Acute myeloid leukemia in adults: mast cell leukemia and other mast cell neoplasms
Richard M. Stone, MD  Charles A. Schiffer, MD
Charles A. Schiffer, MD
Overview
Acute myeloid leukemia (AML) is the most common variant of acute leukemia occurring in adults, comprising approximately 80% of acute leukemia cases diagnosed in individuals of age >20 years. Remarkable advances in transfusion medicine, treatment of infections, development of potent antiemetics, improved chemotherapeutic approaches, and increased use of safer allogeneic transplantation have led to an improved outcome at least in younger patients. Moreover, a more sophisticated understanding of pathophysiology, particularly in the area of genomics, may soon lead to less toxic, patient-specific, and more effective therapies. At present, approximately 80% of younger (age <60 years) adults and 30%–50% of all older patients achieve complete remission (CR) defined as a morphologically normal bone marrow with reasonable neutrophil and platelet counts and no evidence of extramedullary disease. Varying with patient age and other biologic factors, 10–70% of these complete responders can be expected to achieve long-term survival with the likelihood that most of these individuals are cured of their disease. However, AML is largely an intrinsically chemoresistant disease, the outcome in older adults has changed little, and the chemotherapeutic approach has remained stagnant.
Introduction
Acute myeloid leukemia (AML) affects adults of all ages, but is particularly common in older adults. The median age of patients with AML is approximately 70 years (Figure 1). AML can present as either a de novo leukemia without an apparent antecedent illness or an evolution from marrow disorders such as myelodysplasia, myeloproliferative neoplasms, aplastic anemia, and Fanconi anemia, or after the administration of therapy for other types of cancers or nonmalignant disorders.1–4 AML presenting without prior marrow disease or anti-neoplastic therapy is termed de novo, whereas other types are considered secondary. Such terms may be less important predictors of response to therapy and long-term outcome than the mutational profile reflecting the biological heterogeneity of this condition.5 Proper care of patients with AML is a multidisciplinary effort, benefiting from a team approach. Expertise in transfusion medicine, infectious disease, placement and care of indwelling catheters, nutrition, and antineoplastic drug pharmacology are required, as well as sophisticated diagnostic laboratory facilities and psychosocial counseling for both patients and their families are needed. Optimally, allogeneic stem cell transplant, currently used in patients up to age 75, should be available. These disciplines are described elsewhere in this book, but their critical importance in the care of the leukemia patient cannot be overestimated.
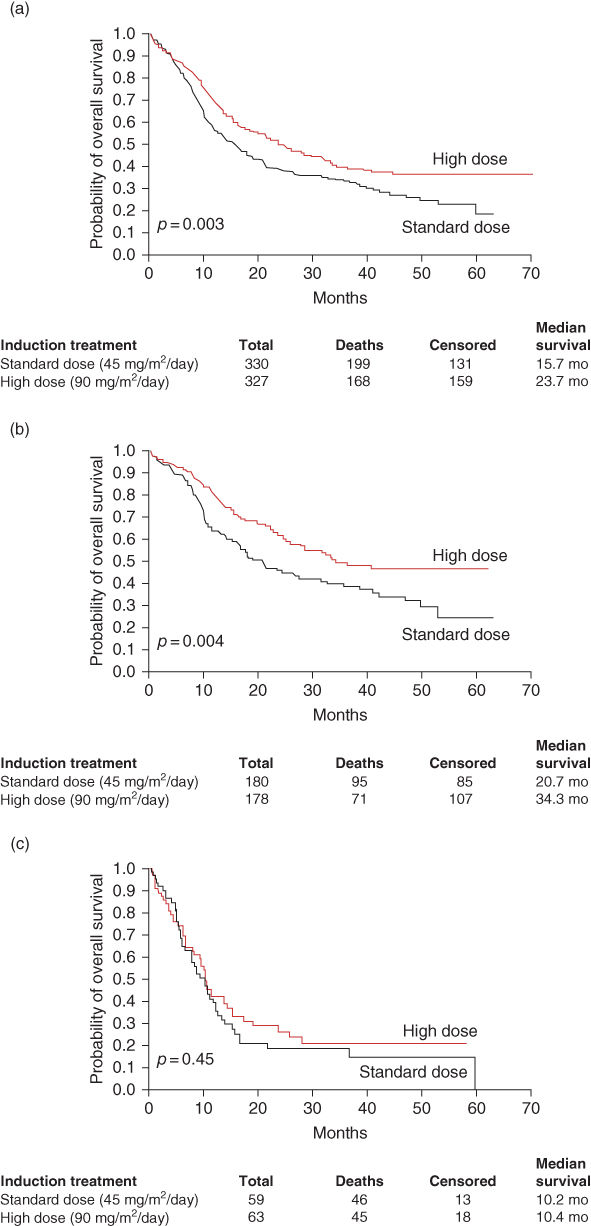
Figure 1 Data from ECOG demonstrating that overall survival in AML patients less than age 60 is approximately 40% and varies according to the cytogenetic risk and daunorubicin dose.
Source: Fernandez 2009.4 Reproduced with permission of NEJM.
Pathogenesis and etiology
Pathophysiology
The pathophysiology of AML can be partially explained by the acquisition of genetic changes in hematopoietic stem cells that both promote self-renewal and impair normal hematopoietic differentiation, resulting in an accumulation of immature cells. The genetic landscape of de novo AML has been recently characterized.6 An average of five mutations are found in an individual’s leukemia cells and “only” 30 mutations are recurrent (observed in more than 3% of patients). Of the 30 recurrent mutations, several are relatively common, including gain-of-function mutations, which are targets for therapy in active development. FLT3 is a transmembrane tyrosine kinase, which is mutated in approximately 30% of patients with AML.7 Approximately three-quarters of these mutations are length or internal tandem duplication (ITD) mutations, in which the protein is elongated in the juxtamembrane region by three to more than 100 amino acids. This type of mutation is associated with an adverse prognosis because of a high relapse rate. Both the ITD mutation and the less common tyrosine kinase domain point mutation (usually D835Y) cause ligand-independent constitutive activation of the receptor. There are several FLT3 inhibitors in active development, both as single agents and in combination with chemotherapy in mutant FLT3 patients. A second common mutation is a point mutation in the NPM1 shuttle protein.8 Such mutations, particularly if occurring in patients with a normal karyotype without an associated FLT3 mutation, confer a favorable prognosis.9 Mutations in the Ras guanine nucleotide-binding protein occur in approximately 20% of patients with AML and are associated with increased proliferation.10 Attempts to inhibit posttranslational modification, a required step for activation of Ras, have been largely unsuccessful in AML.11 Ras pathway interruption, such as with MEK inhibitors (useful in melanoma and other solid tumors), shows promise.12 Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 occur in approximately 20% of patients with AML (∼10% for each case).13 While the prognostic significance of these mutations is unclear, they cause the neomorphic production of 2-hydroxygluterate (2HG) rather than the usual reaction product, alpha-ketogluterate. 2HG levels may correlate with disease activity.14 Inhibition of IDH1 or IDH2 with small molecules may prove to be a fertile therapeutic intervention. Mutations in transcriptional machinery and mutations in enzymes, which are epigenetically active (posttranslational modification of the DNA) potentially leading to profound effects on gene expression, are common in AML.6 DNMT3, mutations occurring in approximately 20% of patients with AML,15 confer an adverse prognosis, although may be associated with a response to dose-intensified daunorubicin during induction.16 The presence of some of these so-called epigenetic modifying enzymes such as ASXL1, EZH2, and TET2 is often associated with AMLs that have arisen from a myelodysplastic syndrome (MDS) prodrome. TP53 mutation confers a bleak prognosis.17 On the contrary, CEBPα (biallelic) mutations are favorable.18
Genetic complexity of AML is an important feature of disordered molecular pathophysiology. The epigenetic profiles19 of AML can differ widely and perhaps suggest a specific therapy. Specific epigenetic profiles are found in distinct cytogenetic subsets of AML. Moreover, because of either mutations or altered patterns of genome regulation, messenger RNA20 and indeed microRNA expression patterns offer both pathophysiological insights and prognostic information. For example, microRNA expression patterns, particularly with moieties that control immunological regulation, have been found to play an important role in AML.21 Integration of the vast potential array of data including genomic lesions, epigenomic changes, and RNA and microRNA expression patterns yield an enormous amount of information and will be the subject of intense research in the future. The overexpression of many specific genes such as BAALC22 or WT123 has been shown to confer an adverse prognosis, but their independent significance remains to be confirmed.
Genetic changes in AML were first recognized by the identification of genes at cytogenetic break points involved in balanced translocations. Many of these chromosomal abnormalities, for example, t(8;21), t(15;17), and inv(16), are associated with specific AML subtypes and are of prognostic importance. The fusion proteins generated by the translocation generally result in disruption of transcription factors, which is believed to be critical in myeloid differentiation.24–27 Murine experiments indicate that transfection of mutated genes that primarily alter cell differentiation, such as AML1/ETO resulting from the t(8;21) translocation, produces abnormal hematopoiesis, but is not sufficient to generate frank AML.27 Initial murine experiments suggested a “two-hit” hypothesis, in which mutations affecting both differentiation and proliferative signaling pathways are needed to result in AML.28 The mechanisms that account for poor prognosis in patients with complex cytogenetics remain to be elucidated. Findings in 5q-MDS suggest a mechanism by which haploinsufficiency can lead to a myeloid malignancy.29 Many patients with the so-called monosomal karyotype (two monosomes or one monosomal plus one stromal abnormality) have p53 mutations, and this abnormality likely accounts for the dismal prognosis in this disease subset.17
Exposure and risk
Although more acquired genetic lesions that lead to leukemia are being defined, DNA damage from a known cause comprises only a small fraction of patients with AML. Leukemia occurs with increased frequency after exposure to nuclear bomb30 or therapeutic radiation,31 after certain types of chemotherapy,32, 33 and with heavy and continuous occupational exposures to benzene or petrochemicals.34, 35 There are two types of chemotherapy-related leukemias: (1) the classic alkylating agent-induced type, in which the leukemia is usually preceded by a myelodysplastic prodrome and is characterized by clonal abnormalities, often with loss of chromosome 5 and/or 732 and (2) an epipodophyllotoxin/topoisomerase II inhibitor-associated type with a shorter (median 2 years vs 5 years) incubation period, often with myelomonocytic or monocytic differentiation and abnormalities at the 11q23 region.33 Recent studies have challenged preconceived notions about the origin of therapy-related leukemia: (1) some patients who have been exposed to chemotherapy have genetic5 or cytogenetic lesions36 and clinical behavior indistinguishable from de novo AML; (2) preexisting p53 mutant hematopoietic clones due to the “normal” stochastic accumulation of mutations may be selected for survival after genotoxic therapy and thus predispose to the development of AML.37
Familial AML
AML is not usually a familial or inherited disorder, which is a fact worth noting to concerned families. There appears to be an increased incidence of acute leukemia in the first 6 months after diagnosis in the identical twin of an affected child.38 Familial syndromes have been described in which the incidence of leukemia was increased.39–41 In some of these families, different types of leukemias and other cancers have been found, whereas in other potentially more informative families, such as a large family in which more than or equal to seven cases of erythroleukemia or myelodysplasia developed in three successive generations,41 the morphologic or clinical characteristics of the leukemia have been similar, suggesting a common, heritable genetic mutation. A family has been described in which three patients with AML had identical inherited mutations in CEBPA,42 a gene involved in granulocytic differentiation, while another large kindred with a familial platelet disorder with a predisposition toward AML had been shown to be associated with mutations in the gene encoding CBF-α (formerly AML1).43 Finally, there seems to be a predisposition toward evolution to AML in individuals with inherited polymorphisms in the receptor for granulocyte colony-stimulating factor (G-CSF).44 AML is also more common in certain inherited conditions identified by an inability to repair DNA damage (e.g., Fanconi anemia)45 or with defective telomerase machinery.46
Prognosis
As noted, AML is a genetic and biological heterogeneous disease; thus, it is not surprising that a patient’s prognosis at the time of diagnosis can vary widely from a cure rate of 80–90% in acute promyelocytic leukemia (APL) to virtually zero in those patients with monosomal karyotype and/or p53 mutations in their malignant cells. A reasonably accurate prognosis can be derived from the knowledge of three factors (in addition to the obvious importance of comorbid disease and frailty in the elderly) to estimate the likelihood of long-term favorable outcome in patients with AML: (1) age; (2) cytogenetics; and (3) genetics. In general, the likelihood of remission, the chance of getting through standard remission induction without treatment-related mortality, and the likelihood of not relapsing vary inversely with age.47 For practical reasons, patients above 55–65 years of age are considered older adults with AML; however, the ability to withstand cytotoxic chemotherapy varies between individuals. The Eastern Cooperative Oncology Group (ECOG) Performance Status scale offers some help in this regard, but there is a trend toward using a more detailed and comprehensive geriatric assessment to determine a patient’s ability to withstand chemotherapy.48 In addition to the increased likelihood of occurrence of comorbid diseases with aging, other considerations are the more limited stem cell reserve in the older population49 and the inevitable decline in hepatic and renal function. Moreover, the fact that leukemia in older adults tends to be more biologically aggressive with an increased ratio of adverse chromosomal abnormalities to favorable abnormalities50 and the increased likelihoods of the so-called myelodysplasia-related genetic abnormalities and an antecedent clinical marrow stem cell disorder are associated with a relatively poor outcome. The prognosis is sufficiently adverse that a different treatment approach is generally employed in older adults with AML.
For the last 40 years, we have relied upon chromosomal findings at diagnosis to establish prognosis and in some cases to guide therapy. Numerous studies have divided newly diagnosed younger adults with AML into three prognostic categories.51, 52 Approximately 15% were found to have more favorable prognostic abnormalities (not including APL) including core-binding factor translocations such as t(8;21) and pericentric inversion of chromosome 16. Such patients have a high complete remission (CR) rate and relatively low relapse rate, although at least two-thirds are destined to relapse with an increased likelihood of an adverse outcome in those with an associated mutation in the c-KIT tyrosine kinase gene.53 Approximately 15% had unfavorable chromosomal abnormalities,50 including complex karyotypes (generally greater than three, although in some classifications five distinct chromosomal abnormalities) or the so-called monosomal karyotype (two monosomies or one monosomy plus one structural abnormality).54 Patients in this adverse prognostic category taken together have a 15% likelihood of long-term disease-free survival, but those with monosomal karyotype are destined to fare even worse.55 This is partially explained by high incidence of TP53 gene mutations in this subgroup.17 About 70% of patients are considered to have an intermediate prognosis with an approximate long-term survival chance of 40%. Some of these patients have well-characterized abnormalities such as trisomy 8 or translocations involving the 11q23 region (the MLL gene) from chromosome 11.
However, the vast majority of the intermediate prognostic group comprised those with a normal karyotype. Significant effort has been made in the last 10–20 years to divide patients with normal karyotypes into prognostic subgroups. At present, the following three genes have been generally used to provide information required for such a classification: CEBPα, NPM1, and FLT3. Patients with biallelic CEBPα mutations in the leukemia cells tend to exhibit a high expected disease-free survival of 80%; however, only a small number of patients have such an abnormality.18 The presence of FLT3 ITD (ITD) mutation producing variations in the length of this transmembrane tyrosine kinase is considered adverse, while a mutation in the NPM1 nuclear shuttle protein is considered favorable. When considering the possible combinations of these two mutations, only the subgroup of patients with an NPM1 mutation without a concomitant FLT3 ITD mutation are destined to exhibit high survival rates.9 This is the only patient group within the intermediate chromosomal prognostic category that is recommended to undergo a postremission approach based on chemotherapy only.9 A recent study suggests that only those with NPM1 mutant and FLT3 ITD wild-type genetic patterns as well as an IDH1/IDH2 mutation had a high overall survival16 but this remains to be confirmed by other studies. DNMTA3 mutations also imply an adverse prognostics,15, 56 similarly to those who have MDS-associated abnormalities such as EZH2 and ASXL1 mutations.57
In older adults, there is a higher incidence of complex or adverse cytogenetic findings. However, a small number of older adults have favorable karyotypes and should be treated with conventional induction and consolidation regimens if their other medical conditions permit.50 In addition, a large minority of older patients with a normal karyotype at the time of diagnosis who have an NPM1 mutation without an associated FLT3 ITD mutation can be expected to exhibit a higher survival rate than older adults who lack this genetic/chromosomal pattern.58
Table 1 depicts a modified European LeukemiaNet (ELN) classification scheme59 showing the frequency and general prognosis of those younger adults with various combinations of cytogenetic and genetic findings. This list will likely change over time as other genetic abnormalities are introduced into the equation.
