Acute myelogenous leukemia (AML) represents a heterogeneous group of hematologic malignancies arising from the transformation and expansion of an early myeloid stem or progenitor cell. The term leukemia originated with Virchow, who, in 1845, recognized a clinical entity characterized by too many white blood cells (WBCs), leading him to name the condition white blood, or leukemia.1 Of some historical interest is that Dr.
John Hughes Bennett’s report of a case of leukemia preceded Virchow’s description by ˜6 weeks.1 However, Bennett had concluded that the condition was secondary to an infection and referred to it as pyemia. The term myelogenous, or myeloid, derives from the terms myelos, meaning marrow and genesis, meaning birth.
The original cases of Virchow and Bennett probably represented what we now know to be either chronic lymphocytic or myelogenous leukemia. The first likely case of acute leukemia was reported by Friedreich and was believed to be lymphocytic.2 It would take the identification of the myeloblast as a precursor cell for granulocytes by Naegeli in 1900 to set the stage for the description of the first cases of AML or what was originally termed acute nonlymphocytic leukemia.3 Even during the first half of the 20th century, reports describing different types of myeloid leukemia made it clear that this was not one but a variety of distinct disorders, all deriving from a bone marrow precursor myeloblast. Initially, cases of monocytic leukemia were described, followed by myelomonocytic leukemia.3,4 Cases of erythroleukemia, megakaryoblastic leukemia, and acute promyelocytic leukemia (APL) were subsequently described in 1917, 1931, and 1957, respectively.5,6 and 7 During the mid-1970s, the French/American/British (FAB) classification system was developed and defined the major categories of AML as M1 through M7.8 More recently, the World Health Organization (WHO) provided a new classification system that utilizes genetic, immunophenotypic, biologic, and clinical features in addition to morphology.9
The description and classification of AML moved more rapidly than the development of effective treatments. During the mid-1800s, Virchow used diet therapies, ferric iodide, and application of abdominal and foot baths.1 In 1865, Lissauer used arsenic (Fowler solution) to treat patients with leukemia, but with little success.1 Radiation therapy was used in the late 1800s, mostly as a form of palliation for chronic leukemias.1 In 1938, Forkner stated, “Although leukemia is a fatal disease much can be done to add to the comfort, and promote the general health of sufferers from the chronic forms of the disease. Unfortunately acute leukemia does not respond satisfactorily to any form of treatment.”1 In 1948, Farber demonstrated that the use of the antimetabolite aminopterin could produce transient remissions in children with acute lymphocytic leukemia (ALL).10 The pioneering work resulted in the National Cancer Institute developing screening programs for other possible antitumor therapies during the 1950s. During the 1960s, several chemotherapeutic agents, particularly cytarabine and anthracyclines, were developed and used in the treatment of AML. During the 1970s, clinical trials demonstrated that combining these two agents would result in long-term remissions for 10% to 15% of patients with AML. The subsequent introduction of more intensive remission induction regimens and post-remission therapy increased the need for more rigorous supportive care measures, and the development of bone marrow transplantation led to current cure rates of about 50%.11,12
EPIDEMIOLOGY
Approximately 6,500 children <20 years of age develop acute leukemia annually in the United States, and AML represents approximately 15%, resulting in approximately 600 new cases/year.13 The remaining cases of acute leukemia in children and adolescents are lymphoblastic leukemia. Essentially the opposite ratios exist for adults, with AML accounting for about 80% of acute leukemia and lymphoblastic the remaining 20%. With the exception of a peak in incidence of AML in infants, the incidence of AML is relatively constant until early adolescence following which it continues to rise slowly through young adulthood and beyond 50 years of age the incidence rises dramatically.14
Some variation in the incidence of AML in children has been reported among different racial and ethnic groups. For example, black children have an incidence of 5.8 cases/million, compared to 4.8 cases/million in white children.15,16 Children and young adults of Hispanic background have the highest annual incidence at 9 cases per million with the major subtype being APL.17,18,19 and 20 The annual incidence of AML in children from Japan, Australia, and Zimbabwe has been reported as 8, 8, and 11 per million.21 The increasing incidence of secondary leukemia, resulting from chemotherapy and treatment for other malignancies, is a problem of increasing significance in pediatrics.22,23,24,25,26,27 and 28
CELLULAR AND MOLECULAR ORIGINS OF ACUTE MYELOGENOUS LEUKEMIA: HEMATOPOIETIC HIERARCHIES
The determination of the AML stem cell is not solely of biologic interest but has profound significance for understanding the causes of leukemia and potentially the development of curative therapies. This section discusses the cellular and molecular determinants of AML.
