Acute myeloid leukemia, acute myelogenous leukemia, and acute nonlymphocytic leukemia are equivalent terms for a hematopoietic neoplasm that is uniformly characterized by the presence of a malignant clone of myeloid cells in the bone marrow with maturation arrest at the level of blast. Acute myeloid leukemia (AML) is the term used in the World Health Organization (WHO) classification of the myeloid neoplasms1,2,3 and is the current recommended term.4
AML may follow a myelodysplastic syndrome (MDS) (Chapter 79) or a myeloproliferative neoplasm (MPN; Chapters 80, 81, 82 and 83). AML should be distinguished from acute lymphoblastic leukemia (ALL; Chapter 74), in which blasts are of lymphoid rather than myeloid lineage. AML should also be distinguished from chronic myelogenous leukemia (CML; Chapter 81), in which, in the initial phase of the disease, myeloid cells are expanded but do not exhibit maturation arrest. In addition, CML is a uniform disease, whereas AML has heterogeneous clinical, morphologic, immunophenotypic, cytogenetic, and molecular features. Chapter 77 addresses presentation and therapy of AML in children. Chapter 78 addresses the biology, presentation, and management of the acute promyelocytic leukemia (APL) subtype of AML in adults and children.
AML occurs at any age, but is more common in adults, with increased frequency as age advances. Patients typically present with manifestations of anemia, neutropenia, and/or thrombocytopenia resulting from impaired hematopoiesis due to replacement or suppression of normal marrow elements by malignant blasts. The total white blood cell count is variable, ranging from leukopenia to hyperleukocytosis with an elevated blood blast count that represents a medical emergency.
The malignant cell in AML is a blast that most often shows myeloid or monocytic differentiation, but shows erythroid or megakaryocytic differentiation in approximately 5% to 10% of cases. The myeloid blast can be identified by the presence of granules and/or Auer rods by Wright Giemsa staining, by Sudan black, myeloperoxidase (MPO), chloroacetate esterase, or nonspecific esterase cytochemical staining, and/or by an immunophenotype demonstrating expression of myeloid antigens. Immunophenotyping by flow cytometry is currently in widespread use4,5 and serves to define lineage in the absence of defining morphologic and cytochemical features.6 In addition, ultrastructural features characteristic of AML may be demonstrated by electron microscopy,7 although this modality is not generally a component of clinical diagnostics.
Immunophenotyping by flow cytometry has shown that AML is markedly heterogeneous, variably expressing antigens expressed on stem cells and on more mature myeloid cells, also with infrequent co-expression of lymphoid antigens.8 Moreover AML generally exhibits aberrant patterns of antigen expression or co-expression in relation to normal myeloid cells.9,10 Detection of aberrant phenotypes by flow cytometry has been applied toward monitoring of residual disease.11
Cytogenetic abnormalities are present in leukemia cells of most patients with AML,12 and are currently the most powerful predictors of treatment outcome.13,14 Because of the importance of cytogenetics in diagnosis and prognosis in AML, several recurrent cytogenetic abnormalties, including t(8;21)(q22;q22), inv(16) (p13.1q22), or t(16;16)(p13.1;q22), t(15;17)(q22;q12), t(9;11) (p22;q23), t(6;9)(p23;q34), inv(3)(q21q26.3), or t(3;3)(q21;q26.2) and t(1;22)(p13;q13), have been incorporated into the WHO classification of AML.2 Moreover, cytogenetic abnormalities have been classified as favorable, intermediate, and unfavorable with regard to AML treatment outcomes,13,14 and this information is used to assist in the choice of initial and subsequent therapies. New diagnostic tools, including fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), comparative genomic hybridization (CGH), and microarray analysis, have improved the sensitivity of detection of genetic abnormalities and the ability to subclassify AML and to detect residual disease. Diverse molecular abnormalities are also being demonstrated in AML and predict treatment response, particularly in AML with a normal karyotype. Molecular abnormalities are also serving as a basis for development of targeted therapies.
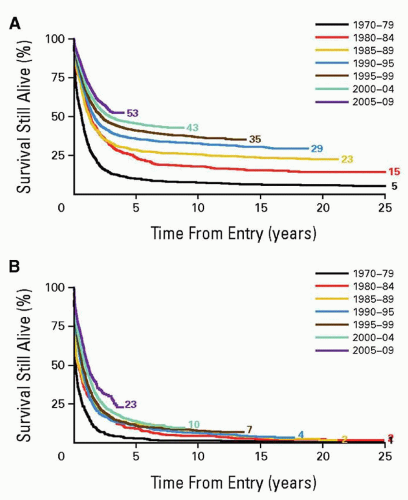
Advances in therapy have changed cure rates in AML from less than 20% in 1960 to 1980 to 40% to 70% for selected groups in the 2000s. The cure rate of APL is very high with the addition of all-trans-retinoic acid (ATRA) to chemotherapy and, in some regimens including in the relapse setting, use of arsenic trioxide (Chapter 78). For other types of AML, outcomes have improved significantly for younger, but not older, adult patients (Fig. 75.1). The highest cure rates have resulted from allogeneic stem cell transplantation (alloSCT) in first remission but, with improved survival of patients receiving high-dose cytosine arabinoside (cytarabine or ara-C) post-remission chemotherapy, the question of what is the best post-remission therapy in AML remains unanswered, and stratification based on patient and leukemia characteristics is indicated. Novel targeted approaches are currently being tested in AML, and their ultimate roles remain to be determined.
FIGURE 75.1. Change in overall survival of AML patients with time. A: Age 15 to 59 years. B: 60 or more years. (From Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol 2011:29(5):487-494.)
In this chapter, epidemiologic, clinical, biologic, cytogenetic, and molecular features of adult AML are addressed in the context of therapeutic principles and prognosis.
HISTORICAL PERSPECTIVE
The terms “weisses Blut” and “leukämie” were first used by Virchow in the middle of the 19th century, following initial descriptions of patients with an excess of white blood cells and splenomegaly at autopsy.15,16 Ebstein introduced the term “acute leukemia” in 1889 to differentiate rapidly progressive leukemias from more indolent “chronic” leukemias. Naegeli identified the myeloblast in leukemia in 190017 and divided leukemias into “myeloid” and “lymphocytic.” During the first half of the 20th century, most of the subcategories of AML were identified by light microscopy with the aid of cytohistochemical stains and were described based on resemblance to normal hematopoietic precursors. Reschad and Schilling described acute monoblastic leukemia in 1913;18 DiGugliemo described acute erythroleukemia in 1917;19 Von Boros and Karenyi described acute megakaryocytic leukemia in 1931;20 and Hilstad described APL in 1957.21
The initial classification of AML developed by the French-American-British (FAB) cooperative group in 197622 was based on morphology. It was subsequently expanded to include new morphologic and immunogenotypic subsets, notably minimally differentiated disease with myeloid antigen expression.6 The WHO classification, published in 1997 and updated in 2002 and 2008,1,2,23,24 incorporates clinical and cytogenetic data, distinguishes therapy-related AML and AML with MDS-related changes from de novo AML, and uses several recurring structural cytogenetic abnormalities to define AML subtypes.
Numerous cytogenetic abnormalities, both structural and numerical, have been identified in AML, beginning with the t(8;21)25 and t(15;17)26 translocations. Several of these recurring abnormalities were incorporated into the WHO classification, serving to define new subtypes.1,2,24 Cytogenetic findings in AML are the strongest predictors of treatment outcomes.13,14,27,28,29,30,31,32,33 and 34 Structural cytogenetic abnormalities have been characterized at the molecular level, resulting in increased understanding of AML pathogenesis, and establishment of potential therapeutic targets, as well as molecular markers for quantitative monitoring of residual disease.11 Increasing numbers of gene mutations are also being described in AML, also enhancing our understanding of its molecular pathogenesis, providing new prognostic categories,35,36,37,38,39,40 and 41,42,43,44,45,46,47,48,49,50 and identifying new therapeutic targets.
The European LeukemiaNet has developed a new prognostic classification of AML incorporating cytogenetic and molecular data.51,52
Full sequencing of cytogenetically normal AML genomes was first reported in 200853 and 2009,54 demonstrating 10 and 12 genes with acquired mutations, respectively. Finally, microRNAs, or small noncoding RNA molecules that hybridize to target messenger RNA species and regulate their translation, have also been found to have distinct expression patterns in AML, with prognostic significance, and may represent new therapeutic targets.55,56,57
EPIDEMIOLOGY
It is estimated that 13,780 individuals (7,350 men and 6,430 women) were diagnosed with AML and 10,200 died of AML in the United States in 2012.58,59 Lifetime risk of AML based on rates from 2007 through 2009 is 0.39%, which means that 1 in 254 men and women born today will be diagnosed with AML during their lifetime.