Table 1 Cytogenetics- and genetics-based risk
| Risk group | Features | Approximate 4-year survival (%) | Approximate prevalence (%) |
| Very high | Monosomal karyotype (two monsomies or one monosomy plus balanced translocation) | 10 | 6 |
| High | Complex (>3 abormalities) or unfavorable (-7, 7q-, -5, 5q-, 3q, or t(6;9)) cytogenetics | 20 | 12 |
| Intermediate | Normal cytogenetics (with FLT3 mutation or FLT3-ITD not present/NPM1 wild type/CEBPα wild type) or other karyotype | 35 | 25 |
| Inversion 16 or t(8;21) with c-KIT mutation | 40 | 5 | |
| Normal cytogenetics (with FLT3-ITD not present/NPM1 mutation) | 50 | 25 | |
| Favorable | Normal cytogenetics (with CEBPα biallelic mutation) | 60 | 5 |
| t(8;21) or inversion 16 (with c-KIT wild type) | 65 | 10 | |
| Very favorable | t(15;17) | 85 | 12 |
Source: Stone 2013.59 Reproduced with permission of the American Society of Clinical Oncology.
Morphologic classification and clinical and laboratory correlates
The diagnosis of AML depends on the examination of well-prepared specimens of peripheral blood and bone marrow. Both bone marrow aspirates and biopsies should be evaluated. Although the biopsy is usually not helpful in identifying individual cells, it provides the best assessment of cellularity, can occasionally identify aggregates of leukemic cells that are not seen on aspirate, and is necessary to evaluate marrow fibrosis. In 1976, a group of morphologists from France, the United States, and Great Britain suggested a classification system designed to standardize definitions of the sometimes clinically and biologically distinct subtypes of AML and acute lymphoblastic leukemia (ALL).60 This French, American, and British (FAB) classification has been serially modified, initially in 2002,61 and in 201662 by a World Health Organization (WHO)-sponsored undertaking, which represented efforts to improve concordance among different observers and incorporate new findings from immunologic, cytogenetic, and molecular studies.
The diagnosis of AML requires that myeloblasts constitute 20% or more of bone marrow cells or circulating white blood cells (WBC), generally evaluated on Wright or Wright–Giemsa stained smears. The FAB classification required >30% blasts to distinguish AML from myelodysplasia. Although this change was based on a study which showed that the outcome could be based solely on the blast count,63 it is unclear that this is biologically well founded. The difference in eligibility criteria should be considered when comparing older AML trials with more recent studies. Neoplastic promyelocytes, monoblasts or promonocytes, and megakaryoblasts are included in this percentage, and their presence defines the various morphologic subtypes described below. An assortment of histochemical stains may be used to aid in subclassification and distinguish AML from ALL. Monoclonal antibodies directed against antigen groups [termed cluster designations (CD)] considered to be restricted to cells committed to myeloid differentiation are also helpful in making this diagnostic distinction. Antibodies against CD11b, CD13, CD14, CD33, and C117 are used most commonly.64 These antigens are found on normal hematopoietic elements, are not leukemia-specific, or unique to different AML FAB subtypes. In general, they do not correlate with prognosis, with the possible exception of CD34 antigen,65 which is detected on undifferentiated hematopoietic progenitors and can be found on the blasts of patients with either AML or ALL. It is suggested that patients with AML whose blasts strongly express CD34 have an inferior outcome because of chemotherapy-resistant leukemia, particularly in patients with less morphologically differentiated leukemias in which other myeloid-associated antigens are less strongly expressed.66
The FAB and WHO nomenclatures classify the subtypes of AML according to the normal marrow elements that the blasts most closely resemble. This does not indicate, however, that the leukemic event exclusively involves the cell lineage that is most prominently represented morphologically. Until recently, the involvement of other hematopoietic lineages could be inferred only by the presence of prominent morphologic abnormalities in these other cell lines. In patients with myelodysplasia or erythroleukemia, there is usually morphologic evidence of trilineage dysplasia with the inference that the initial cell that was malignantly transformed was a hematopoietic precursor with capability of multilineage maturation.67 Fialkow and colleagues,68 on their study of female patients with X chromosome-linked polymorphisms of glucose-6-phosphate dehydrogenase, were able to demonstrate involvement of myeloid, but not erythroid or megakaryocytic progenitors in some patients with AML. This first observation was proved prescient; however, at present, we believe that most hematopoietic elements emanate from a disordered stem cell, with multilineage involvement being quite common. Moreover, AML should be considered oligoclonal, rather than monoclonal; clonal heterogeneity exists at diagnosis; and the predominant clone can change with time, given the selective pressure exerted by chemotherapy. For example, there is evidence for persistence of certain subclones at remission as well as the emergence at relapse of clones, which have acquired “progression” mutations (or these mutations were present in very small subclones at diagnosis)5 and that a small clone at diagnosis could be the one to lead to relapse.
Representative examples of different subtypes of AML are shown in Figures 2–11. The immunologic, cytogenetic, and (where they exist) clinical correlates of these morphologic subtypes are reviewed in Table 2.
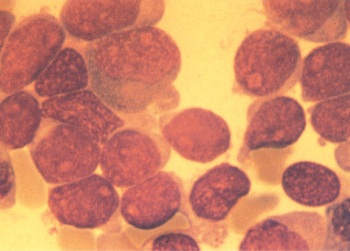
Figure 2 M0. Marrow blasts from patients with this undifferentiated type of acute myelogenous leukemia can have variable amounts of agranular cytoplasm. Cells are peroxidase- and Sudan Black-negative and can be confused with FAB M7 or FAB L2. Myeloid commitment of these blasts can be confirmed by immunophenotyping with antibodies against myeloid antigens and/or demonstration of ultrastructural peroxidase-positive granules using transmission electron microscopy.
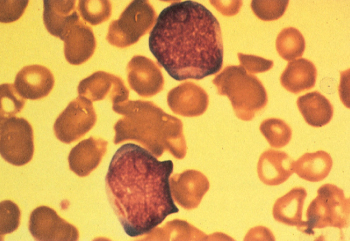
Figure 3 One of the blasts from a patient with M1 acute myeloid leukemia contains a prominent Auer rod.
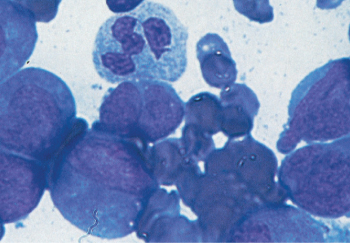
Figure 4 M2. Leukemia is characterized by evidence of continued myeloid differentiation with myelocytes and more mature myeloid elements present.
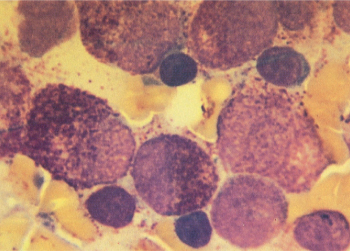
Figure 5 M3. Promyelocytic leukemic cells usually have spherical nuclei with heavily granulated cytoplasm. Extracellular granules are often noted, and blasts with multiple Auer rods (not shown) are common. This leukemia has typical 15;17 translocation and a characteristic clinical picture of disseminated intravascular coagulation.
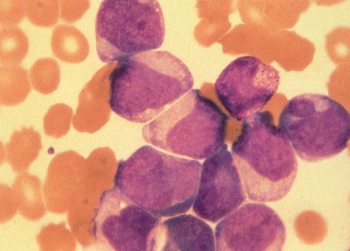
Figure 6 M4. Myelomonocytic leukemia has blasts with both myeloid and monocytoid appearance.
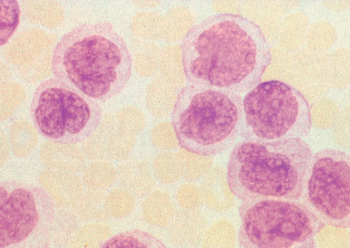
Figure 7 M5. Monocytic leukemia. Prominent nuclei filled with nucleoli in some cells, light granulation, and large amounts of lightly basophilic cytoplasm give these cells the appearance of promonocytes.
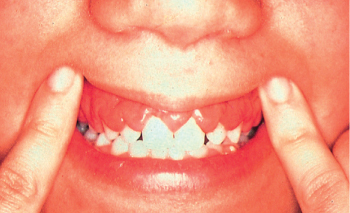
Figure 8 M5. Gingival hypertrophy due to infi ltration by leukemic cells in acute monocytic leukemia.
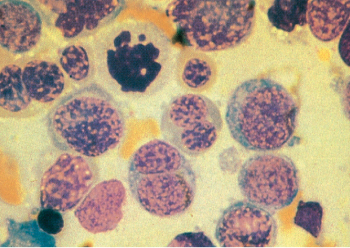
Figure 9 M6. Erythroleukemia is characterized by the presence of bizarre megaloblastic and often multinucleated erythroid precursors. Karyorrhexis is seen in some cells. The somewhat arbitrary distinction between FAB M6 and myelodysplastic syndrome with excess blasts in transformation is made by quantifi cation of the fraction of myeloid blasts.
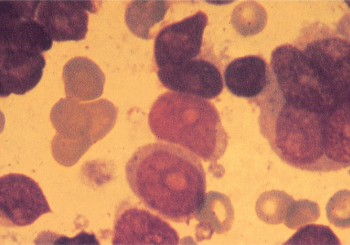
Figure 10 M7. Megakaryocytic leukemia. Blasts in this category are often morphologically undifferentiated. The presence of multinucleated cells, dysplastic micromegakaryocytes, and cytoplasmic budding can be useful diagnostic clues. The diagnosis is confirmed by immunophenotyping or ultrastructural studies.
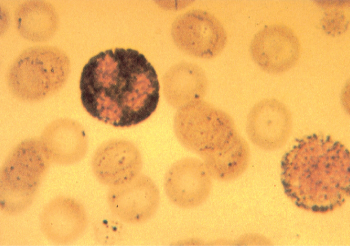
Figure 11 Typical granular staining with Sudan Black B of a blast and a neutrophil from a patient with FAB M1 acute myeloid leukemia.
Table 2 Recurring karyotypic and molecular abnormalities in AML
| Cytogenetic abnormality | FAB morphology | Affected genes | Median age | Approximate incidence in de novo AML | Prognostic effects | Comments |
| t(8;21) | M2 | AML1/ETO | 30 years | 5–7% | Favorable | Auer rods usually present |
| t(15;17) | M3 | PML-RARa | 40 | 5–8% | Favorable-high cure rate with ATRA-based therapy | DIC |
| t(11;17) | Similar to M3 | PLZF/RARa | ? | <1% | Poor response to ATRA-based therapy | |
| abn 16q22 | M4 with eosinophilia | CBFA/MYH11 | 35–40 | 5% | Favorable | High reinduction rate post relapse |
| abn11q23 | M5 | MLL + many partners | >50 | 3% | Poor except t(9;11) | Hyperleukocytosis, extra medullary disease |
| +8 | Varied | >60 | 5–10% | Poor | ||
| del 5, del 7, 5q-, 7q-, or combinations | Varied; common in FAB M6 | >60 | 15–20% | Poor | Common in patients with secondary AML, prior MDS | |
| Inv 3 | Abnormal megakaryocytes | Ribophorin/EVI1 | ? | <1% | Poor | Increased platelet count; other abnormalities common (del 5, del 7) |
| +13 | Varied; sometimes undifferentiated | Probably >60 | ∼1–2% | Poor | Higher frequency of hybrid features | |
| t(6;9) (p2;q34) | M2/M4 with basophilia | DEK/CAN | ? | <1% | Poor | Prominent basophilia |
| t(9;22) | Usually M1 | BCR/ABL | Probably >50 | ∼1% | Poor | Splenomegaly |
| t(1;22) | Often M7 | MOZ/CBP | Infants | <1% | Poor | Organomegaly |
| t(8;16) | M4,5 | KAT6A/CREBP | ? | <1% | Poor | Erythrophagocytosis, often threap-related |
| Molecular abnormality | ||||||
| Fms-related tyrosine kinase gene mutations | Varied—most common in CN-AML; can be found with (6;9); t(15;17) | Internal tandem repeat or point mutation of | ? | ∼30% in CN-AML | Adverse | |
| Nucleophosmin (NPM1)—(5q35) mutation | Varied | Nucleophosmin (NPM1); often found with other mutations | ? | ∼35% of AML, ∼50% of CN-AML | Favorable DFS except when associated with mutation | |
| CEBPα gene | Varied | Mutation results in decreasing levels of CEBPα (CCAAT entamer binding protein) | ? | ∼15% of CN-AML | Favorable when biallic with FLT3 ITD mutation | |
| Overexpression of BAALC (brain and acute leukemia cytoplasmic) protein | Varied | Overexpression of BAALC | ? | Studied most extensively in CN-AML | Adverse—further studies needed | |
| Partial tandem duplication of MLL (mixed lineage leukemia) gene | Varied | Affects HOX gene function | ? | ∼8% of CN-AML | Unclear—further studies needed | |
| IDH1, IDH2 | varied | isocitrate dehydrogenase | ? | IDH1 in 10%, IDH2 in 18% | variable | specific inhibitors in clinical trials |
| TET2 | varied (common in MDS) | DNA methylation | >60 | 15% | adverse | increased with older age |
| DNMT3A | varied | DNA methylation | >60 | 20% | adverse | increased with older age |
| ASXL1 | varied | epigenetic regulation | ? | 6% | adverse | associated with other mutations |
Abbreviations: AML, acute myelogenous leukemia; ATRA, all trans retinoic acid; DIC, disseminated intravascular coagulation; FAB, French, American, and British; MDS, myelodysplastic syndrome.
Peripheral blood
Most patients with AML present with anemia (median hemoglobin 8 g%), thrombocytopenia (median platelet count 40,000–50,000/μL), and leukocytosis (median WBC count 10,000–20,000/μL). The red blood cell morphology is usually relatively normal. Large, sometimes hypogranular, platelets can be observed, and functional defects can contribute to hemorrhagic manifestations. Most patients are neutropenic, and morphologic abnormalities (nuclear hyperlobulation, hypogranulation, Pelger–Huet anomaly) are often noted in the remaining neutrophils. Careful examination can detect blasts in most patients, although it can be difficult to distinguish among leukemia subtypes (or occasionally even to be confident of the diagnosis of acute leukemia) in patients with a low number of circulating blasts. In occasional patients, marked leukopenia at presentation (the so-called aleukemic leukemia) may obscure the diagnosis until a marrow examination is performed.
AML with recurrent genetic abnormalities
Clinical findings associated with t(8;21), inv(16), and t(15;17) have been appreciated for decades and are described in the sections on M2, M4 with eosinophilia, and M3 (APL) later. AML with t(8;21) and inv(16) are referred to as “core binding factor” (CBF) leukemias because of the molecular abnormalities in transcription produced by these translocations. Mutations of the C-KIT tyrosine kinase receptor, which result in a constitutive proliferative signal, are found in approximately 25% of patients with CBF AML with data suggesting a poorer outcome in patients with this additional mutation as well as in whom the protein is overexpressed.53
The 2008 WHO classification added inv3(q21;q26.2), t(3;3)(q21; 26.2), and t(1;22) (p13;q13) [RBM15-MLK1] usually found in infants (see M7 later) as well as t(6;9) (p23;q23)[DEK-NUP214] to this group. The translocation t(6;9) is very uncommon, may occur more frequently in younger patients, and can be found in association with a variety of AML morphologies, often with prominent basophilia.69 FLT3 ITD mutations are found in approximately two-thirds of patients with t(6;9).70 The outcome with chemotherapy only is poor, and allogeneic transplantation should be performed if possible.