Normal hematopoiesis occurs through a series of complex changes that facilitate multipotential hematopoietic stem cells to both expand and differentiate into various mature blood cell types. Because AML is derived from an abnormal immature hematopoietic precursor cell, these leukemias also have the capacity to expand and to show characteristics of limited differentiation. Thus, myeloid leukemias retain many of the molecular and cellular phenotypic characteristics of their normal hematopoietic origins, providing the means to distinguish subtypes of the disease and define potential leukemic stem cell compartments. For example, although most myeloid leukemia cells often express growth, survival, and differentiation receptors for specific cytokines such as KIT, FLT3, and granulocyte-macrophage colony-stimulating factor receptor, some subtypes also express more lineage-specific surface receptors, such as those for granulocyte colony-stimulating factor (G-CSF) and erythropoietin. The same is true for the expression of differentiation markers that characterize various myeloid lineages such as megakaryoblastic, erythroid, or monocytic. These diverse phenotypic characteristics of different subtypes suggest significant heterogeneity of both the genetic changes and the cell of origin in AML.
Some of the earliest biologic tools used to define the cellular compartment in which leukemic stem cells arise included the use of X-linked glucose 6-phosphate dehydrogenase isoenzyme analysis in female patients with chronic myelogenous leukemia (CML) and then AML.29,30 Subsequently, karyotypic abnormalities were examined in the maturing colony-forming units to evaluate which lineage (colony-forming unit stem, colony-forming unit granulocyte-macrophage, colony-forming unit megakaryocyte, colony-forming unit granulocyte, colony-forming unit eosinophil, etc.) contained the aberrant chromosomal marker.31 These studies revealed that although CML arises in a very early pluripotential hematopoietic cell, there are cases of AML that arise in more mature pluri- and unipotential progenitor cells.31
By depleting samples of AML with antibodies directed against lineage-specific surface antigens, a very small percentage of lineage-negative (Lin–) cells were isolated that had the capacity to generate AML at a higher percentage than more mature leukemic cells when transferred to immunodeficient mice. These leukemogenic CD34+, CD38–, Lin– cells (termed self-renewing leukemia-initiating cells) were in most cases rare, having a frequency of as few as 0.2 to 200/106 mononuclear cells.32,33,34,35 Of further significance was that the frequency of the self-renewing leukemia-initiating cell did not correlate with age, sex, or FAB classification, with the exception of some cases of APL.33 In the case of APL, a significant percentage of cases appeared to be derived from a more committed progenitor cell.33,36 Subsequent investigations identified both CD34+ and CD34– lineage-negative self-renewing leukemia-initiating cell populations that were present at extremely low frequency, demonstrating that the AML stem cell is in many cases derived from a very immature hematopoietic precursor cell.37
When these results are placed into the context of the hematopoietic differentiation schema, the conclusion can be made that there must be primary genetic changes that occur in a very primitive self-renewing stem cell and that the nature of those genetic changes in part determines the subtype of AML. For example, a t(8;21) abnormality may lead to M1 AML with minimal differentiation, whereas an inv(16) abnormality may result in an M4Eo subtype. The initiating events may primarily affect the ability of the leukemic stem cell to differentiate but retain the ability to self-replicate.38,39 Subsequent genetic changes, such as mutation pathways regulating apoptosis, cell survival, and proliferation, may further change the phenotype of the leukemia as well as provide the proliferative potential for the leukemia. These might be considered “driver” and “modifier” mutations, respectively. Mutations that do not affect the phenotype may be considered “passenger” mutations. This latter type of genetic change involves mostly secondary mutations that affect the function of growth and survival factor receptors, such as FLT3 or KIT. By themselves, most single mutations are insufficient to cause leukemia. However, when present together in the same cell, they can cooperate and lead to the development of AML. A slightly different alternative way of understanding the etiology of AML in the context of gene mutations is to consider genetic changes such as AML-ETO translocation as a Type I mutation and FLT3-ITD or KIT point mutations as Type II mutations that in combination result in the proliferation of progenitors with limited and aberrant differentiation.40
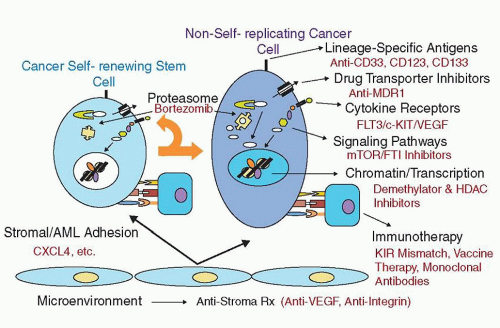
FIGURE 77.1. Therapeutic targets being tested in acute myelogenous leukemia (AML). The schema shows different pathways and/or approaches that have been or are being tested in clinical trials, including those that may affect the leukemia-initiating cell.