TABLE 75.1 AGE DISTRIBUTION OF ACUTE MYELOID LEUKEMIA (AML) AT DIAGNOSIS
Age (Years)
AML Incidence (%)
<20
6.0
20 and 34
6.6
35 and 44
6.6
45 and 54
11.8
55 and 64
15.5
65 and 74
20.1
75 and 84
23.3
>85
10.2
The incidence of AML increases with age. AML accounts for 80% to 90% of cases of acute leukemia in adults, but less than 15% of cases of leukemia in children younger than 10 years and 25% to 30% in those between 10 and 15 years.60,61 From 2005 through 2009, the median age at diagnosis of AML was 66 years.62Table 75.1 shows the age distribution for diagnosis of AML.58
The increased incidence of AML in the elderly is likely related to a combination of improved diagnosis, recognition of AML after MDS, and longer life expectancy, with consequent increased environmental exposures. Based on the number of patients diagnosed from 2005 through 2009 in 18 Surveillance Epidemiology and End Results (SEER) geographical areas, the age-adjusted incidence of AML is 3.6 per 100,000 subjects per year.58 The incidence is higher in males than in females and higher in whites than in blacks (Table 75.2).58,61 There is an increased risk for Eastern European Jews and a decreased risk for Asian populations.63
The overall 5-year relative survival for AML from 2002 through 2008 in 18 SEER geographic areas was 23.4%.58 As with incidence, mortality rates in AML vary with age, gender, and race. Table 75.3 summarizes the age distribution of AML-related death.58 The median age at death from AML was 72 years in 2005 to 2009,64 and the age-adjusted mortality rate was 2.8 per 100,000 subjects per year.58Table 75.4 summarizes mortality rates by race and gender.58 From 2002 through 2008, 5-year relative survival by race and sex was 21.3% for white men, 24.6% for white women, 23.2% for black men, and 24.8% for black women.58
RISK FACTORS
Risk factors for AML include both exposures that result in DNA damage, and congenital diseases and gene polymorphisms associated with impaired repair of DNA damage. Epidemiologic studies have identified genetic, environmental, and occupational factors that may contribute to the pathogenesis of AML.60,63,65 In addition, AML arising following cytotoxic therapy for prior malignancies or benign conditions, termed therapy-related AML (t-AML), is an increasing problem in the face of successful therapy for lymphomas and solid tumors, as well as leukemias, and decreasing the risk of this devastating complication is an important goal in the evolution of oncology treatment regimens and strategies.
TABLE 75.2 ACUTE MYELOID LEUKEMIA (AML) INCIDENCE RATES (PER 100,000 INDIVIDUALS) BY RACE AND BY GENDER
Race/Ethnicity
Male
Female
All Races
4.3
3.0
White
4.5
3.1
Black
3.5
2.8
Asian/Pacific Islander
3.5
2.8
American Indian/Alaska Native
2.5
3.1
Hispanic
3.5
2.7
TABLE 75.3 AGE DISTRIBUTION OF ACUTE MYELOID LEUKEMIA (AML)-RELATED DEATH
Age (Years)
AML-Related Death (%)
<20
2.2
20 and 34
3.2
35 and 44
3.8
45 and 54
8.1
55 and 64
15.7
65 and 74
24.6
75 and 84
29.9
>85
12.4
Identified occupational, envitronmental, lifestyle-related, and medical risk factors for development of AML are discussed below. Multiple factors may contribute and interact. A recent case-control study of risk factors for de novo AML at MD Anderson Cancer Center demonstrated multiple factors, joint effects, and differences by sex and WHO subtype.66 Heavy smoking (≥30 pack-years; odds ratio [OR], 1.86) and low-level (OR, 2.87) or moderate/high-level (OR, 4.13) occupational solvent exposure increased the risk of AML in men, whereas obesity (OR, 1.62) and low-level (OR, 2.73) or moderate/high-level (OR, 3.90) occupational solvent exposure increased the risk in women.
Occupational Exposures
Occupational exposures are risk factors for the development of AML. In recent literature, an increased risk of AML has been reported in workers manufacturing, or exposed to, rubber, paint, embalming fluids, pesticides, ethylene oxide, petroleum, poultry, munitions, automobiles, nuclear power, plastics and electrical wiring, as well as gasoline station attendants, beauticians, barbers, and cosmetologists.67 Most of these reports are based on retrospective and cross-sectional studies, which makes establishing causal relationships difficult. Older literature also implicated employment in shoe-making, painting, furniture manufacturing or repair, paper mill, clothing or textile industry, chemical manufacturing, printing, nursing, and biologic laboratories,67 and it can be speculated that enforcement of regulations pertaining to occupational exposures has altered the occupational risk profile. Of note, an association with hobbies, including artistic painting, car repair, ceramics or pottery, furniture repair, model building, photography, shooting or hunting, or wood working, has not been reported.67,68,69,70,71 and 72
TABLE 75.4 ACUTE MYELOID LEUKEMIA (AML)-RELATED DEATH RATES (PER 100,000 INDIVIDUALS) BY RACE AND BY GENDER
Race/Ethnicity
Male
Female
All Races
3.7
2.2
White
3.8
2.3
Black
2.7
1.9
Asian/Pacific Islander
2.4
1.6
American Indian/Alaska Native
2.6
1.2
Hispanic
2.3
1.5
The most fully characterized occupational exposure associated with AML is to the aromatic hydrocarbon benzene.73,74 and 75 Benzene is absorbed through the skin and lungs and can accumulate in the body fat and neurologic tissues. Toxicity is related to cumulative dosage, and the risk of leukemia was high before safety controls were put into place in the workplace. Chromosome damage can occur at 1 to 10 ppm, and leukemogenic risk is considerable at 124 to 200 ppm. In surveys of factories in China, the leukemogenic risk was four to seven times higher in workers exposed to benzene than in the general population, and the average latency was 11.4 years.76,77 A dose-response pattern was suggested, with the highest risk in Chinese workers exposed at constant levels of 25 ppm or higher.77 Increased risk of AML with occupational exposure to benzene was confirmed in a recent systematic review and meta-analysis,78 and occupational benzene exposure was found to be associated with specific aneuploidies.79
Environmental Factors
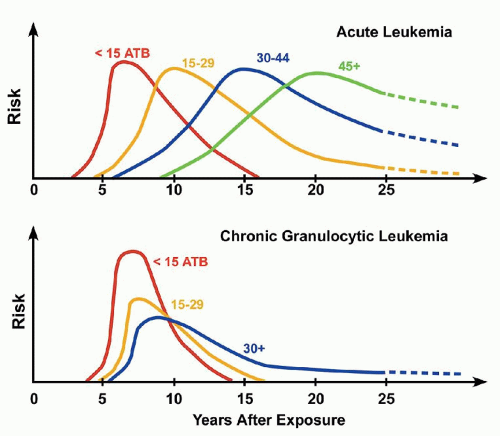
Ionizing radiation is carcinogenic primarily via induction of double-strand DNA breaks. The risk of leukemia correlates with radiation dosage and age at exposure, with a more rapid peak early in life (<15 years), as well as a more rapid decline than in those exposed at older ages (Fig. 75.2).80
Fallout from atomic tests and exposure to nuclear reactors has been a concern in the second half of the 20th century and the start of the 21st century. Atomic bombs were released over Hiroshima and Nagasaki in 1945, and an excess risk of leukemia was reported in 1952.81 The excess relative rate, per Gray (Gy), of AML among Japanese atomic bomb survivors was best described by a quadratic dose-response function that peaked approximately 10 years after exposure, but with the effect persisting for more than five decades.82 Whereas little radioactivity was released into the environment at Three Mile Island, there was extensive exposure to radioactivity after Chernobyl,83,84 and increased AML incidence has been reported in Chernobyl clean-up workers.85 Of note, one Gray is the absorption of one joule of ionizing radiation energy, per kilogram of body matter.
FIGURE 75.2. Effect of age at exposure and temporal pattern of developing leukemia according to cell type (acute vs. chronic granulocytic leukemia). ATB, at time of bomb. (From Okada S, Hamilton HB, Egami N, et al., eds. A review of thirty years of Hiroshima and Nagasaki atomic bomb survivors. J Radiat Res 1975;16(Suppl):1-164.)
Radon, cosmic radiation, and nonionizing radiation have been implicated as possible etiologic factors in AML but are unlikely to contribute a major risk.
Lifestyle-related Factors
Lifestyle-related risk factors for AML include smoking, obesity, and use of some hair dyes. Smoking has been repeatedly identified as a risk factor for AML in an extensive literature.86,87,88,89,90,91 and 92 Cigarette smoke contains more than 4,000 chemical compounds, of which 60 have been found to be carcinogenic and leukemogenic, including benzene, urethane, nitrosamines, and polyaromatic hydrocarbons such as benz[a]anthracene, benzo[a]pyrene, benzo[b]fluoranthene, benzo[c]phenanthrene, benzo[e]pyrene, and benzo[j]fluoranthene. Meta-analyses have estimated a relative risk for AML of 1.3 to 1.5 in smokers.87,93 The risk of developing AML is two to three times higher in male smokers who have exceeded 20 pack-years.86,94 Smokers of more than 40 cigarettes per day who develop AML have an increased incidence of unfavorable cytogenetic abnormalities, including -7/7q-.88,92 In a recent case control study at MD Anderson Cancer Center, a history of heavy smoking was associated with AML in men, and current smoking was associated with an increased risk of AML in women, and there was a joint effect of smoking and solvent exposure.66
Obesity has been reported as a risk factor for AML.66,95,96 and 97 In a large cohort of male US veterans, obesity was associated with a higher incidence of AML in both white and black populations.96 Similarly, among over 40,000 women ages 55 to 69 years in the Iowa Women’s Health Study, the risk of AML was increased among women who reported being overweight or obese (relative risk, 1.9; 95% confidence interval [CI], 1.0 to 3.4; relative risk, 2.4; 95% CI, 1.3 to 4.5; P(trend) = 0.006, respectively), compared with women of normal weight.97 Findings were similar in a Canadian population.95
Finally, there is evidence of an association between use of permanent, but not nonpermanent, dark hair dye and AML.98
Therapy-related Acute Myeloid Leukemia
Antineoplastic therapy is associated with an increased risk of subsequent development of AML, and AML diagnosed following cytotoxic therapy for a prior malignant or benign condition is considered to be t-AML.