AML with MDS-related changes
Addition of this category shows that a substantial fraction of AML, particularly in older patients, evolves from a prior myelodysplastic disorder. This group includes patients with a prior history of MDS, those with >50% dysplasia in at least two cell lines and patients with the so-called “MDS cytogenetics,” including, -5, -7, i(17)/t(17p), -13, del 11q, del (12p), del 9q and those with complex karyotypes, which may include these changes as well as the presence of marker chromosomes.67 These leukemias seem to arise in a very early hematopoietic stem cell and tend to have low response rates with short durations of response. It seems likely that those patients in this category, only based on morphological abnormalities, may exhibit a variety of oncogenic pathways based on mutational profile and thus may not have a uniformly poor prognosis.5
Therapy-related AML
This category includes patients whose AML followed treatment with chemotherapy and/or radiation therapy for other disorders. Morphology and karyotypes are often similar to those observed in MDS with the addition of a group of patients with abnormalities of 11q23 associated with prior treatment with topoisomerase II inhibitors and often with a short interval until the development of AML.32, 33 Therapy-related AML tends to be more resistant to chemotherapy, and allogeneic transplant should be considered in patients who achieve remission. It is important to note that some patients with therapy-related AML can have inv(16), t(8;21), and t(15;17) (APL) or genetics typical of de novo AML and respond well to standard approaches for these subtypes, although perhaps not as well as those with these karyotypes with de novo disease.5, 36
Extramedullary AML (aka myeloid sarcoma)
Occasionally patients will present for medical attention because of lesions identified to be composed of myeloblasts by histochemical staining, but without apparent bone marrow involvement. Masses can involve the skin, gastrointestinal tract, ovaries, the central nervous system (CNS), and virtually every body organ. There have been only few systematic studies on the management of such patients, although there is a risk of high eventual systemic relapse rate without treatment.71 Most clinicians consider induction and consolidation in medically fit patients after diagnosis. Despite such “early” treatment, the recurrence rate is high; the role of stem cell transplant is unclear, but it is reasonable to consider transplantation to maintain the remission. Some of these patients have a t(8;21) chromosomal translocation, but in this setting, may not have the same favorable prognostic input as found in more typical t(8;21) AMLs.72
AML not otherwise specified
This category includes the large group of patients whose AML does not fall into the previously listed groups.
FAB classification
Although the “old” FAB classification is not used currently, clinical–pathological correlates can be discussed. Wright–Giemsa stained peripheral blood or bone marrow aspirate smears can prefer AML to ALL. In general, the blasts from patients with AML are larger, with more abundant cytoplasm and more prominent, often multiple, nucleoli. The definitive diagnosis depends, however, on the presence of Auer rods, which are linear bundles of myeloid-containing granules. Cytochemical stains, such as myeloperoxidase, which is present in 73% of blasts, can diagnose AML.
M0: Minimally differentiated AML
As shown in Figure 2, some patients have blasts that resemble myeloid blasts, but are negative at the light microscopic level when examined with myeloperoxidase, Sudan Black B, or other histochemical stains. The myeloid nature of these leukemias can be detected, however, by immunologic means or by election microscopy of peroxidase-stained preparations. Electron micrographs reveal ultrastructural peroxidase-positive granules, whereas immunologic phenotyping shows reactivity with antibodies directed against myeloid antigens and nonreactivity with antibodies that characterize lymphoid differentiation.73, 74 The cells are often reactive with antibodies directed against CD34. In general, terminal deoxynucleotidyl transferase (TdT) is absent, but can sometimes be detected in a minority of blasts.
Approximately 7% of patients with untreated AML have minimally differentiated AML (M0 AML), which is relatively resistant to treatment.75 This undifferentiated leukemia can easily be confused with ALL, and it is therefore critically important to obtain data of immunophenotyping of blasts from patients with morphologically undifferentiated leukemias. Other than resistance to chemotherapy, this M0 variant does not appear to be associated with specific clinical findings. Many M0 patients have complex karyotypic abnormalities.75 No distinctive cytogenetic pattern has been noted, except trisomy 13, which has been reported to occur in some patients with morphologically less differentiated leukemias.76
M1: Myeloid leukemia without maturation
The blasts from patients with M1 morphology have round nuclei with moderate amounts of sometimes lightly granulated cytoplasm, which can contain Auer rods (Figure 3). In contrast to M2, there is little evidence of myeloid maturation, with <10% of cells beyond the level of the promyelocyte. There is no particular age, gender, clinical feature, or characteristic cytogenetic abnormality associated with this morphologic variant.
M2: Myeloid leukemia with maturation
In contrast to M1, there is obvious continued maturation in the myeloid series with the presence of promyelocytes, myelocytes, and often more mature myeloid elements. Granulation is generally more obvious; Auer rods are often prominent (Figure 4). Approximately 20–25% of patients with M2 AML have a characteristic translocation between chromosomes 8 and 21 [t(8;21)(q22;22)]; this translocation is seen almost exclusively in patients with M2 and Auer rods. Such patients have a lower median age (∼30 years), very high initial complete response rate (>85% in most series), lower relapse rate, and increased long-term disease-free survival, particularly when treated with high-dose cytarabine-based consolidation therapy,77 except for a subset with an activating mutation of KIT or adverse gene signature.78 The incidence of extramedullary granulocytic sarcomas, often in unusual sites, may be increased in patients with t(8;21) M2 AML, whose blasts express the adhesion molecule CD56 on their surface. These can present as discrete tumor masses, sometimes in paraspinal locations, confer a poor prognosis, and are distinct from the gingival and cutaneous involvement found in monocytic leukemia.71
M3: Acute promyelocytic leukemia
APL is one of the most distinctive subtypes of AML with regard to morphologic, clinical, cytogenetic features, and response to differentiating agent therapies, such as all-trans retinoic acid (ATRA) and arsenic.79 In most patients, the morphologic diagnosis is straightforward, with the marrow being replaced by blasts that resemble unusually heavily granulated progranulocytes. The nuclei are spherical, with obvious nucleoli, and the cytoplasm is filled with multiple, large, and often coalesced azurophilic granules (Figure 5). Auer rods are usually observed, and multiple Auer rods (the so-called faggot cells) are frequently noted. In a minority of patients, the blasts are hypogranular, and sometimes granules can only be visualized by an electron microscope.80 This hypogranular variant often has cells with bilobed or lobulated nuclei, which can sometimes be confused with monocytic variants of AML. In contrast to the typical leukopenic presentation of APL, patients with the hypogranular variant tend to have higher white cell counts. In both types of APL, staining with either Sudan Black B or myeloperoxidase is strongly positive. Class II human leukocyte antigens (HLA DR), which are found on all hematopoietic precursors, are not usually detected on the surface of the malignant progranulocytes. The explanation for and biologic implications of this finding are not known. By contrast, CD33 is consistently strongly expressed.81
Patients with APL tend to be somewhat younger, with a median age of 30–40 years, although it is observed in patients of all ages. APL accounts for approximately 10% of AML and may be more prevalent in Latinos82 and obese people.83 It is almost uniformly characterized by hypofibrinogenemia, variable depletion of other coagulation factors, elevated levels of fibrin degradation products, and increased consumption of endogenous and transfused platelets. The granules contain potent procoagulants, and the rate of disseminated intravascular coagulation (DIC) is generally increased following lysis of blasts by chemotherapy,84 often with increased bleeding, although the problem can be rapidly ameliorated with the use of ATRA.85 In some patients, there is evidence that accelerated fibrinolysis may be the primary event triggering coagulopathy.86 APL is associated with the highest frequency of hemorrhagic morbidity and mortality, the latter usually related to intracranial hemorrhage, emphasizing the need to initiate ATRA at the first thought of APL.87 Before DIC is controlled with ATRA, severe hypofibrinogenemia (<100 mg/dL) may require supplementation with cryoprecipitate, and thrombocytopenia should be managed with the aggressive use of platelet transfusions.
Almost all patients with APL have a characteristic translocation involving chromosomes 15 and 17 [t(15;17) (q22;q12)],88 which may be accompanied by additional cytogenetic abnormalities, such as trisomy 8.89 Reverse transcription polymerase chain reaction (RT-PCR) can be used to detect the fusion transcript, is useful for assessing minimal residual disease (MRD),90 and permits the proper classification of a patient with clinically and morphologically typical APL and an apparently normal karyotype. The break point on chromosome 17 is in an intron of the retinoic acid receptor alpha gene. A gene that has been termed PML, also with DNA-binding capability, is translocated from chromosome 15, resulting in the formation of a fusion protein that functions in a dominant manner to block transcription of genes controlled by RAR-α, probably by nuclear corepressor activity. Retinoic acid treatment relieves the corepressor activity,91, 92 allowing transcription of genes involved in differentiation.93 There is also interest in the use of histone deacetylase inhibitors in patients with APL as another means of enhancing gene expression.94 FLT3 ITD mutations can be detected in approximately one-third of patients with APL, are associated with higher WBC counts and M3 variant morphology, but unlike the case for non-APL AML, does not always seem to be associated with inferior outcome.95, 96 A group of patients with a leukemia similar in morphology to APL, but with alternate translocations such as t(11;17)(q23;q21) have been described. Although RAR-α is rearranged, these patients fail to respond to ATRA. A novel zinc-finger gene termed PZLF from chromosome 11 is translocated to RAR-α, rather than the PML gene from chromosome 15, creating a fusion protein that does not allow the ATRA-mediated release of transcriptional corepressor activity.97
Historically, the remission rate in patients with APL treated with anthracycline-based chemotherapy was quite high. Initial drug resistance was very unusual, and most treatment failures were related to hemorrhagic or infectious deaths. APL is uniquely sensitive to single agent anthracycline therapy, which can produce CR rates >80%.98 In contrast to other types of AML, remission can be attained with chemotherapy in APL without producing bone marrow aplasia.99 Posttreatment bone marrows frequently remain cellular with abnormal progranulocytes, with follow-up marrows demonstrating disappearance of these cells and return of normal hematopoiesis without additional chemotherapy. DIC does not reappear despite the persistence of morphologically abnormal cells. Undoubtedly, this unique feature of APL is related sensitivity to agents that have a differentiating and noncytotoxic mechanism of action (see section titled “Therapy of Acute Promyelocytic Leukemia (APL)”), and is now cured in at least 80% of patients with minimal or no chemotherapy with the use of ATRA and arsenic trioxide in combination.100
M4: Myelomonocytic leukemia
Myelomonocytic leukemia is characterized morphologically by a mixture of myeloid and monocytic elements and represents approximately 15–20% of newly diagnosed patients with AML. According to the FAB criteria, >20% of the leukemic cells must be monocytic in morphology to distinguish this variant from FAB M1 and particularly from FAB M2. The monocytic elements often resemble partially differentiated monocytes with lightly granulated, grayish cytoplasmic, and folded nuclei, which are frequently seen in the peripheral blood (Figure 6). Monocytic derivation can be confirmed by staining with nonspecific esterases such as α-naphthyl acetate and α-naphthyl butyrate.
There is no distinct clinical picture associated with this variant, perhaps because this classification encompasses a wide spectrum of patients owing to the generous morphologic criteria for inclusion. The median age tends to be somewhat higher, and there may be an increased incidence of hyperleukocytosis and extramedullary leukemic involvement, as can be seen with monocytic leukemia. There is no particular cytogenetic clustering, and it is impossible to accurately predict short- or long-term outcome in patients with M4.
M4EO: Myelomonocytic leukemia with eosinophilia
Approximately 5% of patients with de novo AML have typical morphologic features of myelomonocytic leukemia in the presence of variable numbers of dysplastic eosinophils at various stages of maturation. The distinctive eosinophils usually represent only 5–10% of the cells of the marrow.101 In general, these cells contain large basophilic granules in addition to typical eosinophilic counterparts. Occasional patients with M2 morphology with eosinophilia have also been described.
M4Eo tends to occur in patients of younger age (median 35–40 years) and is associated with an excellent prognosis.102 CR rates are high (generally >85%), and failure because of initial drug resistance is unusual. In some series, this variant represents the subtype with the most favorable long-term prognosis. Mutations of the C-KIT tyrosine kinase receptor have recently been described in some patients with CBF AML, with data indicating a poorer outcome in this subset.53, 78 In addition to long initial CRs, second remissions, which are often quite sustained, are generally easier to accomplish in patients with FAB M4Eo.103 Older series indicated the possibility of achieving a high rate of CNS relapse in patients with bone marrow eosinophilia. With more intensive regimens using higher doses of cytosine arabinoside (ara-C), CNS relapse in AML is unusual, and patients with FAB M4Eo do not require prophylactic CNS therapy.
All patients with FAB M4Eo exhibit a cytogenetic abnormality, involving chromosome 16 at band q22. In most patients, the cytogenetic changes involve a pericentric inversion (inv16), although translocations between the two chromosomes 16 with homologous deletions at 16q22 have also been noted.104 This break point involves a fusion between the CBF-β chain and the gene encoding the smooth muscle myosin heavy chain. The fusion protein thus generated may use nuclear corepressor activity (in the form of histone deacetylase), which prevents transcription of genes required for myeloid differentiation in a manner analogous to the CBF-α ETO fusion in t(8;21) M2 AML.105 Although it is evident that patients with inv(16) leukemia respond very well to intensive chemotherapy [3-year DFS rate of >60–70% in those receiving high-dose ara-C (HIDAC)],102 the reason for this is unclear.
M5: Monocytic leukemia
Two variants of monocytic leukemia have been described; in both, >80% of the blasts are of monocytic derivation. Less common is the so-called M5a, in which the monocytic blasts have spherical nuclei and small amounts of sometimes deeply basophilic cytoplasm without evidence of morphologic differentiation. In monocytic leukemia with differentiation (M5b), at least 20% of the blasts resemble promonocytes with folded nuclei and abundant, lightly granulated cytoplasm, generally without Auer rods. The nuclear folding can often be quite marked with rarification of the nuclear chromatin (Figure 7). Phagocytosis of other hematopoietic elements by these cells is frequently noted in bone marrow preparations. These monocytic elements stain prominently with nonspecific esterase that is inhibited by fluoride.
Although observed in patients of all ages, monocytic leukemias are somewhat more common in older adults. Patients with FAB M5 have higher blast counts at diagnosis, and problems with hyperleukocytosis are most common in this morphologic variant (see section titled “Complications”).106 In addition, the incidence of extramedullary leukemia is highest in M5, particularly in those with evidence of morphologic differentiation.107 For example, it is common for patients to present to the dentist with gingival hypertrophy (Figure 8). Skin infiltration is common at both diagnosis and relapse, and it generally represents the initial site of recurrence, sometimes while the bone marrow is still morphologically normal. Other less common areas of extramedullary involvement include the gastrointestinal tract, conjunctiva, and the CNS. It is likely that extramedullary infiltration is related to active migration of the leukemic promonocytes to these sites. These partially differentiated cells are capable of migration to skin windows in vivo as well as phagocytosis of microorganisms and adherence to nylon fibers in vitro.108
Serum levels of lysozyme are elevated in most patients with AML, but are generally much higher in patients with monocytic leukemia.109 Lysozyme can affect renal tubular function, and severe, symptomatic hypokalemia can occur in patients with FAB M4 and M5 leukemia. This problem generally resolves with cytoreduction, but can also produce hypokalemic side effects of vomiting and diarrhea.
