of paclitaxel and docetaxel—polyoxyethylated castor oil and polysorbate-80, respectively—can reverse taxane resistance; however, whereas the plasma concentrations of polyoxyethylated castor oil achieved with paclitaxel are sufficient to reverse MDR, sufficient modulatory concentrations of polysorbate-80 are not achieved with docetaxel in the clinic.291, 292 In general, strategies aimed at reversing resistance to taxanes and other MDR substrates in the clinic have not been successful, but most have involved MDR modulators that reduce taxane clearance and increase toxicity (e.g., verapamil, cyclosporine A), thereby confounding the interpretation of the inherent effects of these agents on MDR modulation. However, MDR and/or MRP modulators that do not affect taxane clearance and toxicity do not appear to enhance anticancer activity.293, 294
Although the potency of cabazitaxel is similar to that of docetaxel in cancer cell lines and human tumor xenografts sensitive to docetaxel, cabazitaxel retains relevant activity in docetaxel-resistant cancer cell lines and human tumor xenografts that possess the MDR phenotype, which is due to the lower substrate affinity for Pgp. Resistance ratios range from 1.8 to 10 for cabazitaxel compared with 4.8–59 for docetaxel.203, 204 The extent to which the differences in Pgp substrate affinity account for cabazitaxel activity following failure of docetaxel in castrate-resistant prostate cancer is not known.
Similar to the vinca alkaloids, several human cell lines rendered taxane resistant by treatment with high drug concentrations for protracted periods have structurally altered α- and β-tubulins and an impaired ability to polymerize tubulin dimers into microtubules (see sections titled “Vinca Alkaloids, Introduction and Indications” and “Vinca Alkaloids, Mechanisms of Resistance”).106, 107, 295–307 These cells lack normal microtubules in their interpolar mitotic spindles and have an inherently slow rate of microtubule assembly when grown in the absence of drug. The continuous presence of the taxanes is required for microtubule assembly to proceed. Furthermore, these mutants are also collaterally sensitive to the vinca alkaloids. In some experimental systems, paclitaxel-resistant cells had mutated β-tubulin alleles, predominately involving β1-tubulin, with mutations involving the putative taxane binding sites; specifically, leucines at positions 215, 217, and 228 were mutated to histidine, arginine, or phenylalanine.297, 299, 300, 306 Low-level expression resulted in drug resistance, whereas high-level expression of these mutations caused impairment of microtubule assembly, cell-cycle arrest, and failure to proliferate, all of which were reversed by incubation with paclitaxel. Even though mutations of tubulin isotype genes, gene amplifications, and isotype switching have been reported in taxane-resistant cell lines, there is little indication that mechanisms are clinically relevant and, if they do occur, they are likely to be rare events.306 In a study that examined paclitaxel-naive and paclitaxel-resistant samples from nonsmall cell lung cancer and ovarian cancer, ovarian cancer xenografts, and ovarian cancer cell lines, no βI-tubulin mutations were identified.306 However, silent polymorphisms in exon 1 of βI-tubulin were detected at a very low frequency, suggesting that the polymorphisms are not relevant.306
Alterations in tubulin content, tubulin isotype profiles, and tubulin polymerization dynamics confer resistance to the taxanes in preclinical studies, and available evidence indicates that these mechanisms may explain variability responsiveness to the taxanes in the clinic.306 As previously discussed, there are multiple α- and β-tubulin isotypes that are encoded by different genes that are located on different chromosomes and that have tissue- and cell-specific expression patterns. These tubulin isotypes can undergo posttranslational modifications, which can alter the interaction of microtubules with MAPs and change their function. Predictive molecular modeling, which has been used to examine how the affinity of tubulin-binding agents for different β-tubulin isotypes varies, may explain differential sensitivity. For example, paclitaxel is thought to diffuse through nanopores in the microtubule to reach its binding site on the interior-facing lumen of the