AML most commonly develops following treatment with alkylating agents or topoisomerase II inhibitors, but nucleoside analogs, antitubulins, and radiation are also associated with t-AML.99,100,101
Radiation used for medical purposes may be leukemogenic. Smith and Doll reported a fivefold increased risk of leukemia in patients with ankylosing spondylitis receiving a single dose of pelvic radiation; the risk peaked 3 to 5 years after radiotherapy.102 A twofold increased risk has been reported after pelvic radiation for menorrhagia, a treatment commonly used during the 1930s and 1940s.103 Small increases in risk have also been reported after radiation for tinea capitis104 and peptic ulcer disease.105
t-AML presentation and treatment response differ based on the setting in which it arises and the likely causative agent. Most therapy-related leukemias occur 3 to 10 years after initial therapy, with a longer latency for alkylating agents (5 to 9 years) than for topoisomerase II inhibitors (6 months to 5 years).99,101 Alkylating agents cause point mutations, as well as chromosome deletions and unbalanced translocations.100 Topoisomerase II inhibitors result in loss of a critical enzyme involved in DNA replication, leading to balanced chromosomal translocations usually involving 11q23, and less frequently 21q22, with the formation of fusion genes.106
The prognosis for t-AML is poor, with a median survival of 7 to 10 months and usually less than 10% 5-year survival.100 Patients with chromosome 5 and 7 abnormalities have the worst prognosis. Alkylating agent-associated t-AML, generally characterized by deletions or monosomies of chromosomes 5 and/or 7 or complex karyotypes, frequently evolves from t-MDS, and has a low complete remission rate and short disease-free and overall survival. In contrast, topoisomerase II-associated t-AML, most commonly with 11q23 translocations, is typically not preceded by t-MDS and has a high complete remission rate, but short disease-free and overall survival. More rarely, patients treated with antineoplastic agents may develop t-AML with t(8;21), inv(16), or t(15;17).101,107 An early report suggests similar prognosis for t-AML and de novo AML with t(8;21) or inv(16),108 whereas a more recent report suggests a worse outcome for t-AML than for de novo AML with t(8;21).109
Alkylating agent-asociated t-AML has been extensively described. Cumulative drug dose is a primary determinant in causing leukemia, and alkylators differ in their leukemogenicity. Mechlorethamine and melphalan are more leukemogenic than cyclophosphamide. Germline mutations in tumor suppressor genes (NF-1,TP53), genetic variations in enzymes (cytochrome P450, glutathione S-transferase, and NAD(P)H: quinone oxidoredutase 1) that are involved in drug metabolism, and genetic variations in genes such as XRCC1 and hMSH2, encoding DNA repair proteins, can all contribute to an increased risk for t-MDS/t-AML.110,111 and 112 Alkylating agent-associated t-AML was initially best characterized following treatment of Hodgkin lymphoma (HL), one of the first malignancies for which curative therapy was developed,113,114 and 115 but has now been described after therapy of multiple neoplasms, including but not limited to breast cancer, multiple myeloma, ovarian cancer, and non-Hodgkin lymphoma (NHL),101,116,117 and 118 as well as in nonneoplastic disorders such as collagen vascular diseases.101
In HL, radiation can be a contributing factor, but the risk in patients treated with radiation alone is usually less than 1.0%, compared to 1.5% to 5.0% for those who received both radiation therapy and chemotherapy, particularly when chemotherapy is administered as salvage therapy for relapse after radiation.119,120 The risk of leukemia after HL ranges from an 11- to a 136-fold increase over that in the general population.120 With the increased use of autologous transplantation as salvage therapy for lymphomas, t-MDS/t-AML is being recognized in 5% to 15% of patients and is associated with a poor prognosis.101,121 The time of leukemia onset after alkylating agent exposure has ranged from 1 to 28 years and is most commonly in the 5- to 9-year range.122 The risk is greater and the latency is shorter in older patients with HL (>50 years of age).123 Splenectomy has been suggested as a contributing factor to leukemia in some, but not all, studies.122,123 Elevation in the mean corpuscular volume (MCV) may be an early sign of development of myelodysplastic changes, and up to two thirds of patients who develop leukemia have a preceding myelodysplastic phase that lasts approximately 6 months.122,124 Clonal cytogenetic abnormalities are often complex; the most common single abnormality is monosomy 7 (-7), followed by del(5q) and -5.122
Topoisomerase II inhibitors, particularly etoposide and teniposide, were recognized as leukemogenic agents in survivors of lung cancer,125,126 germ cell cancer,127 ALL,128 neuroblastoma,129 and osteosarcoma130 in the 1980s. Large cumulative doses and prolonged courses have been implicated as increasing the risk of leukemia. The latency period is short, with most cases occurring between 6 months and 5 years after initial therapy. There is no myelodysplastic phase, and the majority of cases are myelomonocytic (FAB M4) or monoblastic (FAB M5). The most common cytogenetic abnormality involves a translocation of the mixed lineage leukemia (MLL) gene at chromosome band 11q23.122 Over 40 partner genes that encode different proteins are involved in MLL translocations. Patients with AML and 11q23 abnormalities after topoisomerase II inhibitors tend to be chemosensitive but are rarely long-term survivors because of a high relapse rate.122
Breast cancer is treated with both alkylating agents and topoisomerase II inhibitors, as well as antitubilins. Cytogenetic abnormalities in t-AML after breast cancer therapy are heterogeneous and include those associated with either alkylating agent or topoisomerase II inhibitor therapy.131 Use of G-CSF has been implicated in increasing the risk of t-AML following breast cancer adjuvant therapy131,132,133 and radiation therapy may also contribute.131,134,135 and 136
Nucleoside analogs have also been implicated in t-AML. Fludarabine in chronic lymphocytic leukemia is associated with t-AML, particularly when combined with chlorambucil137 or mitoxantrone.138 Nucleoside analogs are also associated with an increased risk of t-AML in Waldenström macroglobulinemia.139 Azathioprine is associated with t-AML, frequently with chromosome 7 abnormalities, in patients with rheumatologic disorders and in solid organ transplant recipients.140 6-Mercaptopurine is also associated with t-AML.141 In addition, MDS/AML with dysplasia and chromosome 7 abnormalities has been seen in patients treated for de novo AML and may represent t-MDS/t-AML associated with cytarabine.142
An increasing literature is also documenting t-MDS/t-AML occurring following therapy for APL with ATRA and diverse chemotherapy drugs. Many of these cases of t-MDS/t-AML have chromosome 5 and/or 7 abnormalities, but others have 11q23 translocations, and miscellaneous abnormalities are also seen.143
Although less leukemogenic than chemotherapy, radiation therapy is leukemogenic, as evidenced by an increased leukemia risk in patients receiving radiation in the past for ankylosing spondylitis,102 menorrhagia,103 tinea capitis104 and peptic ulcer disease.105 The risk of leukemia (latency period of 2 to 11 years) is approximately 2 times higher in patients who have received either radium implants or external beam radiation for cervical, ovarian, or endometrial cancer.144,145 and 146 Similarly, a twofold increase in risk has been reported in breast cancer patients receiving adjuvant radiotherapy, compared to a tenfold increase in risk after chemotherapy; combined radiation and chemotherapy resulted in a seventeenfold risk.131,136 Radiation therapy has been associated with only a small increase in risk of leukemia in patients with HL and NHL, unless the radiation is extensive and encompasses a large volume of bone marrow.115,147
In a recent report, patients who developed MDS/AML following radiation alone were found to have a lower incidence of high-risk karyotypes and longer survival than those who developed MDS/AML following chemotherapy or chemotherapy and radiation. Karyotypes did not differ from those in de novo MDS/AML, suggesting that post-radiation MDS/AML may not represent a direct consequence of radiation toxicity and may warrant a therapeutic approach similar to de novo disease.148
Medical exposure to radioactivity is leukemogenic. In a recent meta-analysis, the relative risk of leukemia in thyroid cancer survivors treated with radioactive iodine (RAI) was 2.5 (95% CI 1.13, 5.53, p = 0.024).149 A standardized incidence ratio (SIR) of 5.68 (95% CI, 2.09 to 12.37) was recently reported in patients with low-risk (T1N0) well-differentiated thyroid cancer, with the excess risk of leukemia significantly greater in patients aged <45 years (SIR = 5.32; 95% CI, 2.75 to 9.30) than in older patients (SIR = 2.26; 95% CI, 1.43 to 3.39).150 Increased leukemia risk has also been previously reported following therapy with radioactive phosphorus (32P) for polycythemia vera, as well as following exposure to Thorotrast, a radioactive contrast agent containing thorium (232Th).151
Chromosome Instability or Defective DNA Repair
The incidence of AML is increased in patients with syndromes characterized by chromosome instability or defective DNA repair,152 notably Bloom syndrome,59 an autosomal recessive disorder with excessive chromosomal breakage, including quadriradial formation, and increased sister chromatid exchanges, caused by alterations in a gene on chromosome 15q26.1 encoding a protein with helicase activity that is central to DNA repair.153
An increasing recent literature also associates polymorphisms in genes encoding detoxifying enzymes and DNA repair proteins with incidence of AML, as well as with treatment outcomes. Glutathione S-transferases (GSTs), including GSTM, GSTP, and GSTT, are detoxification enzymes involved in metabolism of carcinogens. A recent meta-analysis of the association of GST polymorphisms with risk of AML supported a significant risk of AML in the presence of null genotypes of GSTM1 and GSTT1.154 Associations with treatment outcomes were also suggested.155,156
Polymorphisms in the RAD51 and XRCC3 genes, encoding proteins involved in repair of DNA double-strand breaks via homologous recombination, have been associated with increased risk of development of AML, and the association is additionally strong for t-AML and in the setting of coincident polymorphic deletion of the GSTM1 detoxification gene.110 Other work implicates polymorphisms of the XRCC1 gene, encoding a protein involved in base excision repair,157 and of RAD51111 in the risk of t-AML, and of polymorphisms in the xeroderma pigmentosum group D (XPD) DNA repair gene, encoding a protein involved in nucleotide excision repair, in AML with 5q and 7q chromosome deletions.158 Additionally, XPD polymorphisms have been associated with AML treatment outcome, and ERCC1 and XRCC3 polymorphisms with treatment toxicities.159
Heritable Genetic Factors
Heritable syndromes associated with increased risk of AML are usually recognized in childhood and are discussed in Chapter 77. In addition to Bloom syndrome,59 they include Fanconi anemia (FA),160 familial platelet disorder (FPD),161,162 and 163 Schwachman-Diamond syndrome,164,165 amegakaryocytic thombocytopenia, which may be X-linked, as well as Blackfan-Diamond syndrome, familial aplastic anemia,166 and Kostmann syndrome.167 Severe congenital neutropenia (SCN), with mutations in the neutrophil elastase (ELA2) gene, when treated with G-CSF increases risk for AML, associated with acquired somatic mutations in the G-CSF receptor.168
Inherited mutations of tumor suppressor genes predispose patients to malignancies, including those of myeloid origin. The Li-Fraumeni syndrome, described in 1969, is an autosomal dominant cancer family syndrome with an increased risk for sarcomas, breast cancer, and other neoplasms, including leukemia, due to germline mutations in the TP53 gene.169,170
CLINICAL PRESENTATION
AML presenting symptoms and signs are related to failure of normal hematopoiesis, resulting in anemia, neutropenia, and thrombocytopenia. The most common complaint is nonspecific fatigue or malaise that may have been present for several months. Pallor and weakness are caused by anemia. Fever is common and is the presenting feature in 15% to 20% of patients, often associated with sweats, and may result from infection secondary to neutropenia or from leukemia itself. Hemorrhagic signs and symptoms, including petechiae, epistaxis, and easy bruising, may be found in up to one half of patients at diagnosis. Petechiae correlate with the severity of thrombocytopenia, and ecchymoses with the presence of disseminated intravascular coagulation (DIC), which is most common in APL, but also occurs in other AML subtypes. Weight loss is present in up to 50% of patients, but is usually not severe. Bone pain occurs in less than 20% of patients. Organomegaly and adenopathy have been reported in up to one half of patients with AML, but are less common than in ALL.