In addition to the initial problems presented by complications of hyperleukocytosis, patients with monocytic leukemia tend to have lower complete response rates related to drug-resistant disease. Although previous studies indicated that CR durations tend to be shorter with very low rates of long-term disease-free survival, an analysis of a large number of patients treated by the ECOG suggested similar outcomes to other morphologic subtypes of AML when other risk factors are taken into account.110 A variety of cytogenetic abnormalities can be detected, although the most common findings involve abnormalities of chromosome 11 at band q23. This break point, at what has been termed the mixed lineage leukemia (MLL) gene, can be involved in leukemias of myeloid or lymphoid origin as well as in those following therapy with epipodophyllotoxins and other drugs directed at topoisomerase II.111 The MLL gene, also called All-1 or HRX, may combine with at least 16 different genes in balanced translocation.112 MLL is homologous to a gene important in the development of Drosophila and includes DNA-binding elements. The t(9;11) translocation is relatively common, involving the MLL gene, which may in fact confer a better prognosis than formerly thought, with high initial CR rates.113, 114 There is an association between M5b with extensive erythrophagocytosis and the t(8;16)(p11;p13), a translocation involving the CBP class of translocation factors that are positive regulators of myeloid differentiation. 115
M6: Erythroleukemia
Erythroleukemia, often called Di Guglielmo syndrome previously, is a variant of AML, in which morphologic abnormalities of erythropoiesis are most prominent.116 Cases of pure erythroleukemia, in which the predominant malignant cell is clearly identified as a pronormoblast, are rare. Rather, this is a disease of the myeloid stem cell with marked dysplastic changes in all three hematopoietic lines. Together with the increase in myeloid-appearing blasts, there is persistence of morphologic abnormalities in the erythroid series with profound megablastosis, multinuclearity, karyorrhexis, increased number of mitoses, and staining with periodic acid–Schiff (PAS), often in a block pattern (Figure 9). Increased iron stores are usually observed, often with ringed sideroblasts. These changes are morphologically identical to those observed in patients with myelodysplasia, and many observers feel that most cases of erythroleukemia are biologically similar, if not identical, to patients with refractory anemia with excess blasts. This contention is supported by the very poor response to therapy in both groups, the tendency for the disease to occur in patients of older age, and the presence of similar cytogenetic abnormalities (complex karyotypic abnormalities, loss of part, or all of chromosomes 5, and/or 7, and marker chromosomes).116 Nonetheless, in order to be consistent in terms of protocol entry and reports of clinical trials, the FAB and WHO groups, somewhat arbitrarily distinguished among M6, myelodysplasia (RAEB), and other FAB subtypes with significant numbers of erythroblasts by quantification of the number of erythroblasts and myeloblasts. Most of these patients will now be placed in the “AML with MDS-related Changes” in the most recent WHO iteration. M6 is defined by the presence of >30% blasts among nonerythroid cells when >50% of the marrow nucleated elements are erythroid. Antibodies against glycophorin A are lineage-specific for erythroblasts, but immunologic phenotyping is rarely needed to identify these morphologic subtypes.
M7: Megakaryocytic leukemia
Morphologic abnormalities of megakaryocytopoiesis, usually characterized by the presence of mono- or binucleated micromegakaryocytes, are common in many variants of AML and can be particularly prominent in patients with M6 or myelodysplasia. A minority of these patients have thrombocytosis and abnormalities of chromosome 3 [inv(3) (q21;q26)]. This cytogenetic abnormality is often found in association with other chromosomal deletions, with a variety of primary morphologies (M1, M2, M4), and in patients with a prior background of MDS.117 These patients have a poor response to initial treatment and low overall survival (OS). Thrombocytosis is present not only in patients with the inv(3) karyotype, but also in those with AML at the time of diagnosis.118 The gene at the chromosome 3q21 break point is associated with the activation of EVI1 transcription factor.119
Diagnosis of FAB M7 is reserved for patients in whom the predominant leukemic cell is of megakaryocytic lineage.120 In some patients, there is evidence of megakaryocytic dysplasia or multinucleated cells that strongly point toward principal involvement of the megakaryocyte (Figure 10), whereas in others, the leukemia is undifferentiated morphologically, with variable amounts of agranular cytoplasm, and it is sometimes confused with M1 or ALL. Sudan Black B (Figure 11), myeloperoxidase, and α-naphthyl butyrate stains are negative, whereas PAS and acid phosphatase may be positive, usually in a diffuse, speckled pattern. However, histochemical staining is nondiagnostic and the definitive diagnosis depends on the detection of platelet-specific peroxidase by either ultrastructural techniques or the demonstration of a variety of platelet antigens (usually glycoprotein IIb/IIIa [CD41] or von Willebrand factor) on the surface of the blasts.121 At times, the diagnosis can be quite difficult to confirm, particularly because of the increased marrow reticulin in most patients, rendering the marrow fibrotic and inaspirable. Careful evaluation of peripheral blood blasts is necessary in such patients. It is likely that most patients with acute myelosclerosis in fact have acute megakaryocytic leukemia, which should not be confused with the late stages of primary myelofibrosis, and, indeed, prominent splenomegaly is not a clinical feature of M7. Although it is an uncommon variant of AML, most series suggest that this subtype is associated with a very poor prognosis. Prolonged aplasia is common following induction chemotherapy, and, because of the marrow fibrosis, it is often difficult to follow the results of therapy with repeated marrow aspirations. There have been relatively few cytogenetic evaluations of this variant, and except for the inv(3) karyotype and cases of t(1;22) (p13;q13) [RBM15/MKL1 gene fusion] found in infants,122 no consistent abnormality has been identified.
Acute panmyelosis with myelofibrosis
This is a very rare variant of AML felt to derive from the hematopoietic stem cell, in which the marrow demonstrates a marked increase in reticulin fibers with evidence of morphologically abnormal trilineage hematopoiesis and a variable number of blasts with an immature myeloid immunophenotype.123, 124 Patients usually present with pancytopenia and constitutional symptoms. It can sometimes be difficult to distinguish acute panmyelosis with myelofibrosis (APMF) from acute megakaryocytic leukemia or MDSs with myelofibrosis. APMF responds poorly to standard chemotherapy.
Mixed phenotypic leukemia
There are certain cases of acute leukemia that defy easy categorization, because the cells may have features of both lymphoid and myeloid derivation. The WHO 2008 monograph on classification of hematopoietic and lymphoid tissues uses the term “mixed phenotype acute leukemia” (MPAL) to encompass these heterogeneous groups of neoplasms. MPALs are defined by immature cells which display cytochemical or immunophenotypic features of both myeloid and lymphoid lineages (biphenotypic), or there are two different populations of leukemia cells: myeloid and lymphoid (bilineal). The difference between bilineal and biphenotypic cells generally does not alter the diagnostic therapeutic approach. Although prior systems such as the EGIL (European Group for Immunological Characterization of Acute Leukemias) were used,125 the most recent algorithm is based on the 2008 WHO monograph.126 Such entities tend to have features of either B-ALL and AML or T-ALL and AML and are called B-myeloid or T-myeloid diseases, respectively. Entities with evidence of derivation from both lineages are specified if there is a known recurrent genetic lesion such as Philadelphia chromosome-positive leukemia, MLL-rearranged leukemia, or AML-defining balanced translocation such as t(8;21). Technically, MPALs also exclude secondary leukemias, leukemias with FGFR1, mutations, and CML in blast crisis.127
The essential features of an MPAL is the specific expression of certain lineage-defining markers in two categories. Although the algorithm is slightly complex, CD3 expression is evidence of T-lymphoid derivation, and CD19 together with one or two other markers indicate B-lymphoid origin. Myeloid origin can be determined by a set of monocytic immunophenotypic markers or most commonly by myeloperoxidase expression. TdT is characteristically seen in ALL; however, because 25% of AML patients express TdT, this is not considered a lineage defining abnormality. It is also important to indicate that many AMLs may have lymphoid antigens detectable by flow cytometry, but do not meet the criteria for biphenotypic leukemia. Such cases should be considered in the AML prognostic and therapeutic rubric.127
The literature does not yield clear guidelines for the treatment of MPALs. In particular, patients with BCR/ABL-positive disease should be treated by chemotherapy together with a tyrosine kinase inhibitor. Most other MPALs should be treated with an ALL regimen, although controversy certainly exists particularly if there is prominent expression of myeloperoxidase or even Auer rods. A regimen that combines AML- and ALL-type chemotherapy is also possible, but may be more toxic than a more typical ALL regimen. Given the relatively resistant nature of MPALs, it is reasonable to consolidate any remissions achieved with an allogeneic stem cell transplant.
Presenting signs and symptoms
Patients with AML generally present with symptoms related to complications of pancytopenia, including combinations of weakness, easy fatigability, infections of variable severity, or hemorrhagic findings such as gingival bleeding, ecchymoses, epistaxis, or menorrhagia. Occasionally patients present with prominent extramedullary sites of leukemia usually related to either cutaneous or gingival infiltration by leukemia cells. Bone pain is infrequent in adults with AML, although some individuals describe sternal discomfort or tenderness, occasionally with aching in the long bones, particularly of the lower extremities. In general, it is difficult to determine the time of onset of AML precisely, at least in part because individuals have different symptomatic thresholds for choosing to seek medical attention. It is likely that most patients have had more subtle evidence of leukemia for weeks, to perhaps months, before diagnosis.
The findings on physical examination are variable and generally nonspecific. If fever is present, an infectious site must be vigorously sought and treated empirically with broad-spectrum antibiotics. A large number of patients have fever related solely to the underlying leukemia, which abates with appropriate chemotherapy. Examination of the skin can reveal pallor, infiltrative lesions suggestive of leukemic involvement, cutaneous sites of infection, which may be either primary or embolic, or, most commonly, petechiae or ecchymoses related to thrombocytopenia and/or coagulopathy. Examination of the fundus reveals hemorrhages and/or exudates in the majority of patients (see section titled “Ophthalmic Complications”). The conjunctivae may be pale, according to the magnitude of the anemia. Careful examination of the oropharynx and teeth is important because of the infrequency of leukemic involvement. Palpable adenopathy is uncommon in patients with AML, and significant lymph node enlargement is rare. Similarly, hepatomegaly and splenomegaly are uncommon and, if found, may suggest the possibility of ALL or chronic myeloid leukemia in blast crisis. None of these findings is a diagnostic of acute leukemia, and the final diagnosis and categorization depends on appropriate evaluation of the peripheral blood and bone marrow.
Because of the rigorous nature of the chemotherapy required for the successful treatment of AML, particular attention should be paid to other medical problems that could complicate management of the patient. A history of congestive heart failure or other heart disease may preclude therapy with anthracyclines and mandates careful monitoring of the large amounts of intravenous fluids, including antibiotics, blood and platelet transfusions, hydration for nephrotoxic antimicrobial agents, and sometimes parenteral nutrition, given during the 3–4 weeks of chemotherapy-induced pancytopenia. Prior transfusion for other disorders or multiple previous pregnancies may presage difficulties with platelet transfusions or herald the occurrence of transfusion reactions after red blood cell or platelet administration. Careful appraisal for possible drug allergies is critical, because virtually every patient will require antibiotic therapy. A history of prior herpes simplex infections (or the presence of an elevated antibody titer) provides justification for prophylactic administration of acyclovir.128 In premenopausal women, menses should be suppressed with a GNRH agonist or estrogens and/or progestational compounds until thrombocytopenia is resolved. Considerations of fertility preservation should be discussed with selected patients. Sperm-banking in males wishing to have a family should be considered, although many patients are either oligo- or azospermic at the time of diagnosis. GNRH antiagonist therapy, by shutting down the pituitary–ovarian axis during chemotherapy, may preserve fertility in females with childbearing potential.129
Once the diagnosis (Table 3) is established, the physician and staff must present the goals of therapy and the side effects of treatment to the patient and his/her family. For almost all patients, this discussion can rightfully emphasize the potential benefits of treatment with regard to both the short- and long-term outcomes. It is appropriate and necessary to repeat this discussion and counsel later during the patient’s course of hospital stay. In general, for younger patients, induction therapy followed by postremission treatment with intensive chemotherapy and/or allogeneic transplant is appropriate. For older patients (>60–65 years), there may be merit to the standard, more aggressive approach, but hypomethylating agent therapy should be discussed.
Table 3 Initial diagnostic evaluation
History and physical examination—In addition to an overall comprehensive evaluation, emphasis should be placed on the following:
|
Bone marrow aspirate and biopsy
|
Blood chemistries
|
Coagulation studies
|
| Chest radiograph, electrocardiogram, left ventricular ejection fraction if clinically indicated |
| HLA typing (patient and family); lymphocytotoxic (anti-HLA) antibody screen |
| Herpes simplex and cytomegalovirus serology |
| Lumbar puncture (only if symptomatic) |
Abbreviation: HLA, human leukocyte antigen.
Therapy: general overview
The therapy of AML has traditionally been divided into stages: induction, postremission therapy of varying intensity and duration, and postrelapse therapy. In newly diagnosed patients with AML, the goal of induction therapy is to achieve CR, which then permits the administration of subsequent therapy that for most patients is designed to maximize the rate of disease-free survival and cure. CR is defined primarily on morphologic grounds and includes the development of a bone marrow containing less than 5% blast elements, no signs of extramedullary leukemia, and return of normal neutrophil (>1500/μL) and platelet (>150,000/μL) counts. Even if the bone marrow contains <5% blasts, patients are not considered to be in remission if distinctive morphologic signs of leukemia, such as Auer rods, are noted. Low hemoglobin levels and the presence of symptoms unrelated to leukemia no longer exclude CR, as they are often treatment-related and slow to normalize.
It has been assumed that CR is accomplished because the cytotoxic chemotherapy markedly decreases the number of cells in the leukemic clone, thereby allowing repopulation of the bone marrow by residual normal progenitors, whose proliferation had been suppressed. This explanation is supported by observations that cytogenetic abnormalities present in the original leukemia cells cannot be detected in patients in remission. However, in some patients, intensive chemotherapy appears to eliminate the block in differentiation such that the apparently normal cells seen in the bone marrow and peripheral blood during a CR are actually progeny of the leukemic clone.130, 131 as determined by X chromosome-linked polymorphisms (glucose-6-phosphate dehydrogenase isoenzyme or restriction fragment-length polymorphisms) or the presence of disease-specific mutations in apparent normal cells at the time of CR.132 The frequency of such “clonal CRs,” the possible association with particular subtypes of AML or remission duration, the mechanism by which this important biologic phenomenon occurs, and a number of important technical issues need to be further defined.
Induction therapy: general principles
Induction therapy is designed to produce rapid clearing of leukemic cells from the peripheral blood with subsequent marrow aplasia. The only exception to this principle occurs in patients with APL (FAB M3), in whom remission can be achieved despite the persistence 2–3 weeks later of what appear morphologically to be viable leukemia cells,133 although, especially given the use of ATRA/arsenic-based therapies, multiple marrows in APL need not be done. Approximately 1 week after standard induction therapy (the so-called “3 + 7”; 3 days of anthracycline, usually daunorubicin and 7 days of continuous infusion cytarabine) is completed (generally 2 weeks after the initiation of treatment), bone marrow aspirates and biopsies are done to evaluate the magnitude of cytoreduction. If the marrow is profoundly hypoplastic, one waits for count recovery. If the marrow is not hypoplastic and only leukemia cells are noted on the day 14 marrow, then a second course of therapy is generally administered. If high-dose cytarabine-based therapy was used as induction chemotherapy, multiple marrow examinations are not recommended.
At times, particularly if the “day 14” marrow is hypocellular, it can be difficult to distinguish between residual leukemia cells and normal undifferentiated hematopoietic progenitors. In this instance, it is advisable to delay retreatment and perform another marrow aspirate in a few days. If there is no evidence of further maturation, then a second course of treatment is indicated. The presence of erythroid precursors, juvenile megakaryocytes, or increased peripheral blood platelet or neutrophil counts serve as indicators to delay a second course of treatment as normal regeneration is occurring. With standard regimens, approximately 30% of patients with AML require two courses of treatment to enter remission. Despite these guidelines, there remains considerable variability and imprecision as to when a second course of therapy is indicated.