Genome-wide sequencing has confirmed the above models but also revealed many additional subkaryotypic abnormalities including gene mutations encoding classes of proteins involved in epigenetic patterning41,42,43 and RNA splicing.44,45 Such studies have also helped to define the heterogeneity of AML and its clonal evolution during treatment.46,47 Of interest, although many of the same mutations observed in older adults with AML have been found in children with AML, there is a growing body of evidence that shows significant differences also exist.48,49 and 50
These results have important implications not only for understanding the etiology and pathogenesis of AML but also for the development of more effective treatments (Fig. 77.1). The causes of generating genetic mutations contributing to AML involve both inherited and environmental factors. In pediatrics, the former class of factors is particularly important.
PREDISPOSING FACTORS AND PATHOPHYSIOLOGY
Inherited Predisposition Syndromes
Abnormal Chromosomal Number
Trisomy 21, or Down syndrome (DS), represents the most common inherited condition that predisposes to the development of leukemia. The overall risk of developing leukemia has been estimated to be about 14-fold above that of the general population.51,52 Although older children with DS have a similar frequency of ALL and AML, within the first 3 years of life, AML, and particularly acute megakaryoblastic leukemia (AMKL), predominates.52
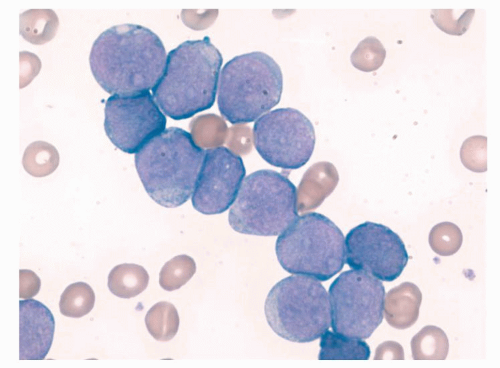
Patients with DS also have an increased predisposition to develop a condition known as preleukemia or transient myeloproliferative disorder (TMD). Approximately 10% of newborns with DS develop TMD. Although clinically indistinguishable from congenital leukemia (Fig. 77.2), TMD, as the name suggests, is usually self-resolving. It is importantly to note that even children who are mosaic for trisomy 21 but phenotypically normal share the increased risk of developing TMD and subsequent leukemia. Approximately 20% to 30% of children whose TMD resolves still develop AMKL.
The close association of trisomy 21 with TMD and AMKL suggests that predisposing genetic events exist. RUNX1 (AML1), which is located on chromosome 21 and known to be involved in some subtypes of AML, has been implicated etiologically, but no definitive evidence has yet demonstrated a specific mutation or gene dosage effect leading to AML. However, other investigations have demonstrated the interesting finding that TMD and AMKL are both characterized by mutations of the GATA1 gene that result in the introduction of a premature stop codon, truncating GATA1 before the amino-terminal activation domain and reducing its transcriptional activation ability.53,54 The same GATA1 mutation has been observed in the blasts from TMD as well as in AMKL.53,54,55 and 56 These results argue strongly that mutations in the GATA1 hematopoietic transcription factor are an early event in the development of TMD and AMKL in children with DS. It remains unclear why a majority of children with DS and TMD show regression of their disease.