Leukemia Cutis
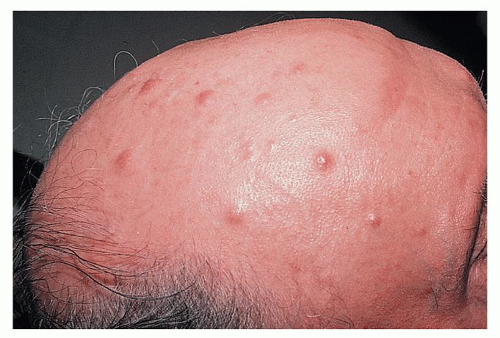
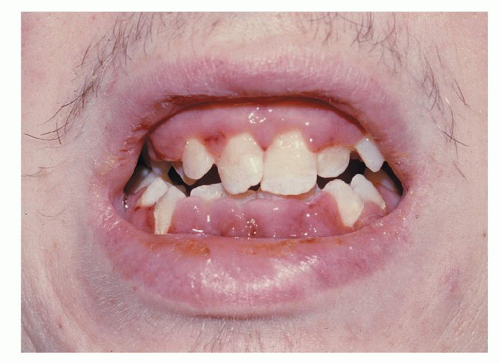
Leukemic skin infiltration, or leukemia cutis, occurs in up to 13% of patients with AML during the course of the disease, most commonly in those with a monocytic component. Skin lesions are often nodular and violaceous (Fig. 75.3), are painless, and may be widespread or localized. Widespread lesions are distinguished from other exanthems by being raised and palpable. Diagnosis is confirmed by biopsy. Skin lesions may precede the diagnosis of AML, or may occur concurrently with AML or as an extramedullary relapse. They are radiosensitive, but patients should usually be treated with systemic chemotherapy.171 Benign skin lesions associated with AML include Sweet syndrome172 and pyoderma gangrenosum.173 These are painful and are responsive to corticosteroids. These diagnoses are also confirmed by skin biopsy. Leukemia cutis is frequently associated with involvement of other extramedullary sites, including the central nervous system (CNS).171 Gum infiltration is also characteristic of acute monocytic leukemia (Fig. 75.4).
Myeloid Sarcoma
Myeloid sarcoma, or granulocytic sarcoma, myeloblastoma, or chloroma is an extramedullary tumor that occurs in 2% to 14% of cases of AML.174,175 and 176 The term chloroma derives from a green appearance due to expression of MPO. The tumors are usually localized, frequently in bone, periosteum, soft tissues, lymph nodes, or skin. Common sites are the orbit and the paranasal sinuses, but other sites reported include the gastrointestinal tract, genitourinary tract, breast, cervix, salivary glands, mediastinum, pleura, peritoneum, and bile duct.175 Myeloid sarcomas may occur at diagnosis of AML, may precede the diagnosis, or occur as an extramedullary relapse. They have also been seen in association with MDSs or MPNs and usually predict transformation to acute leukemia.174,175 and 176 The diagnosis is suggested by presence of eosinophilic myelocytes in hematoxylin and eosin-stained biopsy sections. Imprint preparations can be helpful. The diagnosis can be made if Auer rods are detected or if myeloid origin is confirmed by cytochemical or immunohistochemical methods. Although granulocytic sarcomas are radiosensitive, systemic chemotherapy is warranted in most cases.177
FIGURE 75.3. Leukemia cutis manifesting as subcutaneous nodules. (Courtesy of Dr. Michael Smith, Division of Dermatology, Vanderbilt University Medical Center.)
FIGURE 75.4. Swollen and spongy gums in a patient with acute leukemia. (Courtesy of Dr. Stuart Salmon, Division of Hematology/Oncology, Vanderbilt University Medical Center.)
Central Nervous System Leukemia
The incidence of CNS disease at diagnosis of AML is difficult to determine because lumbar puncture is not generally performed.178 Meningeal disease has been reported to develop in up to 16% of adults with AML.178 The increased use of high-dose cytarabine (HiDAC), which crosses the blood-brain barrier, lessens the risk of CNS leukemia in AML, as evidenced by a 2.2% incidence in a review of 410 patients from a single institution.179 CNS disease is associated with hyperleukocytosis and the AMoL variants.180 It is often asymptomatic, but may be associated with headache, nausea, and/or cranial nerve palsies, particularly V and VII. Ocular involvement may result in blindness and suggests meningeal involvement. Intracerebral masses were reported in FAB M4Eo in association with inv(16)(p13q22) and M5 subtypes,181 but also appear to be less common with increased use of HiDAC. Prophylactic central nervous system (CNS) therapy is not given routinely to adult patients with AML, but some clinicians advocate prophylaxis in patients with AMoL or with presenting white cell counts greater than 100,000 cells/mm3. Diagnostic lumbar puncture with prophylactic intrathecal chemotherapy may also be advocated prior to hematopoietic stem cell transplantation.
Other Organ System Involvement by Acute Myeloid Leukemia
Other organ systems may be involved. Cardiac abnormalities are usually related to electrolyte imbalances, particularly hypokalemia, but may result from direct involvement of the conduction system or infiltration of vessel walls.175 Pulmonary symptoms occur in patients with leukostasis, infections related to neutropenia, or hemorrhage. Gastrointestinal symptoms also include infections, particularly perirectal abscesses and typhlitis, which is a necrotizing colitis related to leukemia infiltration of the bowel wall. Management of typhlitis is supportive, including antibiotics and nasogastric suction, but surgical intervention is sometimes unavoidable.182 Obstructive jaundice has occurred secondary to myeloid sarcoma.
Hypocellular Acute Myeloid Leukemia
Hypocellular AML occurs in 5% to 10% of patients with AML and is defined by the presence of AML in a hypocellular bone marrow, with the definition of hypocellularity varying between 5% and 40%.183 Hypocellular AML is typically seen in older patients, and should be distinguished from MDSs and aplastic anemia. Howe et al. demonstrated a CR rate of 73% in a series of 29 patients with hypocellular AML.184 Although hypocellularity alone should not exclude a patient from receiving chemotherapy, caution should be exercised in treating these patients, and transplantation strategies should be considered.
LABORATORY FINDINGS
Presenting blood counts vary widely among patients with AML.185 The leukocyte count is elevated in more than half of patients, but is greater than 100,000 cells/mm3 in less than 20%. Blasts are usually present in the peripheral smear or in a buffy coat smear. Auer rods and Phi bodies are considered pathognomonic of AML. Phi bodies are fusiform or spindle-shaped rods similar to Auer rods that require special stains for hydroperoxidases.186
Cytopenias result from hematopoietic failure and contribute to symptoms and signs. Anemia is common in AML and is predominantly normochromic and normocytic. Reticulocytopenia is generally present, but nucleated red blood cells may be seen. Neutropenia is present in most AML patients. A normal neutrophil count is more common in patients with monocytic variants of AML. Giant lysosomes are rarely noted in neutrophils of patients with AML; their presence has been called a pseudo-Chédiak-Higashi syndrome.187 Thrombocytopenia is usually present and may be severe at diagnosis. Thrombocytosis is rarely identified, and is characteristic of AML with abnormalities of the long arm of chromosome 3, involving the EVI1 gene.