microtubule. It has been predicted that this movement is mediated by the formation of a hydrogen bond that involves serine 275 in all β-tubulin isotypes, except βII- and βVI-tubulin.306 Paclitaxel-resistant tumors have been shown to have high levels of class I, III, and IVa isotypes of β-tubulin.20, 100–107, 295–306, 308, 309 High intratumoral levels of the ßIII-tubulin isotype, which is a minor component of cellular β-tubulin, increases dynamic instability, and impedes microtubule assembly, have been identified in cell lines with acquired taxane resistance, as well as in taxane-resistant tumors sampled from patients.100–106, 298–307, 310–322 The absence of ßIII-tubulin is associated with more rapid rates of microtubule assembly in vitro, whereas overexpression is associated with a decreased rate of microtubule assembly.316
The differential functional aspects of the various tubulin isotypes have not been fully elucidated, and the many posttranslational modifications that tubulins undergo add another layer of complexity. Pro- and antiapoptotic proteins, including the TP53 and Bcl2, are tethered or transported by microtubules. The induction of apoptosis by antimicrotubule agents is mediated through the intrinsic apoptotic pathway, principally by Bcl2 family members.23, 306, 317, 318 The proapoptotic protein Bim is tethered to microtubules, and, after treatment with paclitaxel, Bim translocates from microtubules to mitochondria.306, 319 Cancer cell lines with high Bim expression are more susceptible to the cytotoxic effects of the taxanes than cells with low expression. How differential expression of β-tubulin isotypes or posttranslational modifications affect the ability of antimicrotubule agents to bind, transport, and release of factors that induce apoptosis is not entirely clear; however, the expression of specific β-tubulin isotypes and MAPs has been related to drug resistance in a range of cancers, including lung, ovarian, breast, and prostate cancers.20, 100–107, 295–306, 308, 309
Aberrant proliferative signaling may contribute to taxane resistance by raising the cell’s threshold for taxane-induced apoptosis. Insulin-like growth factor-1, for example, appears to protect responsive breast cancer cell lines from the cytotoxic effects of the anthracyclines and taxanes, possibly by activating the phosphatidylinositol 3-kinase pathway and inducing inactivation of antiapoptotic factors.320 Other mediators that may influence the cell’s threshold for drug-induced apoptosis include TP53, HER-2, aurora 2-kinase, survivin, and BRAC1. The centromere-associated serine/threonine kinase, aurora 2-kinase, which is involved in centrosome, biopolar spindle formation, and chromosomal kinetochore attachment to the mitotic spindle separation (see section titled “Targeting Mitotic Motor Proteins and Kinases”), appears to override the mitotic assembly checkpoint and induce taxane resistance.21, 22, 306, Also, overexpression of survivin, a member of the inhibitor of apoptosis family of proteins, inhibits caspase activity and apoptosis induced by antimicrotubule agents. The disruption of the tumor suppressor gene, BRAC1, which maintains genomic stability through DNA repair and appears to be involved in hereditary breast and ovarian cancers, appears to play a role in conferring resistance to paclitaxel, and the inducible expression of BRAC1 may enhance apoptosis induced by paclitaxel.306, 321 The mutational loss of the TP53 tumor suppressor does not appear to confer resistance to microtubule-polymerizing agents in contrast to DNA-disruptive agents.306
MAPs play a role in resistance by impeding apoptosis following treatment with the taxanes and other antimicrotubule agents. For example, tau and MAP2, which are principally expressed in neuronal tissues, as well as the principal nonneuronal MAP, MAP4, bind to and stabilizes microtubules against depolymerization.28 Tau expression relates to paclitaxel resistance in breast cancer, which seems to be through steric hindrance as tau masks the outer microtubule wall and limits access of paclitaxel to the inner luminal surface of the microtubule.316, 322 MAP4 confers microtubule stability, and induction of TP53 leads to decreased MAP4, resulting in reduced sensitivity to paclitaxel, albeit increased sensitivity to VBL.323, 324
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