FIGURE 77.2. Photomicrograph of a peripheral blood smear from a patient with Down syndrome and transient myeloproliferative disease.
An increased risk of developing AML in patients with Klinefelter syndrome (XXY) and Turner syndrome (XO) has also been reported, but the numbers of such cases are quite low.57,58
Inherited Marrow Failure and Chromosome Instability Syndromes
There are several inherited syndromes characterized by progressive marrow failure and cytopenias with a high frequency of AML. Fanconi anemia (FA) is an autosomal recessive inherited disorder with common congenital abnormalities including skeletal abnormalities, short stature, microcephaly, cardiac abnormalities, genitourinary tract abnormalities, café-au-lait spots, and mental retardation. Patients with FA have an estimated 15,000 times greater risk than the general population for developing AML and an actuarial risk of myelodysplastic syndrome (MDS) or AML of approximately 52% by 40 years of age.59,60 There are multiple gene defects that give rise to FA and affect distinct but functionally related proteins that regulate DNA repair; the mutations result in a hypersensitivity to genotoxic agents such as mitomycin C or diepoxybutane and chromosomal instability.61 Somatic mutations in several of the FA genes have also been observed in AML outside the setting of FA, thus further strengthening the link of these genes with predisposition for AML.62,63
Severe congenital neutropenia (Kostmann syndrome) represents an important inherited cytopenia of the granulocytic lineage with an increased risk of MDS/AML that increases with age.64 Introduction of G-CSF for the treatment of patients with Kostmann syndrome has been linked to the development of AML. However, it is possible that patients on G-CSF may survive for longer periods, raising the possibility that the development of AML is secondary to an intrinsically increased risk of leukemia in patients with Kostmann syndrome or a combination of this predisposition and G-CSF stimulation. Mutations in the elastase gene have been associated with both cyclic neutropenias and Kostmann syndrome.65,66 The detection of somatic activating mutations of the G-CSF receptor has been observed before the development of overt AML in patients with Kostmann syndrome.67,68 Thus, when a rising WBC count is observed in a patient with Kostmann syndrome who was previously stable on G-CSF, one should consider that autoactivating mutations of G-CSF have occurred and that the patient has developed AML.
Shwachman-Diamond syndrome, inherited in an autosomal recessive fashion, is characterized by pancreatic insufficiency, skeletal abnormalities, neutropenia, and an increased incidence of MDS and AML.63,69,70 Diamond-Blackfan anemia (DBA) is another inherited syndrome characterized by congenital anemia, skeletal and urogenital abnormalities, and an increased risk of developing MDS and AML.60 An association of mutations and/or deletions in both small and large ribosomal subunit proteinencoding genes has established DBA as a ribosomopathy. In addition to showing an increased frequency of AML, patients with DBA also appear to have a predisposition to other cancers, making DBA a true cancer-predisposition syndrome.