DIC is more common in AML than in ALL, and is most common in APL (Chapter 78). DIC is seen at presentation in nearly all patients with APL, and its incidence in AML other than APL is in the 10% to 30% range.188 The cause of DIC is thought to be the release of tissue factor-like procoagulants from the azurophilic granules within the leukemia cells. DIC is manifested clinically by bruising and, when severe, by bleeding from multiple sites. Laboratory findings include thrombocytopenia, hypofibrinogenemia, elevated fibrin split products, and deficiency of coagulation factors, including factor V and factor VIII.189 Other mechanisms, such as excessive fibrinolysis and secretion of interleukin (IL)-1 by AML cells, may contribute to bleeding.190
Hyperuricemia is noted in up to 50% of patients with AML and can also be associated with tumor lysis, although the latter is more common in ALL.191 Hydration and administration of allopurinol and/or recombinant urate oxidase (rasburicase) for markedly elevated uric acid levels or elevated uric acid levels with renal insufficiency, can prevent complications of tumor lysis, which may occur during induction chemotherapy, most often in the setting of hyperleukocytosis.
Serum lactate dehydrogenase (LDH) levels may be elevated, particularly in monocytic (FAB M4/M5) subtypes, but to a lesser degree than is observed in ALL.
Levels of lysozyme are elevated, particularly in variants of AML with a predominant monocytic component (FAB M4/M5). Excess lysozyme (muramidase) may cause proximal renal tubular damage, which results in hypokalemia. Other factors that can contribute to hypokalemia in AML include potassium uptake by rapidly proliferating cells, as well as medications, particularly diuretics and the antifungal antibiotic amphoterecin. Hyperkalemia can occur in association with hyperuricemia and tumor lysis. With improved agents to treat hyperuricemia, hyperphosphatemia with or without hypocalcemia is the most common abnormality associated with renal failure during induction therapy. Patients with AML may have hypocalcemia,192 but hypercalcemia is very rare.193
Hyperleukocytosis, arbitrarily defined as a blood blast count greater than 100,000/mm3, occurs most commonly in monocytic (FAB M4/5) AML and in AML with fms-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD).174,194 Hyperleukocytosis is a medical emergency because it is associated with leukostasis in the lungs and the CNS,195 causing respiratory failure and intracerebral hemorrhage,180,196,197 and 198 which are usually rapidly fatal. Pulmonary leukostasis is manifested by dyspnea, tachypnea, rales, interstitial infiltrates, and respiratory failure.195,199 CNS leukostasis manifests as headaches, blurred vision, somnolence, obtundation, ischemic stroke, and intracerebral hemorrhage.200 Spurious laboratory data associated with hyperleukocytosis include a falsely elevated platelet count because blast fragments are counted as platelets, pseudohypoxemia caused by oxygen consumption by leukemia cells, pseudohypoglycemia caused by glucose consumption by leukemia cells, hypophosphatemia, and falsely prolonged coagulation tests due to low plasma volume.201,202 and 203 Artifactual lowering of the pO2 and glucose may be prevented by placing blood samples on ice and performing the tests without delay.174
To decrease the risk of leukostasis in patients with hyperleukocytosis, the white blood cell count must be rapidly lowered. Therapeutic measures include leukapheresis, administration of large doses of hydroxyurea, and immediate initiation of induction chemotherapy.174,198,204 Red cell transfusions should be minimized initially to avoid overcorrection of the hemoglobin level, because this may produce an increase in blood viscosity that can worsen leukostasis. A hemoglobin goal of 8 g/dl, but not higher, is generally appropriate. In contrast, platelet transfusions are needed to decrease the risk of hemorrhage, because the platelet count may be overestimated, as discussed above. In addition, leukapheresis exacerbates thrombocytopenia and patients may also have DIC. An initial platelet count goal of 50,000/mm3 is generally reasonable. No controlled clinical trials have defined the optimal management of hyperleukocytosis, but retrospective data support the use of leukapheresis.205 Leukapheresis is indicated urgently if symptoms of leukostasis are present.196 Moreover, leukapheresis should be considered early to decrease the risk of leukostasis in patients with hyperleukocytosis, as the onset of leukostasis may be abrupt, and, once initiated, it may be fulminant and irreversible.174 Studies suggest that leukapheresis is associated with decreased early mortality and improved complete remission (CR) rates, but not with improved survival.197,198 Because leukapheresis is only a temporary measure to decrease the risk of leukostasis, hydroxyurea should also be administered, and chemotherapy should be initiated as rapidly as possible.174
BIOLOGIC FEATURES
Heterogeneity
AML is a complex and extremely heterogeneous neoplasm in all respects, including cytologic features, stage of differentiation, antigen expression, cytogenetic findings, gene mutations, gene expression, activation of signal transduction pathways, and response to treatment. Significant advances have been made in our understanding of the complex biology of AML, including identification and characterization of multiple interdependent pathogenetic phenomena and pathways. Increasingly sophisticated research techniques are being applied toward elucidating the complex biology of AML and developing more effective therapies.
Aberrant Immunophenotypes
In the majority of cases of AML, leukemia cells have immunophenotypes that distinguish them from myeloid progenitor cells found in normal marrow. Compared to normal bone marrow myeloid cell populations, AML cells exhibit asynchronous myeloid antigen expression, antigen overexpression, loss of antigen expression and co-expression of nonmyeloid antigens.9 These findings are consistent with aberrant differentiation rather than arrest of normal differentiation. The ability to distinguish AML cells from normal marrow cells based on aberrant immunophenotypes has formed the basis for flow cytometric analysis of AML in peripheral blood or marrows of patients with AML, as well as for detection of residual disease by flow cytometry.206,207 and 208 Immunophenotypes remain abnormal at relapse, but commonly exhibit gain or loss of one or more antigens and/or changes in antigen density.11,209
Leukemogenesis
Leukemogenesis in AML is a heterogeneous, multistep process that results in maturation arrest, altered proliferation, and impaired apoptosis, mainly through genetic dysregulation. A “two-hit” model of leukemogenesis has been proposed for AML.210 In this model, AML is the consequence of at least two classes of mutations. Class I mutations (e.g., BCR-ABL, K-RAS, N-RAS, KIT, FLT3) confer a proliferative and/or survival advantage, whereas Class II mutations (e.g., CBFβ-MYH11, AML1-ETO, TEL-AML1, and PML-RARα fusion genes) result in impaired cellular differentiation and apoptosis. Fusion genes such as AML1-ETO and CBFβ-MYH11, which are involved in the specific structural chromosomal abnormalities t(8;21) and inv(16), respectively, in AML, impair differentiation and apoptosis, but are insufficient to cause AML by themselves.211,212 A second class of genetic changes, such as mutations in the receptor tyrosine kinases FLT3,213KIT,214,215 or N-RAS,213 which deregulate proliferation, are required for development of AML. The limitations of this hypothesis include: (1) lack of identifiable mutations in class I and class II in all cases of AML, (2) frequent epigenetic changes in AML, and (3) unexplained role of newly discovered alterations in metabolic pathways and enzymes such as isocitrate dehydrogenase (IDH) in AML pathogenesis.