Most trials report results as complete response or no response. It is beneficial, however, to more rigorously classify the causes of failure in investigations of prognostic factors or assessment of the cytotoxic activity of different regimens, to achieve remission. One such classification divides nonresponders into those with apparent chemotherapy-resistant leukemia, those who die with aplastic bone marrows in whom the response to chemotherapy cannot be determined, and those in whom either early death or failure to obtain adequate bone marrow studies before death preclude determination of whether persistent leukemia was present. Patients with drug-resistant leukemia include those who survive treatment and those who die but have morphologic evidence of leukemia in the bone marrow or blood. The criteria for CR are generally reached a median of 30–35 days after treatment has begun, although patients achieve adequate levels of circulating neutrophils (>500/μL) and no longer require platelet transfusion (at counts of ∼10–20,000/μL), at least 7–10 days earlier, which may be beneficial in sorting out activity of novel and/or noncytotoxic agents. Other types of responses include PR (50% reduction in marrow blast with adequate count recovery), CRp (same as CR, but platelets are not fully recovered), CRi (same as CR, but with low neutrophil counts), and morphological leukemia-free state (<5% marrow blasts, without mention of normal cell counts). This categorization is relatively simple to perform in most patients and can be beneficial in distinguishing between failures due to drug resistance and inadequacy of supportive care.2, 133
The overall rate of CR with standard chemotherapy in large cooperative group studies is approximately 65%. Patient age and cytogenetics are the most critical clinical variables, with CR rates of 75–80% in younger patients and approximately 50% in patients >60 years of age. The reasons for treatment failure vary according to patient age. With improved supportive care, it is uncommon for patients <50 years of age to die from complications of treatment, and most of the approximately 25% induction failure rate is a consequence of drug-resistant leukemia. By contrast, in patients >60 years of age, failures are due to drug-resistant leukemia and deaths occurring during marrow aplasia as a consequence of reduced end organ tolerance, occurring at overall frequencies of 40% and 10%, respectively.
Although there have been gradual improvements in the CR rates worldwide in younger adults with AML during the past 25 years, much of this can be attributed to better supportive care and not to changes in therapy. With the exception of the use of ATRA and arsenic for patients with APL (see below), relatively few changes in therapy have been made since the introduction of combined therapy with daunorubicin and ara-C, the so-called “7 and 3” regimen.134 This two-drug combination was derived from observations of single-agent activity of either compounds. Daunorubicin is generally administered by intravenous push at doses of 45–90 mg/m2/day for 3 days; ara-C is administered at doses of 100–200 mg/m2/day by continuous infusion for 7 days (Table 4). A series of randomized studies134–140 by the Cancer and Leukemia Group B (CALGB) showed that:
- 1. Results were superior using the 7 and 3 regimen to 5 days of ara-C and two doses of daunorubicin.
- 2. The addition of oral 6-thioguanine (DAT regimen) to the 7 and 3 regimen did not increase the CR rate.
- 3. Continuous infusions of ara-C produced a slightly better outcome than twice-daily short intravenous infusions when combined with daunorubicin.
- 4. Results were not improved when ara-C was administered by continuous infusion for 10 days compared with 7 days.
- 5. Substitution of doxorubicin for daunorubicin produced almost identical CR rates, although mucosal toxicity was higher with doxorubicin.
- 6. There was no overall benefit from doubling the dose of ara-C from 100 to 200 mg/m2/day.
Table 4 Representative chemotherapy regimens for acute myeloid leukemia
| Dose | Route | Days | |
| Induction | |||
| Cytarabine+ | 100–200 mg/m2 | Continuous IV infusion | 1–7 |
| Daunorubicin | 45–60 mg/m2 | IV | 1–3 |
| or | |||
| Idarubicin | 12 mg/m2 | IV | 1–3 |
| or | |||
| Mitoxantrone | 12 mg/m2 | IV | 1–3 |
| Postremission | |||
| Cytarabine | 3 g/m2 q12h (over 3 h) | IV (6 doses) | 1, 3, 5 |
| or | |||
| Cytarabine | 1.5–2 g/m2 q12h (over 1 h) | IV (8 doses) | 1–4 (12 doses) |
| or | |||
| Cytarabine | 100 mg/m2 | Continuous IV infusion | 1–5 |
See text for details about number of courses and patient selection for different regimens.
Other modifications of the 7 and 3 regimen
Most attempts to improve the 7 and 3 induction regimen have not led to increase in OS. Alternative anthracyclines or other agents such as mitoxantrone, rubidazone, aclacinomycin, amsacrine, mitoxantrone, and idarubicin have been used in several trials.141–145 None of these studies showed a survival or disease-free survival advantage with these different agents, perhaps because most are relatively similar in structure and mechanism of action and hence, become susceptible to the same mechanisms of resistance. The largest reported experience has been with idarubicin, where three initial trials showed that induction results are at least equivalent to results achieved with daunorubicin and ara-C.142, 144 An important randomized trial did not show an advantage for idarubicin compared with daunorubicin, in older patients.146 In addition, the duration of myelosuppression was longer in the idarubicin cohorts, calling into question the equitoxicity of the arms. Therefore, the substitution of these compounds did not have a major impact on the long-term disease-free survival rate of patients with AML.
An Australian trial added etoposide to the 7 and 3 regimen.147 The CR rate was similar in older patients when compared with daunorubicin and ara-C only with a possibly modest prolongation of CR duration in younger patients receiving etoposide. This was a relatively small study and the overall disease-free survival in the group receiving etoposide during both remission induction and postremission therapy was similar to other larger studies using daunorubicin and ara-C only. Large phase I studies from the CALGB, in which etoposide was added to the 7 and 3 regimen, had outcomes similar to the past experience with only the two drugs.148
Because HIDAC is a beneficial postremission therapy (see below), several groups have tested the use of this approach during induction in younger patients. Studies have compared standard 7 and 3 to daunorubicin plus intermediate- or high-dose ara-C (2–3 g/m2 for 8–12 doses).149–151 These studies failed to show an increased CR rate for the recipients of HIDAC, although one study documented a more prolonged duration of CR (but no change in OS) in the patients randomized to HIDAC.149 The addition of HIDAC to standard daunorubicin/ara-C during induction has also been studied. Although a small trial showed an 87% remission rate in patients <60 years old,152 a cooperative group trial failed to confirm those positive results.153 Results seem to be equivalent if HIDAC is used either as part of induction or only as postremission therapy.154
New induction strategies
Although it is evident that 3 + 7-based strategies remain the standard of care, new data suggest that many patients (perhaps all of those up to age 65) should be treated with daunorubicin at a dose exceeding the originally used 45 mg/m2. The ECOG 1900 study randomized patients to 45 mg/m2 per day of daunorubicin for 3 days versus 90 mg/m2 for 3 days and found for most subgroups (and even those with high white counts or FLT3 ITD with longer follow-up)155 that the higher dose resulted in prolonged survival. A study in older adults in Europe with a similar design also suggested that a dose of 90 mg/m2 per day was better than 45 mg/m2 at least in those between 60 and 65 years of age.156 A report from a study conducted in the United Kingdom suggested that 60 mg was equally efficient as 90 mg/m2.157 As noted, most studies that have added drugs to the 3 + 7 backbone or substitute HIDAC for standard continuous infusion ara-C in 3 + 7 have not led to an increased OS. However, the Polish Acute Leukemia Study Group showed that the addition of cladrabine to anthracycline/cytarabine was superior to 3 + 7158; however, the CR rate in the control group in these younger adults was lower than expected and these results need to be confirmed by additional studies. Continuous infusion of HIDAC and idarubicin has led to favorable results in nonrandomized phase 2 studies at the MD Anderson Cancer Center.159 Addition of CCNU to the 3 + 7 regimen benefited older adults in a French trial.160 The FLAG IDA regimen, while quite intensive and toxic, led to a high remission rate and disease-free survival rate.161 Another agent that could potentially be added to a 3 + 7-type backbone is gemtuzumab ozogamicin (GO), an antibody toxin conjugate directed against the CD33 antigen expressed on blasts from most patients with AML and which was initially approved as a single-agent treatment for older adults with relapsed AML162 and then withdrawn after randomized trials comparing standard initial induction treatment with or without the addition of GO showed marginal benefit and/or equivocal results.163 However, trials in France164 and the United Kingdom165 have suggested that the addition of relatively small doses of GO to standard chemotherapy may have a benefit in patients up to age 70 with AML. The drug has still not reentered the market until the time of this writing; confirmatory trials in North America will likely be required to show that it is worth using this drug routinely, although results in those with favorable chromosomal abnormalities are encouraging.166
Approach to the older patient and other poor prognostic subgroups
Is it worth administering cytotoxic chemotherapy with a 3 + 7 induction approach to patients who have little chance for long-term benefit? This is still a subject of major debate and controversy relevant to the care of AML patients older than 60–65 years of age and some selected younger patients whose prognosis is very poor based on monosomal karyotype54 or p53 mutations17 if known at the time of diagnosis. For younger adults with known adverse prognostic features, there seems to be little recourse at the moment to using a 3 + 7 regimen or similar induction approach followed by postremission therapy followed by allogeneic stem cell transplant if possible. If a sibling donor, matched unrelated donor, or “favorable” partially mismatched unrelated donor is not available, then strong consideration should be made for haploidentical or umbilical cord blood stem cell transplantation (SCT) in these cases. The degree of chemotherapeutic resistance is profound in such cases and the results with allogeneic transplant remain relatively poor, but still may offer a slightly better outcome than17 a chemotherapy-based postremission approach.
It is possible that some older adults will be better served by using a lower-dose chemotherapy approach to initial therapy than a standard 3 + 7 regimen. For a standard chemotherapy, the daunorubicin dose should be ≥60 mg/m2 in virtually all patients. On the contrary, particularly when adverse prognostic factors are present, at times it may be reasonable to consider lower-dose chemotherapy with drugs such as the hypomethylating agents, azacitidine or decitabine, or in clinical trials, with the slightly more toxic single agent, clofarabine. Although there are many prognostic algorithms for older adults, features that are generally considered to decrease the likelihood of good outcome with 3 + 7-based chemotherapy are those that were used in the clofarabine phase II167 trial, including age > 70 years, antecedent hematological abnormality, comorbid diseases, nonfavorable cytogenetics. A phase 2 trial using 30 mg/m2 of clofarabine for 5 days produced a 35% CR rate with a 10% rate of toxic death. In part because of difficulties in defining a group of patients “unfit for intensive therapy,” this trial did not result in the Food and Drug Administration (FDA) approval and clofarabine is being evaluated in a large ECOG-led phase III trial comparing clofarabine to 3 + 7 in this age-group.
In Europe, low-dose ara-C is another frequently used option for patients who are deemed not fit or at least “inappropriate” for standard induction chemotherapy. Low-dose ara-C seems to be slightly better than hydrea,168 but is used little in the United States. Two important trials have compared hypomethylating agent to other therapies for older adults with AML. One such trial compared decitabine to low-dose ara-C or supportive care and yielded a 25% CR rate.169 Although decitabine is approved for an initial therapy for older AML patients in Europe, it is not approved in the United States, primarily because the pivotal trial did not meet its primary end point of extending OS. A trial reported in preliminary form comparing 5-azacitidine to conventional care regimens for AML patients whose white count was less than 15,000/μL again did not meet its primary end point, but certainly suggested that 5-azacitidine was a viable alternative to low-dose ara-C or supportive care.170 It should be emphasized, however, that the CR rates are much lower with hypomethylating agents and low-dose ara-C than with standard chemotherapy, and that the less-intensive treatments have largely been evaluated only in patients with less-proliferative forms of AML with the likelihood that results would be even poorer than summarized, should these treatments be used in patients with high circulating blast counts or a high marrow blast infiltration; as such induction with a 3 + 7-based regimen ideally in the context of a clinical trial should be the default therapy for the relatively fit older adult under approximately 75 years of age.
Patients to be treated with a hypomethylating agent should probably be managed in the same manner as those with MDS receiving such agents. The patient should continue to receive the hypomethylating agent every 4–6 weeks until toxicity or obvious progression occurs. It certainly seems that prolonged exposure to these agents is an important factor in achieving relatively good long-term results. If a patient does achieve a very good outcome such as a CR rate in response to a hypomethylating agent, then the older adult in question, if under approximately 75 years of age, could yet be considered for a nonmyeloablative allogeneic transplant.171 This usually occurs only in people who elect or are given aggressive chemotherapy, but this is nonetheless a consideration even in those who receive a less-intensive chemotherapy.172
Postremission therapy
Morphologic assessments of CR are subjective and relatively insensitive. A better quantitation of malignant cells present at the time of CR (MRD) could segregate patients into prognostic subgroups. It is estimated that as many as 109 leukemia cells may still be present in patients with apparent morphologic CR. Only few patients can remain in CR for 1–2 years without further treatment, but it is generally accepted that some form of therapy after CR is required to achieve long-term disease-free survival. In a trial conducted by the German Cooperative Leukemia Group, a subset of 37 patients did not receive postremission therapy for a variety of protocol and medical reasons; all of these individuals relapsed.173 The ECOG reported a randomized study in which patients achieving CR were randomized to either no therapy, lower-dose maintenance therapy, or an intensive postremission program.174 All of the patients in the no-treatment arm relapsed rapidly, with a median CR duration of 4 months, resulting in early termination of this arm of the trial. Similar results were noted in a smaller randomized study reported by Embury and colleagues.175 Although timed sequential therapy, in which patients receive additional chemotherapy during the early postinduction recovery phase, has been associated with prolonged remissions in the absence of postremission chemotherapy,176, 177 this treatment regimen is more analogous to consolidation therapy. Thus, it remains a standard practice to administer chemotherapy with or without subsequent SCT after remission is achieved with conventional regimens.
Younger adults are generally treated with curative intent and should receive postremission chemotherapy that includes intensive (or myelosuppressive) “consolidation” chemotherapy and/or SCT. The term “maintenance” is generally referred to lower-dose outpatient therapy administered on an intermittent basis for months to years, patterned on the model successfully used in childhood ALL, but this approach has not been clearly found to be useful in patients with AML.
A variety of agents have been used for postremission therapy, including the agents successfully administered in initial induction, with a particular focus on HIDAC as consolidation, as well as different classes of compounds, some of which have proved activity in AML, and others of which may have had only limited activity. Because a variety of prognostic factors significantly affect ultimate outcome, independent of the type of therapy administered, good results in small nonrandomized studies may reflect inadvertent patient selection and must ultimately be confirmed by multi-institutional applicability.
Overall, in adults, it can be expected that the administration of some sort of intensive postremission chemotherapy will result in a median CR duration of 12–18 months with approximately 20–25% of complete responders remaining as long-term disease-free survivors (Figure 10). A large review of CALGB patients who achieved CR showed a relatively constant relapse rate of 4.7% per month during the first 6 months following CR. The failure rate decreased in subsequent 6-month intervals (3.5% per month in months 7–12 and 2.4% per month in months 13–18), with a flattening of the curves after 3+ years of CR.178 In general, patients relapsing earlier have leukemia that is more drug resistant than those who relapse late.179 Older randomized trials of postremission therapy conducted by the CALGB failed to demonstrate long-term benefits from: (1) an alternate month compared with a monthly schedule of maintenance therapy; (2) 3 years of relatively low-dose maintenance therapy compared with 8 months of a similar program (indeed, there was a modest survival advantage for patients randomized to stop therapy after 8 months); (3) doubling of the dose of ara-C from 100 to 200 mg/m2 during maintenance therapy; and (4) addition of nonspecific immunotherapy in the form of methanol-extractable residue of Bacille Calinette Grain (MER) to maintenance therapy. With regard to the role of maintenance chemotherapy, an older ECOG study demonstrated no benefit from the addition of 2 years of maintenance therapy following two courses of postremission treatment with DAT (daunorubicin, ara-C, thioguanine).180 Studies from Germany reported by Buchner et al.181, 182 suggested a modest prolongation of CR duration when long-term maintenance therapy was administered after a single course of postremission DAT, although with a questionable effect on long-term survival. There was less effect of maintenance in later studies when more intensive postremission consolidation was used before the maintenance.181, 182
Two important randomized trials have shown that HIDAC in the postremission setting is better than lower doses of the drug in younger patients. In an ECOG study, patients randomized to receive one course of very intensive postremission consolidation with a HIDAC-type regimen had a longer median duration of remission than patients receiving 2 years of lower-dose maintenance therapy.183 The CALGB randomized 596 AML patients in CR to receive four courses of ara-C administered at three different dose levels [100 mg/m2 by continuous intravenous infusion (CIV) for 5 days, 400 mg/m2 CIV for 5 days, and 3 g/m2 IV for 3 h q12h on days 1, 3, and 5 (total six doses/course)]. There was no benefit from the higher-dose arms in patients >60 years of age with a median duration of CR of approximately 13 months and with only 10–12% long-term disease-free survival.184 In addition, there was a substantial incidence of CNS neurotoxicity in older patients, manifested primarily as cerebellar dysfunction. Other studies have also failed to show a benefit for higher doses of ara-C in older patients.185 By contrast, patients <60 years of age benefited substantially from the HIDAC regimen in terms of both relapse-free and OS. The long-term results in patients <40 years of age were similar to those reported with autologous or allogeneic bone marrow transplant (BMT).