Neurofibromatosis Type I (NF1) is caused by mutations in the neurofibromin gene, encoding a RAS-inactivating GTPase, and closely associated with an increased incidence of juvenile myelomonocytic leukemia (JMML) and AML.71,72 Noonan syndrome, caused by mutations in the PTPN11 gene, which encodes a SHP-2 tyrosine phosphatase, also has an increased predisposition to JMML.73,74 Mutations in CBL, encoding an E3 ubiquitin ligase, have been reported to be associated with a dominant inheritance and predisposition to JMML.75,76 and 77 Patients with Li-Fraumeni syndrome and Bloom syndrome, involved defects in TP53 and the BLM helicase gene, respectively, have also been reported to have a propensity to develop leukemia, including AML.60,78,79,80 and 81
Additional inherited AML predisposition syndromes include congenital amegakaryocytic thrombocytopenia (defects in the CFFA2 gene and thrombopoietin receptor gene, c-mpl), autosomal dominant macrothrombocytopenia (Fechtner syndrome, MYH9 gene), familial platelet disorder with propensity to myeloid malignancy (FPD/AML, and germline mutations in the RUNX1/CEPB-alpha gene).60,69,82,83,84,85,86 and 87 Other DNA repair/chromosome instability syndromes that can lead to leukemia, although more commonly ALL, include Bloom syndrome, secondary to an inherited defect in the blm helicase gene, ataxia telangiectasia, due to defects in the ATM gene, and Li-Fraumeni syndrome, due to inherited mutations in TP53.60,78,79,80,81
Twins and Familial Cases
The increased frequency of both AML and ALL in siblings of patients with leukemia has been recognized since the early 1920s. The risk for identical twins is high when leukemia first develops during infancy; in most cases, transmission has been shown to be the result of transplacental transfer. Transmission rates have been reported to be 20% to 30%, although other investigators have concluded transmission rates may approach 100%.88,89 and 90 There is also a high concordance of timing of the onset of leukemia. Molecular studies have demonstrated that identical molecular defects characterized the leukemia in both twins.88,91,92 and 93
Clinical follow-up is therefore essential in identical twins when one of them is diagnosed with acute leukemia. These normal twins should be followed approximately every 1 to 2 months until approximately 2 years of age with physical examinations and peripheral blood cell counts. Bone marrow examinations should only be done when clinically indicated. The risk of developing acute leukemia for nonidentical twins has been estimated to be a two- to fourfold increase until about 6 years of age, after which the risk becomes similar to that of the general population. Nontwin familial cases of AML are rare and often associated with constitutional translocations, such as t(7;20) and t(3;6) or monosomy 7.94,95 and 96
Acquired Predisposition
A variety of acquired AML predisposition disorders exist. For example, patients with severe aplastic anemia (SAA) treated with immunosuppressive agents, such as cyclosporin A and antithymocyte globulin (ATG) or cyclophosphamide, as well as with recombinant human G-CSF have been reported to have up to a 20% risk of developing MDS or AML.97,98 and 99 Paroxysmal nocturnal hemoglobinuria is also associated with an increased risk of developing MDS and AML although less frequently than SAA.100 Although AML arising in patients with MDS is a relatively common event in adults, MDS is rare in children and may differ in biologic and clinical characteristics from that observed in adults.101 Acquired monosomy 7 may predispose individuals to developing MDS and AML.102 The acquisition of predisposing conditions or chromosomal abnormalities is often linked to environmental exposures.
Environmental Factors
A wide variety of genotoxic environmental exposures can predispose individuals to AML. For example, in the period after the dropping of atomic bombs on Nagasaki and Hiroshima during World War II, an approximately 20-fold increase in myeloid leukemia was documented, with a peak incidence between 6 and 8 years.103,104,105 and 106 The absence of a documented increase in leukemia in children exposed prenatally to the radiation of the atomic bombs has been reported107 and may be consistent with the absence of definitive evidence that prenatal exposure to x-rays increases leukemia risk.108 There remains no convincing evidence that ultrasound or the effects of living near high-voltage power lines predisposes individuals to leukemia, although reports differ in their conclusions.109,110 and 111
Prenatal exposure to chemical genotoxic agents has been reported to increase postnatal incidence of myeloid leukemia. For example, maternal alcohol consumption has been associated with an increased risk of AML in offspring.112 A significant association of dose-response of prenatal alcohol consumption and the development of AML in offspring was documented.113 However, not all reports have concluded such strong associations.112,114 There has been an association of maternal ingestion of topoisomerase II inhibitors and the development of AML with malignant lymphoblastic lymphoma rearrangements in offspring.115,116,117 and 118 Parental smoking of tobacco or marijuana has also been associated with an increased incidence of AML in offspring although there are reports with dissenting conclusions.119,120 and 121 Some reports have linked cigarette smoking in adults to an increased incidence of AML, making antismoking preventive counseling important, particularly during teenage years.122
Exposure of individuals to environmentally derived genotoxic agents is quite substantial, and several specific examples of such exposures have been shown to be etiologically related to the development of AML. Such exposures include petroleum products, benzene, pesticides, and herbicides,123,124,125,126 and 127 although these studies have not focused on children. Interestingly, in some instances, such as with organophosphate pesticides, children may be at greater risk for accumulating higher levels of the chemicals.128
An increasingly worrisome group of patients are those who develop AML as a result of chemotherapeutic exposures for treatment of their primary cancer or even nonmalignant conditions. For example, exposure to alkylating agents, commonly used to treat patients with brain tumors, lymphomas, and other solid tumors, results in an increased incidence of secondary AML, with a peak incidence at 4 to 5 years but with an at-risk period extending 12 years.129,130 and 131 Exposure to topoisomerase I inhibitors, including anthracyclines, and topoisomerase II inhibitors, including epipodophyllotoxins such as etoposide, is also etiologically linked to the development of AML.132,133,134 and 135 Whereas cumulative dose and schedule of drug delivery may play important roles in the development of AML136 nearly any exposures to such genotoxic agents can result in secondary AML, as demonstrated in a child initially treated for neuroblastoma.137,138 The development of secondary AML in patients treated first for primary cancers may be one of the most compelling reasons to develop alternative and less genotoxic approaches to therapy.