Recent whole-genome sequencing of bone marrow and skin, paired with skin, from patients with AML evolving from MDS revealed persistence of an antecedent founding clone containing approximately 200 to 650 somatic mutations, accompanied by outgrowth or emergence of at least one subclone with several new mutations.216
Clonality
The clonal nature of AML is confirmed by the presence of clonal cytogenetic abnormalities in the majority of cases,12 and has also been demonstrated by analysis of G6PD isoenzymes217 and of restriction fragment length polymorphisms (RFLP).218 Clonal abnormalities may be demonstrated in either a single cell line or in more than one cell line (such as myeloid and erythroid or myeloid and megakaryocytic), indicating that leukemic transformation can occur in either restricted-lineage or multipotential stem cells.217,219 Whole genome sequencing demonstrated that irrespective of blast count, more than 80% of bone marrow cells in MDS and s-AML are clonal.216
Deep sequencing of genomes from AML patients at initial diagnosis and at relapse suggested two major clonal evolution patterns at AML relapse: (1) gain of new mutations by the founding clone in the primary tumor, or (2) expansion of a subclone of the founding clone surviving initial chemotherapy, with gain of additional mutations. Thus AML relapse is associated with new mutations and clonal evolution, perhaps due to the influence of chemotherapy on the founding clone.220
Leukemia Stem Cells
It is generally accepted that AML relapse is caused by survival and persistence of rare chemotherapy-resistant leukemia stem cells (LSC).221 Transplantation of AML cells into immune-deficient mice demonstrated that LSCs are present at a frequency of one in 250,000 cells in the peripheral blood of AML patients, are usually enriched in the CD34+CD38- cell fraction,222 and lack expression of CD71 and human lymphocyte antigen (HLA)-DR,223 but express CD123.224 These cells are primarily in the G0 phase of the cell cycle.225 They are also characterized by activation of the transcription factor nuclear factor-κB (NF-κB), which is not activated in normal hematopoietic stem cells.226 Finally, they express multiple multidrug resistance proteins, including P-glycoprotein (Pgp; MDR1; ABCB1) and breast cancer resistance protein (BCRP; MXR; ABCG2), as do normal hematopoietic stem cells.227,228
Substantial basic and translational research has been performed to better understand the biology of LSCs and ultimately to target them therapeutically.229 LSCs cannot be distinguished from normal hematopoietic stem cells (HSCs) based on the the CD34+ CD38– phenotype,230 but intermediate levels of aldehyde dehydrogenase (ALDH) enzyme activity can distinguish CD34+CD38– LSCs, capable of engrafting immunodeficient mice, from normal HSCs, which showed higher ALDH activity, and presence of CD34+CD38–ALDH intermedite leukemic cells in complete remission after induction chemotherapy was associated with subsequent relapse.231 Moreover, using bioinformatic analysis, 42 and 121 genes were identified as LSC-related and HSC-related signatures, respectively, and 44 genes were found in both signatures and were designated as core-enriched HSC-LSC genes. When LSC- and HSC-related gene signatures were examined in 160 patients with cytogenetically normal AML, both signatures correlated negatively with overall survival even in patients with the prognostically favorable FLT3 wildtype and NPM-1 (nucleophosmin-1) mutant status.230
Hematopoietic Growth Factor Effects
The use of culture systems with phytohemagglutinin (PHA)-conditioned medium from peripheral blood leukocytes in the 1980s allowed growth of clonogenic cells in 80% to 90% of cases of AML,232 likely because of the presence of hematopoietic growth factors. Leukemia cells, like normal cells, generally require growth factors for survival and proliferation. AML cells express receptors for a number of hematopoietic growth factors, including granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), interleukins 1, 3, 4, 6, and 9, Steel factor (KIT ligand), thrombopoietin (MPL ligand), and FLT3 ligand (FLT3L).233,234,235 and 236 Growth factors predominantly cause proliferation, but may also inhibit apoptosis235 and induce differentiation.237 Growth factors may be produced by stromal cells or by AML cells themselves, resulting in paracrine or autocrine stimulation, respectively.238,239 Patients whose AML cells exhibit autonomous growth in vitro have an inferior response to treatment,240,241 and autonomous growth of AML cells is associated with other adverse disease characteristics, including unfavorable cytogenetic risk group,242 multidrug resistance243 and expression of antiapoptotic proteins.244
Proliferative Characteristics
The proliferative characteristics of AML cells, including the percentage of S-phase cells, the duration of S-phase, and the total cell cycle time, are highly variable,245,246 and 247 and proliferation may be either slower or faster than that of normal myeloid cells. AML with a high or a low proliferative rate responds less well to chemotherapy than AML with an intermediate proliferative rate.245 A high proliferative rate may be associated with marrow aplasia after chemotherapy, but rapid regrowth of leukemia,246 whereas nonproliferative AML cells may be chemoresistant.248 S-phase and total cell cycle times are longer in relapsed than in newly diagnosed AML.245
Resistance to Apoptosis
Resistance to apoptosis contributes to both leukemogenesis and drug resistance in AML.249 Apoptosis occurs by two pathways, both of which result in caspase activation: (1) receptor-mediated, involving the tumor necrosis factor (TNF) family of death receptors, and (2) mitochondrial-mediated, regulated by the BCL-2 family of proteins.249 Chemotherapy kills malignant cells through activation of mitochondrial-mediated apoptosis, and altered apoptotic pathways are a mechanism of resistance to chemotherapy. AML cells overexpress BCL-2 and other antiapoptotic proteins, including BCL-x(L), MCL-1, XIAP, and survivin,250,251 which may be up-regulated by the effects of stromal cells250 and cytokines.252 Permeabilization of the mitochondrial outer membrane is blocked by higher levels of the antiapoptotic proteins BCL-2, BCL-x(L), and MCL-1, and facilitated by the proapoptotic proteins BAX or BAD. High BCL-2-to-BAX protein ratios in AML cells are associated with lower complete remission rates and shorter overall survival.253 Expression of APAF-1, an important protein in the apoptosis machinery, is commonly silenced by methylation in AML cells, and may be restored by treatment with DNA methyltransferase inhibitors.254 Expression of the multidrug resistance protein P-glycoprotein (ABCB1) may also inhibit AML cell apoptosis by a mechanism independent of drug efflux.255
Signal Transduction Pathway Activation
Activation of signal transduction pathways may result from mutations in gene encoding growth factor receptors such as FLT3, KIT, or RAS. In addition, aberrant activation of the PI3K/Akt/mammalian target of rapamycin (mTOR) signaling pathway is implicated in leukemogenesis in AML,256,257,258 and 259 in the absence of consistent gene mutations. mTOR is constitutively activated in most AML cases,256,257 and 258 but not in normal hematopoietic stem cells,257 and P13K/Akt/mTOR pathway activation is associated with compromised apoptotic response mechanisms260,261 and 262 and resistance to chemotherapy.256,258,259,260 and 261 Thus PI3K/AKT/mTOR inhibitors have the potential to be both effective and selective in AML.263,264 Multiple signal transduction pathways may be activated in AML and the number of pathways activated is associated with a progressively worse prognosis.265
Microenvironment
AML bone marrow exhibits increased microvessel density, compared to normal marrow,266,267 and 268 and AML cells express vascular endothelial growth factor (VEGF), mRNA, and protein,266 and may also express aberrant VEGF receptors.269 VEGF can act via multiple paracrine,270 as well as autocrine,269 mechanisms to contribute to AML cell expansion and survival in the bone marrow microenvironment.271,272 VEGF secretion is strongly correlated with expression of FLT3 and of FLT3 ligand.273 Inhibition of FLT3 signaling with a small molecule FLT3 inhibitor, an antibody that blocks FLT3 ligand binding or FLT3 receptor siRNA decreases VEGF secretion. FLT3 is likely to induce VEGF secretion through the MAPK (mitogenactivated protein kinase) pathway via its activation of ERK1/2 (extracellular-signal regulated kinase-1 and -2) phosphorylation, rather than through AKT (protein kinase B) or STAT5 (signal transducer and activator of transcription-5),273 suggesting novel treatment strategies.274
The chemokine stromal cell-derived factor-1 (SDF-1) regulates homing and engraftment of leukemia cells in the bone marrow microenvironment and stromal niche. SDF-1 and its receptor CXCR-4 (fusin, LESTR) are expressed by AML cells and promote AML cell survival275 and stroma-mediated chemoresistance. CXCR-4 represents a novel therapeutic target276 for AML, and CXCR4 inhibitors have been tested in combination with chemotherapy in preclinical and clinical studies, with the goal of inducing mobilization of AML cells into the circulation, with promising results.277
CYTOGENETIC AND MOLECULAR FINDINGS
Cytogenetics
Acquired chromosome abnormalities are present in AML cells in most patients and diverse recurrent abnormalities are strongly predictive of treatment outcomes. Cytogenetic abnormalities were first described in AML in the 1960s, and were found to be present in approximately one half of AML patients in the 1970s with development of chromosome banding techniques (Chapter 3). With technical improvements, recent series have reported abnormal karyotypes in 55% to 78% of adults with AML.32,278 Approximately 55% of patients with AML have a single cytogenetic abnormality, including 15% to 20% with gain or loss of a single chromosome as the only change; the remaining 45% have two or more changes. The most common recurring cytogenetic abnormalities in AML include t(15;17), t(8;21), inv(16), +8, +21, del(5q), -7, 11q23 translocations, and 12p11-13 abnormalities.32,278
Correlation of cytogenetics and clinicopathologic data has led to the recognition of distinct subtypes of AML and has helped to identify prognostic groups (Table 75.5).32,278 Techniques such as PCR, FISH, and CGH have allowed characterization of cytogenetic abnormalities at the molecular level, demonstrating frequent involvement of oncogenes and tumor-suppressor genes (Chapter 72).
The best described subtypes of AML are defined by recurring structural chromosomal abnormalities, which primarily consist of balanced translocations, occur more commonly in younger patients, tend to correlate with morphology, and are predictive of treatment outcomes. These include t(8;21), inv(16) or t(16:16), and t(15;17) and their variants. AML with t(8;21) tends to have blasts with maturation, often with azurophilic granules and occasionally with very large granules (pseudo-Chédiak-Higashi granules).24 AML with inv(16) or t(16;16) is usually associated with monocytic differentiation and abnormal marrow eosinophils.24,181 APL with t(15;17) and its variants is suggested by the presence of hypergranular promyelocytes in association with disseminated intravascular coagulopathy (DIC), but may also be present in a variant microgranular (hypogranular) subtype (Chapter 78). Other cytogenetic abnormalities associated with morphology in AML include 11q23 translocations with monoblastic features;24,279 t(6;9) with marrow basophilia;280; abnormalities of 3q21-26 with abnormal platelets and thrombocytosis281 and t(9;22), 14q32, or 11q23 with mixed lineage antigen expression.282,283 t(8;21), inv(16) and t(15;17) are associated with favorable responses to chemotherapy, and 11q23 translocations, t(6;9), 3q21-26 abnormalities and t(9;22) with adverse treatment outcomes. AML with 11q23 abnormalities is associated with a high complete remission rate but short disease-free and overall survival, whereas the other unfavorable abnormalities are associated with low complete remission rates as well as short disease-free and overall survival.