These studies strongly support the use of HIDAC-based consolidation programs in younger patients with AML. It is not known how many courses of such therapy are needed, the optimal dose and schedule of HIDAC, and whether the addition of other active agents will improve on these results. It is likely that ara-C incorporated into DNA is maximal at 1–1.5 g/m2 per dose and a recently completed randomized study from the United Kingdom has shown identical OS when doses of 1.5 g/m2 were compared with the original CALGB doses of 3 g/m2.186 In another randomized study, the use of other potentially noncross-resistant regimens substituted for HIDAC was not superior to three cycles of HIDAC187 and studies from the Medical Research Council (MRC) in Britain, which used multiple postremission courses using a variety of different drugs, produced similar overall outcomes.188, 189 The CALGB data suggest that the benefit from HIDAC was most pronounced in patients with favorable cytogenetic findings [t(8;21) and inv (16)] with much less effect in patients with unfavorable karyotypes typically associated with drug resistance.77 If a chemotherapy-based approach is medically feasible, most clinicians attempt to administer at least three courses of reasonably intensive HIDAC-based therapy postremission regimens. As allogeneic SCT is currently becoming more commonly used, HIDAC-based postremission therapy has been reserved most appropriately for younger patients with inv16, t(8;21) or those with normal cytogenetics with an NPM1 mutation but no FLT3 ITD mutations.9
The decision about the type of postremission therapy must consider the patient’s medical condition and the possible persistence of infection (particularly with fungal organisms) acquired during induction, as well as the ability to provide adequate platelet transfusion therapy, in an attempt to balance the risk of intensive postremission approaches with the potential benefit. Depending on the intensity of the consolidation program, a 5% mortality rate in CR is to be expected and must be carefully explained to the patient, although the use of myeloid growth factors after consolidation can appreciably shorten the duration of severe neutropenia and potentially make the administration of this therapy safer.190, 191
An alternative way to administer high-dose chemotherapy in AML is autologous SCT. Cryopreserved autologous bone marrow can rapidly reconstitute recipients of ablative regimens, although marrow recovery may be delayed compared with allogeneic BMT.192 Experience using cytokine-mobilized peripheral blood stem cells (PBSCs) indicates that the durations of neutropenia and particularly thrombocytopenia are shortened compared with the use of bone marrow.193 The autologous procedure is well tolerated and can be readily used in patients up to 65 years of age and perhaps older. The major disadvantages include the absence of a graft-versus-leukemia effect, as well as concern that viable leukemic progenitors will be administered with the autologous stem cells. A number of purging techniques have been used including monoclonal antibodies directed against myeloid blasts, as well as incubation with high concentrations of cytotoxic agents192 that rather remarkably spare hematopoietic progenitors, although delayed marrow reconstitution is not uncommon.193 It is unknown whether these in vitro manipulations are beneficial. When unpurged autologous BMT was harvested after high-dose consolidation therapy, retrospective comparisons did not suggest an increased rate of relapse. A number of centers and groups have reported relapse-free survival rates >40% following autologous SCT in first CR.194–196 Historically, syngeneic transplant experience in this situation showed relapse rates of approximately 50%, which is probably the best that can be expected with the autologous approach. Other evidence of the potential of this approach derive from reports of apparent cure rates of 20% in patients transplanted in the second and third remissions of AML.192
Allogeneic SCT represents an alternative major therapeutic option for postremission therapy for younger patients, and increasingly, nonmyeloablative SCT is being considered in selected older patients. Syngeneic SCT, using identical twins as donors,197 and allogeneic SCT using HLA-identical siblings,198 were first evaluated in patients with advanced, refractory AML. After demonstrating an approximately 10% rate of long-term disease-free survival in this refractory group of patients and a 40–50% disease-free survival in patients in first remission receiving syngeneic transplants, studies of allogeneic transplantation in first remission AML were conducted.199 Early small series were promising, demonstrating a low rate of relapse with most of the mortality related to complications of acute and chronic graft-versus-host disease (GVHD). With the current advances in supportive care, particularly the use of novel GVHD prophylactic and active therapies, allogeneic SCT is safer and more commonly used.200, 201
In the 1990s several large prospective trials “genetically” assigned patients with histocompatible siblings to allogeneic BMT (usually only in those ≤45 years of age) while randomizing the others to either autologous BMT or chemotherapy.188, 202 In the first of these trials which included 422 patients <45 years of age (median 33 years), disease-free survival was similar in patients undergoing allogeneic BMT (55% projected at 4 years) and autologous BMT (48%) and superior to the chemotherapy group (30%), which received a second course of consolidation chemotherapy with intermediate-dose ara-C and m-amsacrine.202 OS was similar, however, because many patients relapsing after chemotherapy could be successfully re-induced and then undergo SCT in second CR. Many patients did not undergo treatment as randomized, and no information was provided about responses to the three treatments in different cytogenetic risk groups.
Another similarly designed trial performed in France failed to show a benefit for either type of BMT compared with patients receiving intensive postremission chemotherapy.203 A large study conducted in Germany failed to show a benefit from autologous transplantation given as a component of consolidation therapy. The MRC 10 trial conducted in Great Britain188 showed that autologous BMT was beneficial. Patients who were not assigned to allogeneic BMT received three cycles of post-CR chemotherapy and were then randomized to nonpurged autologous BMT or observation. The addition of the autologous transplant prolonged CR duration, but there was no significant difference in OS between the two groups. Allogeneic BMT was not significantly better than autologous BMT; no modality was clearly better than another in different cytogenetic risk groups. The MRC trials were updated with an intent to treat analysis of results in patients with and without matched sibling donors.189 Again, there was no apparent survival benefit compared with chemotherapy for patients who had available donors in whom a transplant could have been performed, and no advantage when patients with donors who were and were not transplanted were compared.
The trial conducted by the North American Intergroup204 found that chemotherapy was at least as good as autologous or allogeneic BMT in leading to cure, based on the intent to treat analysis. As in all the other studies, many patients assigned to either modality of BMT did not receive this therapy for a variety of reasons. Some general conclusions can be drawn concerning the role of allogeneic BMT in the management of patients with AML in first CR:
- 1. Applicability of the technique is somewhat limited by patient age. Mortality from GVHD increases every decade. Improvements in supportive care and the increased use of reduced intensity transplants using nonmyeloablative conditioning regimens and PBSCs allow increased use of transplantation to older individuals. Early side effects are markedly decreased by the nonablative approach, which is medically feasible in most older patients.205 A systematic prospective study showed that because of clinical, administrative, and donor availability issues, only a small fraction of older patients in first CR can in fact proceed to transplantation.206 Nonetheless, because the results are poor with chemotherapy in older adults and are better (at least with those who make it to transplant (35% disease-free survival) with reduced-intensity alloSCT, it is generally accepted as feasible for adults up to age 75.
- 2. Less than one-third of potential recipients have suitable HLA-matched family donors. Alternatives include the use of matched unrelated donors, partially mismatched family donors, haploidentical donors, or cord blood preparations. Administrative delays in identifying suitable donors remain problematic despite the availability of millions of HLA-type donors worldwide.207 This is particularly true for patients from ethnic minority groups with less common HLA types and also indicates that there is considerable selection bias in reports of unrelated donor transplants, because some higher-risk patients relapse while awaiting for a donor to be identified. Nonetheless, with the use of molecular histocompatibility typing, the results following unrelated allogeneic transplant are equivalent to those with matching sibling transplant extending the use of allogeneic transplant for a much larger group of patients with AML in first remission.208, 209 Umbilical cord blood may offer an alternative source of stem cells for those without matched siblings or unrelated donors. The incidence of GVHD is much lower following cord blood transplants despite the fact that many of these transplants use mismatched donors. Engraftment can be delayed, however, and dosage considerations represent an issue for many adult recipients, although the use of double cord transplants has helped to address this problem.210, 211 At present, haploidentical transplantation from partially matched siblings, parents, or children is also available, and hence donors are available for almost all recipients, although the relapse rate may be higher than that with matched donor.212
- 3. Although allogeneic SCT can cure some patients with chemotherapy-resistant disease, there remains an appreciable relapse rate and at least some of the factors predictive of drug resistance and relapse after chemotherapy also apply to SCT recipients.213, 214
- 4. The antileukemic effect following BMT correlates with the occurrence and severity of GVHD, presumably as a result of a graft-versus-leukemia effect. Attempts to attenuate GVHD with a variety of immunosuppressive approaches are associated with a decrease in GVHD, but also an increase in the relapse rate.215 Selective T-cell depletion approaches and other immunologic manipulations of the graft are being evaluated in an attempt to make allogeneic SCT safer without an increased relapse risk.216, 217
- 5. As many as 10–20% of surviving patients may have significant symptoms and impairment of performance status because of chronic GVHD. There is also a small increase in the frequency of secondary tumors in long-term survivors.218
There seems little controversy about the advisability of using a HIDAC-based postremission approach for those with CBF cytogenetic abnormalities and in using an allogeneic transplantation-based approach for those with adverse prognosis based on molecular or karyotypic findings at diagnosis. However, there still remains controversy about handling intermediate-risk patients, particularly those with normal chromosomes. Meta-analysis have suggested that in general, all patients with nonfavorable cytogenetics should fare slightly better with an SCT approach.219–221 The only subgroup in the intermediate prognosis group for whom this is not clear-cut is those with normal cytogenetics and an NPM1 mutation but without a FLT3 ITD mutation. Survival rats of patients treated by a chemotherapy-based approach are higher, and it would be reasonable to reserve SCT for second remission for such patients as well as for those with favorable translocations.
Therapy of relapsed and refractory AML
A number of agents with clear-cut activity when used alone or in combination in patients with relapsed or refractory AML are available, including amsacrine, mitoxantrone, diaziquone, idarubicin, fludarabine, 2-chlorodeoxy-adenosine, etoposide, homoharringtonine, topotecan, carboplatin, and clofarabine. There is considerable heterogeneity among relapsed patients, and a number of factors in addition to the specific drugs used influence the outcome of treatment.222–224 Some consistent trends are evident: (1) Response rates are uniformly low in patients with primary refractory leukemia, in those with short initial CR durations, or in those who relapsed while receiving postremission chemotherapy; (2) leukemias that evolved from a prior hematologic disorder, and patients with poor-risk cytogenetics are particularly resistant to further therapy; and (3) patients in second and subsequent relapses have a poorer prognosis than patients in their first relapse, and the durations of subsequent remissions tend to decrease progressively.
Results from the MRC in Great Britain, which prospectively followed all patients entered on an induction AML trial, are supportive of these conclusions.224 In another study, patients whose initial remission was more than 18 months had a reinduction rate of 64% (37 of 58) compared with a 29% CR rate in 278 patients with shorter initial remissions. Furthermore, the duration of second CR was longer in the former group (8 months vs 3 months). Patient age and cytogenetic findings are also important prognostic factors, emphasizing the higher effect of patient selection on the results of reported phase II trials. In the absence of comparative trials, differing results may very well be the consequence of patient selection rather than the superiority of a particular regimen.
The type and timing of therapy for relapsed patients should be individualized. Patients with other medical problems who have had poor responses to initial therapy have a small likelihood of sustained benefit from reinduction therapy and, indeed, may have their life shortened by intensive therapy. Some of these patients can be supported for many months, with maintenance of a reasonable quality of life, with a more conservative approach using red blood cell and platelet transfusions and oral hydroxyurea to control elevated WBC counts or symptoms such as bone pain. However, there is no potential for long-term benefit. Conversely, younger patients with longer initial responses may derive prolonged benefit from intensive reinduction therapy, if a remission or at least a good response can be followed by allogeneic stem cell transplant.
Recurrence of leukemia is often detected when the patient is asymptomatic, blood counts are normal, and there is modest marrow infiltration by blasts. Although it is logical to begin reinduction therapy at the earliest sign of relapse, when tumor burden is presumably the lowest, there are no data to demonstrate the validity of this approach except possibly as preparation for subsequent SCT. The drawback of early treatment is that reinduction therapy is often unsuccessful and may result in excessive early morbidity and premature patient death, but if long-term survival is the goal (e.g., an allogeneic stem cell transplant), there is no reason to delay.
There are essentially no comparative trials providing guidance about the choice of agents to be used. The initial decision is generally between reuse of drugs that have previously been effective in a given patient, compared with the use of new drugs, some of which may be investigational. If a patient has relapsed while receiving chemotherapy, it makes little sense to use these same agents for reinduction. The oft-quoted guidance is to repeat the induction regimen if the disease-free intent is >1 year, otherwise a HIDAC-based approach (2–3 g/m2 q12h for 8–12 doses) is reasonable. There is no proven benefit of postremission chemotherapy in patients achieving second or third remissions. The drawback of postremission chemotherapy in this setting is that one is potentially depriving patients of time when they would be asymptomatic and discharged from hospital by the administration of therapy of variable toxicity, but no proven long-term efficacy.
It is unclear whether SCT should be offered at the time of initial relapse or whether patients should be reinduced and transplanted when a second remission is achieved. Unfortunately, most patients do not achieve second remission, and some develop medical problems during reinduction therapy that preclude subsequent transplantation. Enthusiasm for transplantation in relapse derives from reports from Seattle indicating that the results of allogeneic transplantation in early relapse were equivalent to or better than those achieved in patients transplanted in second or subsequent remissions.225 It is often a practical problem to identify patients during early relapse and to be able to refer such patients for transplantation rapidly. Nonetheless, it would probably be best to consider allogeneic transplantation for patients in early relapse if a suitable donor is available, particularly in younger patients. Similarly, allogeneic BMT can result in long-term survival in some patients with primary refractory leukemia, and it is advisable to HLA-type patients and their families at diagnosis to allow for this possibility. It is more difficult to use nonrelated donors for these purposes because of delays caused by tissue typing, identification of donors, and stem cell procurement. Currently, most transplant centers have recommended that patients in more florid relapse receive induction chemotherapy first because of poor results with SCT as primary therapy in such patients. Because many patients under 75 years of age have an allogeneic transplant in CR1, many relapses to date have occurred after such a procedure. Relapse after allogeneic SCT are particularly ominous, with a 7% chance of 2-year survival.226 Selected patients who relapse following BMT can derive benefit from donor lymphocyte infusions226 or a second BMT227 and, in general, can receive and tolerate further chemotherapy. Targeted therapies, such as sorafenib in FLT3 ITD mutant AML228, 229 or immune checkpoint inhibitors,230 produce better results in the post-SCT relapse setting.
Patients who achieve second remission after a first remission in which only chemotherapy or autologous SCT was used probably have a higher chance of long-term survival if transplanted in second remission. Hence, relapsed patients who are candidates for allogeneic transplantation should have donor searches done while they are undergoing reinduction treatment. Patients who respond to reinduction chemotherapy have also identified themselves as affected with more chemotherapy-sensitive disease with predicted better outcomes from allogeneic transplantation.