PRESENTATION
The clinical presentation of AML varies greatly with systemic symptoms and severity of illness usually being a result of leukemia cells’ replacement of normal hematopoietic progenitors in the bone marrow as well as their infiltration into various organs. Approximately 1012 leukemia cells have been estimated to be present at the time of diagnosis. The leukemic blasts can invade extramedullary sites, such as soft tissues, skin (leukemia cutis), gingiva, orbit, and brain. Patients typically present with signs and symptoms of neutropenia, anemia, and thrombocytopenia. On occasion, other systems may be involved at presentation, as in the case of coagulopathy seen most commonly in APL, spinal cord compression from chloromas, or end-organ damage due to hyperleukocytosis.
The total WBC count may be low, normal, or high depending upon the number of circulating leukemic cells. The absolute neutrophil count is often less than 0.5 × 109/L and is associated with an increased risk of infections, often life threatening. Blood cultures and broad-spectrum intravenous antibiotic coverage are indicated in any newly diagnosed patient with leukemia and fever. Anemia results in fatigue, lethargy, decreased exercise tolerance, headache, and pallor. Although uncommon at the time of presentation, congestive heart failure (CHF) may occur, particularly with severe anemia, which is usually normocytic and normochromic although evidence of red cell fragmentation may be seen in cases presenting with disseminated intravascular coagulation (DIC); the transfusion of packed red blood cells should usually be done slowly to prevent precipitating or worsening CHF. Thrombocytopenia often leads to bruising and petechiae, and occasionally overt hemorrhage into the gastrointestinal track, lungs, or central nervous system (CNS). Approximately 50% of patients have hepatosplenomegaly and lymphadenopathy. Gingival hyperplasia and leukemia cutis are less frequent but particularly characteristic of myeloid leukemia with monocytic differentiation.
Patients may also present with anemia, which gives rise to fatigue, pallor, and, in extreme cases, hemodynamic instability. The anemia is typically normocytic and normochromic, although evidence of red cell fragmentation may be seen in severe cases of DIC. The median hemoglobin is ˜70 g/L, with a range of 25 to 140 g/L.
Nearly 75% of patients present with a platelet count <100 × 109/L.179 Thrombocytopenia may cause petechiae, purpura, mucosal bleeding, and, rarely, CNS and pulmonary hemorrhage. Thrombocytopenia is exacerbated by coagulopathy, especially in the M3 and M5 AML subtypes. Although the mechanism for DIC is not known in M5 AML, there is convincing evidence that expression of annexin II, a receptor for fibrinolytic proteins, facilitates plasminogen activation by associating plasminogen and its activator, tissue plasminogen activator, at the APL (M3) leukemic blast cell surface.180
Patients with peripheral blast counts >200 × 109/L are at risk for CNS stroke due to hyperviscosity, and benefit from leukopheresis to drop the blast count rapidly.181 Similarly, pulmonary insufficiency may occur in patients with very high leukemia blast counts. Approximately 5% of patients with AML have CNS disease at diagnosis, and a smaller percentage presents with CNS chloromas.182 These patients may have headaches, cranial nerve palsies, focal neurologic deficits, and, rarely, seizures.