11q23 translocations are common in t-AML arising following topoisomerase II therapy,99,122 but they also occur de novo, and are associated with monocytic differentiation. 11q23 translocations are characterized molecularly by rearrangement of the MLL gene at 11q23. The frequency of MLL rearrangements is sevenfold higher (5.3% vs. 0.8%) in patients younger than 60 years of age. The molecular pathogenesis of MLL gene rearrangements probably involves aberrant nonhomologous end joining of DNA double strand breaks. The normal MLL protein is proteolytically cleaved and functions as a transcriptional repressor or activator. Chimeric proteins that are generated from MLL rearrangements include the N-terminal region of MLL, which is involved in protein-protein interactions and transcriptional repression, and their leukemogenic effects appear to occur via activation of clustered homeobox (HOX) genes.284 More than 40 different partner genes for MLL have been identified.24 In t(9;11), t(10;11), and t(11;19), the amino terminus of the MLL gene is fused to one of three homologous genes, AF9, AF10, or ENL, from chromosomes 9q22, 10p12, and 19p13, respectively.285 Survival of patients with de novo t(9;11) has varied among studies, with some groups placing it in an intermediate prognostic group whereas others have reported it as unfavorable.29,278,286 De novo AML with t(6;11) (q27;q13) has a very poor prognosis.287 Additional cytogenetic aberrations have been shown to modify the outcome of pediatric 11q23/MLL-rearranged AML.288 AML patients with MLL gene rearrangement may have a better outcome with more intensive treatment regimens.
TABLE 75.5 SELECTED PRIMARY CHROMOSOME ABERRATIONS IN ACUTE MYELOID LEUKEMIA
Type of Rearrangement
Genes Involved
Hematologic Clinical Features
Prognosis
t(1;3)(p36;q21)
MEL1
Preceded by MDS, M1, M4, dysmegakaryopoiesis
Poor
t(1;7)(q10;q10)
Preceded by MDS, M1, M4, genotoxic exposure
Poor
t(1;11)(p32;q23)
AF1P, MLL
M0, M5
Poor
t(1;11)(q21;q23)
AF1Q, MLL
M4, M5, infants
Poor
t(1;22)(p13;q13)
RBM15, MKL1
M7, thrombocytopenia, hepatosplenomegaly, bone marrow fibrosis
Poor
inv(3)(q21;q26);t(3;3)(q21;q26)
EVI1, RPN1
Preceded by MDS, M1, M4, M6, abnormal dysmegakaryopoiesis, thrombocytosis
Poor
t(3;5)(q25, 1;q35)
MLF1, NPM
M6, megakaryocytosis, Sweet syndrome
Intermediate to poor
t(3;12)(q26;p13)
MDS1, EVI1-TEL
Preceded by MDS, dysmegakaryopoiesis
Poor
t(3;21)(q26;q22)
EVI1, MDS1, or EAP;AML1
No FAB preference, genotoxic exposure
Poor
+4
M1, M2, M4; subcutaneous tumors
Poor
-5/del(5q)
No FAB preference, genotoxic exposure
Poor
t(5;17)(q35;q12)
NPM, RAR-α
M3
Poor
t (6;9)(p23;q34)
DEK, CAN
Preceded by MDS; M2 and M4, bone marrow basophilia
Poor
t(6;11)(q27;q23)
AF6, MLL
M4 and M5; localized infections
Poor
-7/del(7q)
No FAB preference; genotoxic exposure; prior MDS
Poor
t(7;11)(p15;p15)
HOXA9, NuP98
M2 with Auer rods
Intermediate
+8
M2, M4, and M5; can be preceded by MDS; no impact on prognosis when associated with good-risk cytogenetics
Note: See Chapter 72 for description of the genes and Mitelman F, Johansson B, Mertens F, eds., Mitelman database of chromosome aberrations in cancer. http://cgap.nci.nih.gov/chromosomes/mitelman.
Structural abnormalities involving the long arms of chromosomes 5 and/or 7, monosomy 5 and/or 7, and complex karyotypes, defined by the presence of three or more unrelated numerical and/or structural abnormalities, are frequently seen in AML arising from prior MDS and in t-MDS/t-AML associated with alkylating agent therapy.100,122 These abnormalities, whether in de novo AML or t-AML, are associated with low complete remission rates and short disease-free and overall survival. Other numerical chromosomal abnormalities associated with poor treatment outcome include +11, +13, and +21.
The monosomal karyotype (MK), recently defined by the presence of at least two autosomal monosomies or a single autosomal monosomy in combination with at least one structural abnormality, is associated with a particularly dismal prognosis, with a 4% ±1% 4-year overall survival, whereas the 4-year OS of patients with complex, but nonmonosomal, karyotypes was 26% ± 2%. In a subsequent Southwest Oncology Group study of 1,344 adult AML patients, MK was present in 13%, increased in incidence with age, being present in 4% of patients age 30 or younger, but 20% of those over age 60, comprised 40% of the unfavorable cytogenetic risk category, and was associated with a complete remission rate of only 18% and only 3% 4-year survival.289 Transplantation resulted in limited improvement in outcome, with 25% 4-year OS in patients transplanted at the Fred Hutchinson Cancer Research Center.290 Thus transplant is indicated when feasible, but improvement in outcome is modest.
Molecular Abnormalities
AML is characterized by recurrent gene mutations that confer constitutive or aberrant signaling through one or more pathways in the complex signaling network that regulates normal hematopoiesis. The therapeutic implications of targeting different signaling pathways have begun to be exploited in recent years, with several promising small molecules and biologic agents in development (see “Therapy” section). Moreover, prognosis in AML is increasingly able to be assessed by evaluating molecular markers including frequently detected gene mutations.291 This can be particularly useful in dissecting the prognosis of AML with a normal karyotype.49,292 In addition, overexpression of specific genes is associated with unfavorable treatment outcomes. Assays for gene mutations are more readily standardized and more widely applicable than assays measuring gene expression levels. The number of gene abnormalities described in AML continues to increase.
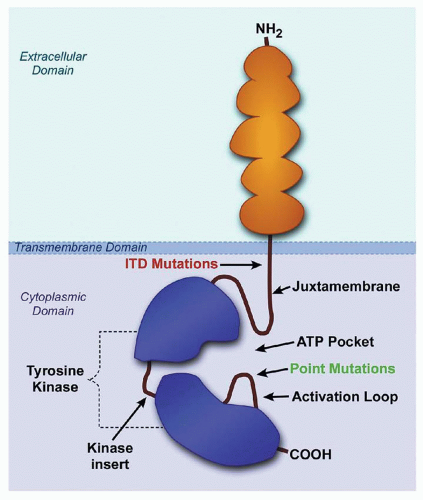
FLT3 is a receptor tyrosine kinase that is expressed on hematopoietic progenitor cells and is activated by binding of FLT3 ligand (FL), stimulating proliferation.210 FL binds to the receptor and induces dimerization, tyrosine kinase activation, and receptor autophosphorylation, followed by initiation of the phosphorylation of downstream signaling proteins. FLT3 is expressed on AML cells in most cases of AML, and is mutated in 20% to 34% of patients with AML35,36,37,293,294 resulting in constitutive activation.210,293FLT3 mutations can be detected by genomic PCR amplification and gel electrophoresis. The most common mutation is a small in-frame internal tandem duplication (ITD) in the FLT3 gene that results in duplication of an amino acid sequence within the juxtamembrane domain of the receptor, which disrupts the autoinhibitory activity of the juxtamembrane domain, resulting in constitutive tyrosine kinase activation (Fig. 75.5). Point mutations in the DNA sequence encoding the FLT3 activation loop, predominantly in the aspartic acid (D) residue at amino acid position 835, occur less frequently.36
FLT3 mutations activate similar transduction pathways as binding of FLT3 ligand to the wildtype receptor, including signal transducer and activator of transcription (STAT) 5 and the RAS/MAPK and PI3K/Akt pathways.293,295,296 and 297 Other effects include myeloid maturation arrest by virtue of suppression of the C/EBPα and PU.1 transcription factors,298 and antiapoptotic effects by virtue of phosphorylation of the proapoptotic protein BAD,298,299 resulting in inactivation of its proapoptotic function.
FLT3 mutations are associated with higher peripheral blast counts, normal karyotype and FAB M5 disease. Presence of FLT3-ITD does not affect CR rate, but predicts high relapse rate and poor survival and is useful in stratifying patients with AML with a normal karyotype into poor risk groups.35,37,293,300,301,302FLT3 point mutations are less prognostically unfavorable than FLT3-ITD.303FLT3-ITDs are of less prognostic significance in AML patients over age 70 years,300,304 likely because they occur in different patient populations than expression of MDR1/Pgp,305 which is a strong adverse prognostic factor in older adult AML patients,306 and because the prognosis of all AML patients over 70 years is poor.307 Prognosis of patients with FLT3-ITD is further worsened by absence of the wildtype allele,38 higher ITD to wildtype allelic ratio, larger size of the ITD300 or presence of the ITD in CD34+CD33-precursors.308 The role of transplantation in overcoming the adverse prognosis associated with FLT3-ITD is under investigation (see “Stem Cell Transplantation” section). Several FLT3 inhibitors are currently being evaluated in clinical trials in AML (see “Therapy” section).
FIGURE 75.5. Simplified diagram of the FLT3 receptor: it contains 993 amino acids and consists of an extracellular ligand-binding domain with five immunoglobulinlike domains, a single transmembrane domain, and a cytoplasmic domain, which is comprised of a juxtamembrane domain followed by the tyrosine kinase domain. Internal tandem duplications (ITD) in the juxtamembrane domain and point mutations at aspartate 835 within the activation loop constitutively activate FLT3. (Drawing by Tim Gilfilen, Medical Art Group, Vanderbilt University.)