Therapy of acute promyelocytic leukemia (APL)
Treatment for APL differs substantially from that for other subtypes of AML in that the use of the so-called “differentiation therapy” is included in the former. In theory, if leukemic progenitors could be forced to undergo terminal differentiation in vivo, then the leukemic clone could lose the capacity for self-renewal and be eliminated, with fewer side effects than those occurring with intensive cytotoxic chemotherapy. With the exception of the HL-60 cell line and blasts obtained from patients with APL, it has been difficult to reproducibly induce fresh leukemia cells to differentiate in vitro.
Limited clinical trials evaluating “differentiating” therapies, including vitamin D and low-dose ara-C, produced disappointing results except for some patients with APL who derived transient benefit from treatment with cis-retinoic acid.231 The differentiation paradigm was verified, however, by dramatic responses in patients with APL reported from China with ATRA administered orally for 30–90 days.232 CRs were seen in >80% of both relapsed and previously untreated individuals. Marrow aplasia did not occur and serial bone marrows showed maturation of the abnormal, hypergranulated promyelocytes.233, 234 DIC resolved promptly, with a profound reduction in the requirement for platelet transfusions following ATRA treatment. Although the hemorrhagic complications are decreased, approximately 20–30% of patients treated with ATRA only develop significant side effects that can include fever, rapidly evolving pulmonary insufficiency, pericarditis, and pleurisy. This syndrome can occur independently of the leukocytosis frequently observed with ATRA therapy and can be fatal. Optimal management includes the prompt administration of corticosteroids at the first signs of the “ATRA” syndrome.235, 236 Resistance to ATRA eventually develops in patients treated in relapse, and virtually all patients treated with ATRA only relapse. Possible pharmacokinetic mechanisms include induction of more rapid metabolism of the ATRA and induction of cytoplasmic retinoic acid-binding protein in normal tissues that bind the ATRA, thereby decreasing the exposure of residual APL cells to the drug.237
Once the efficacy of ATRA in APL was recognized, much effort was made to determine the optimal way to use this agent. Trials conducted on both sides of the Atlantic showed that combinations of retinoic acid and anthracycline-based chemo are better than either agent alone both during induction and postremission therapy.85, 238 A risk score based on platelet and white counts at diagnosis derived from results in these trials, notably from successive Spanish (PETHEMA) trials, indicated that those with the poorest long-term outcome are those with a white count >10,000/μL and those with the best long-term outcomes are those with a white count <10,000/μL and platelet count >40,000/μL at the time of diagnosis.239 Before the introduction of arsenic trioxide, the standard of care could be summarized as anthracyclines plus ATRA, followed by two to three cycles of retinoic acid plus anthracycline as consolidation. Maintenance therapy with oral antimetabolites and ATRA for approximately 1 year after completion of intensive chemotherapy was thought to add an event-free and OS benefit238 and higher doses of ara-C were felt to be beneficial in those with high-risk disease based on a white count >10,000/μL at diagnosis.240 Further, disease recurrence could be predicted by development of molecular evidence of disease (PML-RARα) transcripts by PCR measurement ahead of morphologic relapse.90 Patients who could be rendered molecularly negative with additional therapy could then be salvaged with an autologous transplant,241 whereas those who remained molecularly positive would be better served by an allogeneic transplant. Some of the late relapses were in the CNS,242 prompting calls for routine use of prophylactic intrathecal therapy at least in those with high white count at diagnosis and in all patients in relapse after control of the coagulopathy and thrombocytopenia. There was also some question such as whether this intensive chemotherapy could lead to MDS and/or secondary leukemias.243
The demonstration, again in China, that intravenous arsenic trioxide produces a high rate of CR even in patients with advanced, multiply relapsed disease,244 spurred a major change in our approach to APL. Although the precise mechanism of action and the explanation for its relative specificity for APL remain to be elucidated, arsenic trioxide serves to accelerate apoptotic cell death in APL.245 Recently, tetra-arsenic tetra-sulfide has been shown to be very active in both newly diagnosed and relapsed APL patients with the advantage of oral administration.246 While arsenic trioxide first became the standard therapy for relapsed APL, it is currently routinely used during induction and/or postremission treatment of APL. Investigators from China showed high response rates using ATRA and arsenic as initial treatment,247 while other studies from India248 and Iran249 reported high molecular CR rates, which were durable using arsenic only. Using only ATRA and arsenic in patients not deemed to be candidates for anthracycline-based therapy, with GO added for patients with high WBC or insufficient response, the MD Anderson Group demonstrated a leukemia-free survival (LFS) rate as high as that observed in cooperative trials using more standard therapy.250, 251 The North American Intergroup evaluated the addition of two courses of arsenic as consolidation therapy post remission and showed a survival benefit compared with the standard treatment without arsenic.252 Patients with high-risk disease (WBC > 10K) fared very well with this approach. Australian investigators also showed the value of a combined chemo/ATRA/arsenic approach.253
One of the most dramatic changes in the way we treat APL derived from a Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto (GIMEMA)-led trial which compared a chemotherapy-free based approach (ATRA and arsenic trioxide given together as both induction and post remission therapy) to the aforementioned PETHEMA approach using anthracyclines and retinoic acid (Figure 12)100 The trial was designed as a noninferiority effort and was restricted to patients <70 years of age whose white count was <10,000/μL at diagnosis. Patients assigned to the ATRA/arsenic arm had a 93% event-free and 95% OS, which was statistically superior to the outcome in patients assigned to the chemotherapy-containing arm. As such, the new standard of care for those with white count <10,000/μL at diagnosis is the regimen of ATRA and arsenic as used in the GIMEMA trial. The standard of care for those patients whose white count >10,000/μL has not yet emerged, but it is reasonable to use CALGB 9710252 or the EORTC trial240 approach in such patients as favorable results have been obtained with these regimens in this subgroup. The future of APL therapy certainly involves the minimal use of cytotoxic chemotherapy. It may soon be possible to cure a large majority of patients with this disease by pills, as oral formulations of arsenic trioxide may be able to replace the frequent and relatively cumbersome IV arsenic infusions.
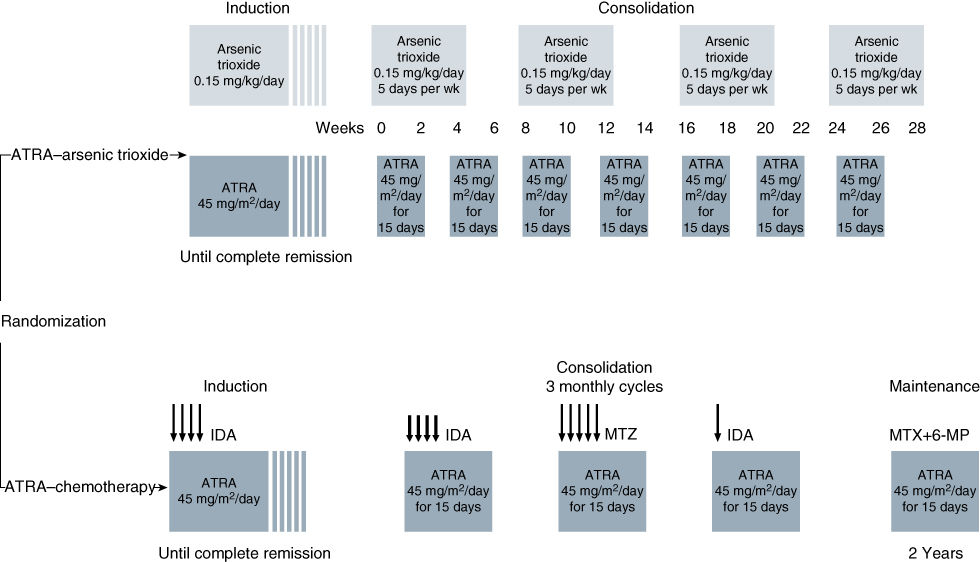
Figure 12 Treatment Groups. In the all-trans retinoic acid (ATRA)–chemotherapy group, the chemotherapy regimen was as follows: idarubicin (IDA) at a dose of 12 mg/m2 of body surface area per day on days 2, 4, 6, and 8 of the induction phase; IDA at a dose of 5 mg/m2/day on days 1–4 of the first cycle of consolidation therapy; mitoxantrone (MTZ) at a dose of 10 mg/m2/day on days 1–5 of the second cycle of consolidation therapy; IDA at a dose of 12 mg/m2/day on day 1 of the third cycle of consolidation therapy; and intramuscular or oral methotrexate (MTX) at a dose of 15 mg/m2/week and oral 6-mercaptopurine (6-MP) at a dose of 50 mg/m2/day, alternating with ATRA at a dose of 45 mg/m2/day, for 15 days every 3 months for 2 years. The vertical lines in the induction therapy boxes indicate variability in the duration of remission induction therapy. The arrows indicate the approximate timing and doses of the different chemotherapeutic agents.
Source: Lo-Coco 2013.100 Reproduced with permission of NEJM.
The major remaining problem in the treatment of APL is the prevention of hemorrhagic death, often the consequence of delays in diagnosis and the initiation of therapy with ATRA. APL should be suspected in patients presenting with hemorrhagic symptoms in association with abnormalities of coagulation including hypofibrinogenemia, accompanied by seemingly modest elevations of the prothrombin time and evidence of accelerated fibrinolysis. ATRA should be initiated empirically in such patients and then discontinued when cytogenetic or molecular studies fail to confirm the diagnosis of APL.
Other supportive care and therapeutic approaches
Hematopoietic growth factors
Because of the higher myelosuppression-associated toxicity and mortality, particularly in older adults with AML, the development of hematopoietic growth factors (HGFs) ameliorated such side effects. Granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF, and interleukin (IL)-3 as well as the thrombopoietic factors, megakaryocyte growth and development factor (MGDF), IL-11, and the newer thrombopoietic agents (romiplostim and eltrombopag) were evaluated. The use of these agents in AML lagged behind that in solid tumors, because of the concern that pharmacologic doses would lead to blast proliferation and a poor clinical outcome. Although adverse effects of this clinical problem have not been proved, the HGFs have not lived up to their promise for other reasons.
Multiple randomized studies have been completed in which older patients received an HGF or placebo following the completion of initial induction therapy, with the goal of increasing CR rate by reducing infectious complications as a consequence of shortened durations of severe myelosuppression.254–259 All trials noted a decrease in the order of 2–5 days in the number of days of neutropenia <500/μL, sometimes in association with a smaller 1- to 2-day reduction in the duration of hospital stay, but with the exception of a smaller trial reported by the ECOG,254 there was no significant difference in the incidence of severe infection, deaths from infections, CR rate, or survival. One randomized trial using G-CSF after induction therapy for older patients with AML showed an increased CR rate for the G-CSF recipients.258 Unexpectedly, the infectious mortality was similar in the two groups of patients, and the reason for the increased CR rate is unclear. There was no difference in OS.
In conclusion, the benefits of growth factors administered after the completion of induction therapy are modest at best.260 By contrast, G-CSF following intensive consolidation therapy has appreciably shortened the duration of neutropenia, albeit without improvement in CR duration or survival.190 Because of the potential for eliminating the need for hospitalization, the use of HGF can be recommended following consolidation therapy. Smaller studies with pegylated thrombopoietin failed to show reduced needs for platelet transfusions during the induction or consolidation treatment of AML.261 The thrombopoietin agonists romiplostim and eltrombopag, each approved in steroid-refractory ITP, have been used in clinical trials in MDS,262, 263 but evidence suggesting leukemia cell stimulation has precluded use in AML and MDS.
Although in vitro evidence suggests that HGFs can increase the cytotoxic effects of ara-C, at least in part by increasing the fraction of cells in S-phase,264, 265 clinical results with the priming strategy have been disappointing. An early study from the MD Anderson Group suggested that GM-CSF administered before and during chemotherapy produced lower response rates and survival than those observed in a historical control group treated with chemotherapy only.266 Most subsequent studies that randomized patients to receive GM-CSF/G-CSF or no marrow stimulants, before, during, and/or after induction chemotherapy267–269 also failed to show an advantage for patients primed with growth factor. Some of these studies used HIDAC, whereas others evaluated more conventional continuous infusion schedules. One of the studies purporting to show an advantage270 could not be confirmed in a follow-up investigation.
Immune modulation
A critical component of the therapeutic benefit associated with allogeneic SCT is related to the graft-versus-leukemia effect. Although this is a complex, multifactorial set of events, further studies may enable the rational application of lymphokines to stimulate the appropriate cells in patients treated with chemotherapy only. Circulating natural killer (NK) cells that can be cytotoxic to leukemia cell lines, and autologous leukemia cells in vitro, can be detected after both autologous and allogeneic transplantation, but not after chemotherapy.271 The numbers of NK cells can be expanded by the posttransplant administration of IL-2,272 although low-dose IL-2 can ameliorate GVHD by expanding T-reg cells.273 Standard or high IL-2 has some antileukemic effect in patients with AML in relapse.274, 275 Pilot trials have documented the feasibility of administering IL-2 in the postchemotherapy setting, but unfortunately, a large randomized trial failed to demonstrate any benefit from the administration of IL-2 after completion of consolidation chemotherapy in older patients with AML.276 A similar trial in younger adults documented a trend toward improved LFS and OS in patients randomized to IL-2, but many patients refused randomization or IL-2 if they are randomized to receive it.277 Another randomized trial showed that a combination of histamine plus IL-2 was beneficial in the late postremission setting,278 but has been criticized because of the heterogeneity of the prerandomization chemotherapy. Vaccine-based approaches using dendritic cell fusions to present leukemia-associated antigens, such as WT1 and PR1, engineered T cells, or transducing AML cells to express proteins capable of stimulating immune response, are under development.279–282
Circumvention of drug resistance
Drug resistance is conferred by properties of a small fraction of leukemia cells, presumably the fraction capable of efficient self-renewal.283, 284 Investigations of these leukemia “stem cells” have been hampered by difficulties in reliably and repeatedly cloning leukemic cells from individual patients to allow serial studies to be performed. In addition, a potential sampling bias exists in that the in vitro results are a reflection only of the characteristics of cells obtained at a single point in time that can grow in an artificial environment. Nonetheless, there is considerable interest in the development of drugs, which may preferentially target these cells.285
The multidrug resistance (MDR) phenotype is associated in most cases with increased amounts of a membrane glycoprotein (p-170), which serves as a pump accelerating the efflux of a wide variety of agents, including the anthracycline antibiotics, vincristine, taxanes, and mitoxantrone.286 Levels of p-170 can be assessed by flow cytometric studies of individual AML cells, Western blots of cell preparations, and indirectly by mRNA levels and gene expression, and are increased in patients refractory to chemotherapy.287, 288 The MDR phenotype is most common in patients with relapsed or refractory disease, older patients, and those with other adverse prognostic factors.289, 290
Incubation with a variety of compounds in vitro, including the calcium channel blockers verapamil, quinine, and cyclosporine A, can reverse the effect of p-170.290 Cardiotoxicity occurs at the doses of verapamil required to produce this effect. Cyclosporine can reverse the MDR phenotype in vitro at drug levels that are likely to be clinically acceptable in terms of toxicity. A randomized trial of the addition of cyclosporin A to an ara-C plus continuous infusion daunorubicin regimen for patients with relapsed AML suggested an OS benefit for adults in the experimental arm.291 PSC-833 is a more potent nonimmunosuppressive cyclosporine analogue, which has been evaluated in randomized trials as an adjunct to standard therapy. Unfortunately, no improvement in CR rates was observed, with some trials showing increased toxicity in the PSC833 recipients.292, 293 Because of their effects on normal tissues such as the liver and kidney, these modulators also affect the pharmacokinetics of antineoplastic agents, such that the dosage of anthracyclines or etoposide usually has to be attenuated if given with an MDR modulator.294 Other MDR modulators have been similarly disappointing.295
Minimal residual disease (MRD)
A number of techniques can detect residual leukemia cells in patients with morphologic CR, including (in approximate descending order of sensitivity) conventional cytogenetics,296 Southern blotting for known gene rearrangements, fluorescent in situ hybridization (FISH), multiparameter flow cytometry, and RT-PCR.297, 298 Serial measurement of MRD is an important component of the management of APL, chronic myelogeneous leukemia, and childhood ALL, but is not sufficiently standardized to be used routinely in patients with AML.299 Technical problems abound, including the requirement for a known and already cloned abnormality when applying molecular techniques and the potential for changes in antigen expression over time when using immunologic monitoring. Only few large prospective studies have been conducted on this topic. Although preliminary data are compatible with the logical premise that persistence of detectable disease presages eventual relapse, false-positive and false-negative rates may be appreciable and could result in incorrect decisions about further treatment. Serial monitoring is cumbersome, and it is hoped that future studies will determine specific time points after completion of therapy when detection of MRD is prognostically significant. It is also likely that different sampling strategies will be needed for different AML subtypes. The key clinical question is whether intervention with further therapy or allogeneic SCT is valuable if applied earlier, before gross relapse. There are no prospective data addressing this issue, but retrospective studies have shown much higher-than-expected relapse rates in allogeneic transplant recipients with detectable MRD. Because of the presence of residual disease, autologous collection and high-dose therapy may be predicted to be of less value in this circumstance.
Small-molecule inhibitors and other novel approaches
While there has not been any newly approved drug in non-APL/AML for many years (not counting GO, which was approved and then withdrawn), many agents are under development. A comprehensive discussion of all such agents is beyond the scope of this chapter; however, it is worth highlighting progress in tyrosine kinase inhibitor therapy as well as a few other interesting agents in developments.
The success of imatinib and other BCR-ABL inhibitors in CML prompted a search for similar gain-of-function mutations in AML, whose inhibition could lead to therapeutic benefit. Gain-of-function mutations in the FLT3 transmembrane tyrosine kinase occur in approximately 30% of patients with de novo AML, consisting most commonly of an ITD mutation, which elongates the juxtamembrane region by between 3 and 100 amino acids; a smaller number of patients have an activating point mutation in the tyrosine kinase domain.7 Both types of mutations can cause ligand-independent activation300 and a myeloproliferative disease in murine models.301 Several so-called FLT3 inhibitors are under development, although none as of yet has been approved by the FDA. Single-agent trials with midostaurin,302 lestaurtinib,303 and sorafenib304 each showed responses in patients with advanced, mutant FLT3 AML. Sorafenib is approved as a vascular endothelial growth factor receptor inhibitor in patients with advanced renal and hepatocellular carcinomas. Midostaurin can inhibit the growth of cells with a tyrosine kinase domain mutation, while most other agents (except for crenolanib)305 cannot and are only being developed in the FLT3 population that has the ITD mutation. Quizartinib, a potent and relatively specific FLT3 ITD inhibitor, has shown a relatively high response rate as a single agent306 and is being compared with chemotherapy in mutant FLT3 ITD patients with AML in relapse; however, an important mechanism of resistance is the outgrowth of clones with an FLT3 TKD mutation.307
Given the relatively rare CR and brief duration of response with single-agent FLT3 inhibitor therapy, there has been a major thrust to develop these agents in combination with chemotherapy. A trial testing the addition of lestaurtinib to chemotherapy in relapsed mutant FLT3 ITD patients was negative,308 but several other clinical trials comparing chemotherapy with or without an FLT3 inhibitor are underway. Moreover, FLT3 inhibitors, particularly sorafenib, seem to be effective in some patients with mutant FLT3 AML who have relapsed after allogeneic stem cell transplant.228, 229
Given that a sizable minority of patients with favorable chromosome (the so-called CBF translocations) AML have either over of c-kit and/or mutations in the c-kit tyrosine kinase, the c-kit inhibitor dasatinib has been added to chemotherapy in this cytogenetic subset of patients with AML. Preliminary results are encouraging and a randomized trial is ongoing.309 Another gain-of-function mutation noted in approximately 20% of AML patients occur in the IDH1 or IDH2 genes whose products catalyze the neomorphic production of 2-hydroxygluterate, which epigenetically affects gene expression in a pro-leukemic manner.310 Preliminary results with IDH1 and IHD2 inhibitors have been encouraging.311 MEK pathway inhibitors in patients with activating mutation of the RAS oncogene12 have also led to responses.
In addition to these genomically based therapies, there are a host of other agents under development, which take advantage of potential biologic differences between AML cells and normal stem cells. AML cells in the marrow niche have their survival promoted by factors elaborated by endothelial and other cells in the marrow stroma.312 Drugs such as the CXCR4 inhibitor, plerixafor, may disrupt the survival signals making AML cells more amenable to killing via cytotoxic agents.313 ABT199 is a BCL2 inhibitor with significant advantage in CLL,314 and it may be used alone or in combination with chemotherapy to promote apoptosis in AML cells. Another agent in development is KPT330, a nuclear export protein inhibitor, which works primarily by preventing expulsion of tumor suppressor genes from the nucleus of cancer cells in general, and in AML cells in particular.315 There are several antibodies and immunoconjugates targeting CD123,316 believed to be specifically expressed on leukemic stem cells. The developmental pathway for any agent in AML is slightly daunting as it has to be determined if it is going to be useful as a single agent in advanced patients and whether it needs to be added in chemotherapy in patients in earlier stages of the disease. In the latter case, given the relatively high initial response rate to chemotherapy in patients, large trials or trials with a novel design are needed.
Complications
Hyperleukocytosis
Leukemic blasts are considerably less deformable than mature myeloid cells317 and are “stickier” than lymphoblasts because of the expression of cell surface adhesion molecules. With increasing blast counts, usually at levels >100,000/μL in the myeloid leukemias, blood flow in the microcirculation can be impeded by plugs of these more rigid cells. Local hypoxemia may be exacerbated by the high metabolic activity of the dividing blasts, with endothelial damage and hemorrhage. Red blood cell transfusions can potentially further increase the blood viscosity and make the situation worse and should be either withheld or administered slowly, until the WBC decreases. Coagulation abnormalities, including DIC, further increase the risk of local hemorrhage. Liberal use of platelet transfusions is recommended, particularly because the platelet count is frequently overestimated because of the presence of fragments of blasts on blood smears, which can be mistakenly counted as platelets by automated blood cell counters.318
Although pathologic evidence of leukostasis can be found in most organs in patients with extremely high blast cell counts, clinical symptomatology is usually related to CNS and pulmonary involvement.319, 320 Occasionally, dyspnea with worsening hypoxemia can occur following therapy and lysis of trapped leukemic cells. Spurious elevation of serum potassium can occur because of the release from WBCs during clotting, and it is sometimes necessary to measure potassium levels on heparinized plasma. Similarly, pO2 can appear falsely decreased because of the enhanced metabolic activity of the WBCs, even when the specimen is appropriately placed on ice during transport to the laboratory. Pulse oximetry provides an accurate assessment of O2 saturation in such circumstances. Hyperleukocytosis is more common in patients with myelomonocytic or monocytic leukemia, and it is possible that the clinical manifestations are exacerbated by the migration of leukemic promonocytes into tissue where further proliferation occurs.
The initial mortality rate for patients with AML and symptomatic hyperleukocytosis is high.320 If patients survive the initial period, they tend to have somewhat lower remission rates and shorter CR durations. Symptomatic hyperleukocytosis in AML (and rarely in ALL) constitutes a medical emergency, and effort should be made to lower the WBC count rapidly. In most patients, rapid cytoreduction can be achieved by chemotherapy, with either standard induction agents or high doses of hydroxyurea (3 g/m2/day). Some centers also advocate low-dose cranial irradiation, including the retina, to prevent further proliferation of leukemic cells in CNS sites, where drug delivery may theoretically be compromised. This treatment is well tolerated, although there are no comparative studies to determine whether the results are superior to chemotherapy only.
In some patients, it is impossible to initiate chemotherapy immediately because of renal insufficiency, metabolic problems, delays in initiating allopurinol therapy to prevent hyperuricemia, or similar considerations. In such patients, emergency leukapheresis has been used to lower or stabilize the white count.321, 322 Although intensive leukapheresis, with procedure times often lasting many hours, can improve pulmonary and CNS symptomatology, there are theoretic and practical limitations to its benefits. It is difficult, for example, for leukapheresis to affect already established vascular plugs, particularly if vascular invasion has taken place. In such cases, chemotherapy is the primary modality, although theoretically, leukapheresis could decrease further accumulation of leukocytes at these sites. Furthermore, it is precisely the patient in whom leukostasis is most likely to occur, that is, the patient with high and rapidly rising blasts counts, in whom the technical limitations of leukapheresis are apparent, in that it is often difficult, even with highly efficient cell separators, to reduce the rising count. In such patients with a high proliferative thrust, cycle-specific chemotherapeutic agents are more likely to be most immediately effective. Leukapheresis is also of modest benefit to patients who develop pulmonary problems during cytotoxic treatment, because in some such patients, the symptoms are related at least in part to a local inflammatory response following leukocyte lysis.323
Central nervous system leukemia
Involvement of the CNS is considerably less common in patients with AML than in both adults and children with ALL. Most clinicians imply that the incidence has decreased even further in recent years, perhaps because of the use of higher doses of ara-C, which can penetrate into the CNS. The incidence of CNS leukemia has been <5% in large AML clinical trials in recent years.324 Therefore, diagnostic lumbar punctures are not routinely indicated in the absence of CNS symptoms, and chemotherapy regimens do not include CNS prophylaxis. Symptoms are consequences of increased intracranial pressure and usually consist of a constant headache, sometimes associated with lethargy or other mental changes. Cranial nerve signs (most commonly cranial nerves III or VI) and, occasionally, peripheral nerve manifestations are secondary to nerve root involvement and can be accompanied by headaches or occur alone.325
The diagnosis is usually suspected clinically and/or by meningeal enhancement on MRI with gadolinium and confirmed by examination of cytocentrifuge preparations of cerebrospinal fluid (CSF) after lumbar puncture. Cell counts can vary from as few as 5 to >1000/μL. Most patients have moderate elevations in CSF protein with a moderate decrease in glucose. Treatment consists of the administration of intrathecal chemotherapy, with the addition of cranial radiation (usually 2400 cGy), to patients who do not respond fully to chemotherapy or in whom cranial nerve involvement is present.325 Either methotrexate (15 mg/dose when administered by lumbar puncture) or ara-C (50 mg/dose) can be used as initial therapy, with a crossover to the other agent in the event of refractoriness or relapse. A typical schedule includes treatment twice to thrice a week until the CSF has cleared, generally occurring after a few injections. Treatment is then given at weekly intervals for two more doses to be followed by monthly administration for 1 year. An Ommaya reservoir to permit intraventricular drug administration is frequently needed because of either difficulties in performing repeated lumbar punctures or concern that in some individuals, the CSF flow does not deliver sufficient amounts of the drug from the lumbar space to the entire CNS.326 Successful therapies with systemically administered agents that penetrate the CNS, such as diaziquone (AZQ), high-dose methotrexate, or HIDAC, have also been reported.327 Slow-release (depo) preparations of ara-C may be an alternative.328 Unfortunately, the relapse rate is high, either concomittantly with bone marrow relapse or independently, even after initial successful therapy.
It has been suggested that the incidence of CNS leukemia is higher in patients with FAB M4 E0 morphology and possibly in patients with monocytic leukemia and high circulating blast counts.329 This may no longer be the case using more contemporary treatment regimens, and prophylactic therapy is not indicated in such individuals. As noted, CNS involvement can occur in patients with APL with high white blood count.
Ophthalmic complications
Essentially every ocular structure can be involved in the leukemias, sometimes dominating the clinical picture in the prechemotherapy era.330 Leukemia cells can infiltrate the conjunctiva and lacrimal glands, producing obvious masses that may require treatment with radiation therapy. Involvement of the choroid and retina is most common, however. A prospective study of 53 newly diagnosed adults with AML documented retinal or optic nerve abnormalities in 64% of patients.331 Hemorrhage and cotton wool spots (a consequence of nerve fiber ischemia) were most frequent, and the occurrence of these findings was unrelated to patient age, FAB type, WBC count, or hematocrit. Initial platelet counts were lower in patients with retinopathy. A total of 10 patients had decreased visual acuity, including five with macular hemorrhages. It was felt that many of the cotton wool spots were either a consequence of or exacerbated by ischemia due to anemia. Definite leukemic infiltrate of the retina could not be confirmed. All patients received aggressive chemotherapy and platelet transfusion support; no patient received cranial or ocular irradiation. All ocular findings resolved in patients achieving CR and there was no residual visual deficit in any patient. Infectious ocular problems were not noted and seem to be uncommon, perhaps because the prompt empiric use of antibacterial and antifungal antibiotics has decreased the possibility of hematogenous spread of infections to the eye.
Pregnancy
AML is occasionally diagnosed in pregnancy, either because of clinical manifestations or as an incidental finding during blood count checks. If detected during the first trimester, termination of pregnancy followed by treatment of the leukemia is advisable. The management of patients diagnosed later in pregnancy, such as late in the second trimester or during the third trimester, is more problematic. If the leukemia is relatively indolent, it is sometimes possible to manage patients conservatively with leukapheresis and/or transfusion with induction of labor and delivery of the fetus as soon as possible.332 There have also been many reports of patients treated with chemotherapy later in their pregnancy.333, 334 The majority of these women have not aborted, and there has been no report of leukemia occurring in children or an increased incidence of abnormalities in infants.
Metabolic abnormalities
Patients receiving therapy for AML can experience a wide range of metabolic problems such as vomiting, diarrhea, impaired nutrition, or renal dysfunction, usually because of side effects from antibiotics, particularly amphotericin B. Some metabolic disorders are related to the leukemic process itself. Hyperuricemia, occasionally accompanied by urate nephropathy with renal insufficiency, is the most frequent metabolic accompaniment of AML. All patients should receive allopurinol (≥300 mg/day) as soon as the acute leukemia is diagnosed, so that chemotherapy can be administered once it is medically appropriate. In most patients, urate nephropathy can be avoided or ameliorated with vigorous hydration and urinary alkalization with systemic or oral administration of sodium bicarbonate. Allopurinol can usually be discontinued within 1 or 2 days after chemotherapy is completed. In occasional patients with markedly elevated levels of uric acid, the use of recombinant urate oxidase, which can rapidly lower levels within a few hours, may be advisable.335 A single dose is almost always sufficient, and the decision about subsequent doses depends on serial monitoring of uric acid.336
Tumor lysis syndrome occurs more frequently in patients with ALL, although some AML patients experience hyperphosphatemia, hypocalcemia, hyperkalemia, and renal insufficiency because of massive leukemic cell death. The inciting cause seems to be the release of large amounts of phosphate from lysed blasts, which coprecipitates with calcium in the kidneys, leading to hypocalcemia and sometimes to oliguric renal failure. Hyperuricemia further contributes to this problem, which is usually self-limited and responds to judicious hydration.
Despite markedly hypercellular marrows, hypercalcemia is extremely unusual in patients with AML. Severe, occasionally symptomatic, hypokalemia is frequent, particularly in patients with monocytic leukemias. The mechanism appears to be renal potassium loss because of tubular damage induced by the high levels of lysozyme often noted in these patients. Aggressive replacement with parenteral potassium is required; the syndrome usually abates after cytoreduction by chemotherapy. Finally, rare patients have been described in whom lactic acidosis has been a constant metabolic accompaniment of the leukemia both at the time of presentation and relapse.337 The mechanism is unclear, although anaerobic metabolism by the leukemia cells at sites of leukostasis has been postulated.
Summary
Largely because of improvements in supportive care and the anticipatory management of complications of the treatment and underlying disease, the overall outlook for adults <60 years of age with AML has improved worldwide. It is equally clear, however, that results have leveled off and that new approaches are needed to increase the fraction of patients cured. Dramatic advances in molecular genetics offer promise both because of increased understanding of leukemia biology and the production of small molecules, monoclonal antibodies, cytokines, and HGF with potential clinical utility. Further clarification of mechanisms of drug resistance with the possibility of enhancement of the effectiveness of currently available drugs is also an exciting and achievable prospect. These strategies, as well as clinical trials designed to assess the appropriate use of SCT and newer more specifically targeted drugs, determine that therapy for AML will be both more successful and less empiric in the future.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