Recently several groups have focused on the important role of the FLT3 ligand (FL) in AML with FLT3-ITD. FL expression increases significantly during induction and consolidation chemotherapy, and AML blasts remain highly responsive to FL at relapse, suggesting a potential role of FL in promoting relapse.309 A treatment strategy involving induction chemotherapy, stem cell transplantation, FLT3 inhibitors, and monoclonal antibodies against FL has therefore been proposed for AML with FLT3-ITD.310
Mutations of exon 12 of the nucleophosmin (NPM or NPM1) gene are the most common single gene mutations in AML, present in up to half of AML cases with normal karyotypes.39,41,311 Nucleophosmin is a nucleocytoplasmic shuttling protein that regulates the p53 tumor suppressor pathway, and NPM1 mutations in AML cells result in cytoplasmic localization of the protein, which can be demonstrated by immunohistochemistry.312NPM1 mutations are found at diagnosis and remain stable throughout the disease course. These mutations are typically found in AML with a normal karyotype,39,40 and 41,42,312,313 and are associated with prolonged event-free and overall survival.42 Co-occurence of NPM1 with FLT3-ITD mutations, but not with other mutations, negates their favorable prognosis,39,40 and 41,45,302,314 (Fig. 75.6). The mechanistic role of NPM1 in the pathogenesis of AML has not been fully elucidated. Perhaps due to favorable prognosis of NPM1-mutated AML, targeted therapies have not been investigated. Mutant NPM1 transcript levels have been validated as a residual disease marker, with cumulative incidence of relapse after 4 years after double induction therapy 6.5% in patients who achieved RQ-PCR negativity compared with 53% in RQ-PCR-positive patients (P < 0.001); and with much better overall survival (90% vs. 51%; P = 0.001) in RQ-PCR-negative patients.315
Mutations in the CEBPA (CCAAT/enhancer binding proteinalpha) gene,316 encoding a transcription factor that is essential for myeloid differentiation, are also a favorable prognostic factor in AML. These mutations are present in approximately 10% of AML patients overall and 15% of those with a normal karyotype,46,48 and are correlated with a distinct DNA methylation profile. Mutations at the CEBPA C-terminus generate dysfunctional proteins, and frameshift mutations in the CEBPA N-terminus create a dominant negative shorter protein. Biallelic CEBPA mutations in AML are associated with better prognosis than for unmutated or monoallelic mutated AML.50,317,318
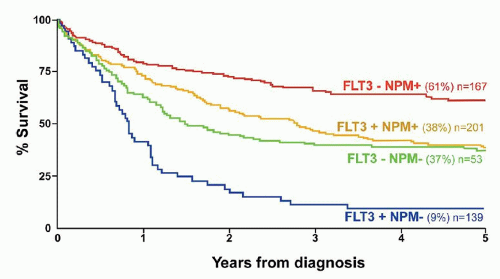
FIGURE 75.6. Clinical outcome in 550 AML patients with a normal karyotype according to FLT 3/ITD and NPM 1 mutant status. Both markers were prognostically significant predictors of survival (p < 0.0001), and together identified three prognostic groups: good (FLT3/ITD- NPM 1+), intermediate (FLT 3/ITD- NPM1- or FLT 3/ITD+ NPM1+), and poor (FLT 3/ITD + NPM1-). (With permission from Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008;111:2776-2784.)
Partial tandem duplication of the MLL gene (MLL-PTD), which is the gene at 11q23 that is involved in 11q23 chromosome translocations, was the first molecular abnormality to be identified in cytogenetically normal AML and to be associated with short disease-free survival,319,320 and 321 although outcomes are better with recent regimens incorporating more intensive post-remission therapies.322
Overexpression of ERG (ets-related gene) adversely affects outcome in AML with a normal karyotype and can be detected in AML with complex karyotypes that have a cryptic amplification of chromosome 21.323
When expression of the BAALC (brain and acute leukemia, cytoplasmic) gene is dichotomized into high and low levels, high expression is associated with a higher rate of primary resistant disease and is an adverse prognostic factor for disease-free survival, cumulative incidence of relapse, event-free survival, and overall survival in AML with a normal karyotype, and predicts adverse prognosis independently from FLT3 mutations.324 High BAALC expression is also highly predictive of shorter disease-free and overall survival in normal-karyotype AML without FLT3 or CEBPA mutations.49 High BAALC expression is associated with FLT3-ITD, absent FLT3-TKD, wildtype NPM1, mutated CEBPA, MLL-PTD, and high ERG expression, but high BAALC expression independently predicted lower complete remission rates when adjusting for ERG expression and age, and shorter survival when adjusting for FLT3-ITD, NPM1, CEBPA, and white blood cell count.325
Overexpression of the EVI1 (ectotropic viral integration site 1) gene on chromosome 3q26.2 has an incidence of approximately 11% and is seen not only in AML involving 3q26, including inv(3) and t(3;3), but also with other unfavorable karyotypes (e.g., -7/7q- and 11q23 abnormalities) and is associated with a poor prognosis.326,327,328EVI1 is not overexpressed in AML with favorable-risk karyotypes or with NPM1 mutations. Stem cell transplantation in first complete remission has a favorable impact on relapse-free survival of patients with AML overexpressing EVI1 (20% to 40% versus 0%).
RAS mutations occur in 10% to 44% of AML, but rarely with FLT3 mutations, and have not consistently predicted outcome.36,329,300 Activation of the RAS/Raf/MEK/ERK pathway has also been demonstrated in AML, promoting AML cell survival and inhibiting apoptosis.330,331
TP53 mutations have a frequency of 10% in de novo AML, but much higher frequency (40% to 50%) in t-AML. They have been associated with abnormalities of chromosomes 5 and/or 7 and with worse overall survival in older patients.300TP53 mutations, or TP53 mutations and 17p-loss of heterozygosity combined, also have independent negative prognostic effects on survival in AML.332
Mutations in the WT1 (Wilms tumor 1) gene occur in 5% to 10% of AML patients, with equal distribution in cytogenetically normal and abnormal AML groups.291 There are reports of both positive333 and negative334 prognostic effects of WT1 gene mutation in AML. WT1 mutation in AML was found to be associated with overexpression of CD96, a leukemia stem cell-specific marker, and of genes involved in regulation of gene expression (e.g., MLL, PML, and SNRPN) and in proliferative and metabolic processes (e.g., INSR, IRS2, and PRKAA1).335
The RUNX1 gene, located on chromosome 21 at band q22.12, encodes a transcription factor that is involved in benign and malignant hematopoiesis. The t(8;21) translocation, which is common in AML, results in a RUNX1 and ETO fusion protein. RUNX1 mutations are identified in 5.6% to 13.2% of AML patients, and are more common in older patients and in men. They are associated with lower lactic dehydrogenase, FAB M0/M1 subtypes, and expression of HLA-DR and CD34, and with lack of CD33, CD15, CD19, and CD56 expression. RUNX1 mutation is a poor prognostic factor, with resistance to chemotherapy and inferior event-free survival, relapse-free survival, and overall survival.336,337,338
The additional sex comblike 1 (ASXL1) gene encodes an enhancer of trithorax and polycomb proteins, which functions as a transcriptional activator or repressor in different cells. ASXL1 mutations are present in AML and were found to be five times more common in older (≥60 years) patients (16.2%) than those younger than 60 years (3.2%; P < 0.001), and to be associated with wildtype NPM1, absence of FLT3-internal tandem duplication and mutated CEBPA, and with inferior complete remission rate and disease-free, overall, and event-free survival in older patients.339
TET hydroxylase enzymes are involved in DNA hypomethylation and demethylation by α-ketoglutarate-dependent conversion of 5-methylcytosine to 5-hydroxymethylcytosine (alcohol moiety), followed by further oxidation to 5-formylcytosine (aldehyde moiety) and to 5-carboxylcytosine (acid moiety), Figure 75.7,.340,341 and 342 Mutations in the 10 to 11 translocation 2 (TET2) gene were identified with frequencies of 22% of myelomonocytic leukemia, 24% in secondary AML, 19% in MDSs, and 12% in MPNs.343 Heterozygous TET2 mutations were found in 7.6% of younger adult patients with AML and were not associated with favorable or unfavorable response to chemotherapy or with overall survival.344
Chromatin conformation can be affected at the DNA level by addition of a methyl group to the C-5 position of cytosine. Cytosine methylation occurs when the cytosine (C) is followed by a guanosine (G) in CpG pairs (p indicates phosphodiester bond). When CpG dinucleotides in the genome cluster together, they form CpG islands, which are located in proximity to gene promoter regions or in other intergenic areas. DNA methylation is catalyzed by the DNA methyltransferase (DNMT) family of enzymes which transfer a methyl group from S-adenosyl methionine to DNA. Hypermethylation of CpG islands in the promoters of tumor-suppressor genes is common in many cancers.
In 2010, Ley et al. found that 22% of 281 de novo AML cases had mutations in DNMT3A that could affect translation.345 These mutations were seen predominantly in patients with intermediate-risk cytogenetic profiles, and not in patients with favorable-risk profiles. The precise effects of these mutations have not yet been elucidated. FLT3, NPM1, and IDH1 mutations were significantly enriched in samples with DNMT3A mutations. DNMT3A mutations were independently associated with poor prognosis. DNMT3A mutations were associated with significantly shorter median overall survival (12.3 months vs. 41.1 months, p < 0.001).
Several subsequent studies have shown that DNMT3A mutations are frequent (approximately 20%) in older patients with normal karyotype AML and are associated with higher WBC and platelet counts, and concurrent mutations in the NPM1, FLT3, and IDH1 or IDH2 genes, and that they independently predict a higher relapse rate and shorter overall survival, but are not associated with a lower CR rate.346,347 and 348,349
Only gold members can continue reading. Log In or Register to continue
 Clinical Flow Cytometry
Clinical Flow Cytometry