DIFFERENTIAL DIAGNOSIS
Whereas the diagnosis of AML is generally straightforward, the differential diagnosis is broad, including both malignant and nonmalignant conditions. Juvenile rheumatoid arthritis, infectious mononucleosis, aplastic anemia, congenital and acquired cytopenias, and the transient myeloproliferative syndrome of DS infants may all mimic AML. AML may be mistaken for MDS or chronic leukemias, including CML, chronic myelomonocytic leukemia, and JMML. Undifferentiated leukemia or FAB L2 ALL may be morphologically difficult to distinguish from megakaryoblastic AML. Metastatic rhabdomyosarcoma or neuroblastoma in the bone marrow may appear as megakaryoblastic or monoblastic AML, especially in the neonate.
The diagnosis of AML is typically made on bone marrow aspirate examination, with special stains, flow cytometry, and cytogenetics providing additional data. On occasion, definitive diagnosis is difficult either because of technical difficulties in obtaining an adequate specimen or because of conflicting data. Repeat marrow aspirate and biopsy may provide a specimen adequate for diagnosis. Touch preparations of the bone marrow biopsy may be used in cases in which bone marrow aspiration is difficult. Increasingly, a diagnosis can be made from peripheral blood using multiparameter flow cytometry, although in some instances significant differences in antigen expression may exist on leukemic blasts in the bone marrow compared to the peripheral blood.
TABLE 77.1 COMPARISON OF KEY CHARACTERISTICS OF ACUTE MYELOGENOUS LEUKEMIA (AML) IN PEDIATRIC VERSUS ADULT PATIENTS
Characteristic
Pediatric ≤ 21 y
Adults >21 and <55-60 y
Cytogenetics
Higher frequency favorable risk cytogenetics
Greater percent of unfavorable cytogenetics and drug resistant markers
Antecedent AML predisposing disorders
Rare (some inherited predisposition)
Common (MDS, MPN, and therapy-related)
Subtype
More M4/M5, DS, TMD
Less M4/M5, no DS or TMD
Extramedullary disease
More CNS disease/leukemia cutis
Uncommon except in monocytic lineage
Induction
ADE
“7&3” standard
Remission rates (after 2 courses)
85-90%
60-80%
CNS Prophylaxis
Yes
Not routine
HSCT
Only for high-risk patients
For some intermediate- and most high-risk patients
ADE, ARAC, daunomycin and etoposide; CN-AML, cytogenetically normal AML; CNS, central nervous system; DS, down syndrome; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; TMD, transient myeloproliferative disorder. Adapted from214,422 and149.
ACUTE MYELOGENOUS LEUKEMIA SUBTYPES
Both FAB and WHO classification systems are used by clinicians treating pediatric AML patients. Generally, both classification systems apply equally well to pediatric and adult patients. However, some important differences exist. FAB subtypes M0, M1, and M2 correspond to AML that is minimally differentiated, without maturation, and with maturation, respectively, are more common in older rather than younger children, with frequencies in children 10 to 15 years of age very similar to reported adult frequencies.139,140 On the other hand, FAB subtypes M5 (acute monocytic leukemia) and M7 (AMKL) are significantly more common in younger children.139,141 Likewise, the increased frequency of M7 AML in young patients is mostly due to the high rate of the M7 subtype in patients with DS.142 Younger children are less likely to have t(8;21) and t(15;17) but more frequently have chromosome abnormalities involving 11q23. M0 AML is defined as AML without morphologic signs of differentiation and by expression of CD13, CD33, and CD117 (c-KIT) and myeloperoxidase by flow cytometry or electron microscopy (Table 77.1).
THERAPY FOR PATIENTS WITH NEWLY DIAGNOSED ACUTE MYELOGENOUS LEUKEMIA
Background
Pediatric AML protocols begin with a remission-induction regimen, followed by a course of consolidation therapy, and, subsequently, intensification courses that may include hematopoietic stem cell transplantation (HSCT). This relatively brief but intensive approach has yielded an approximately 60% to 70% chance of overall survival across different cooperative group protocols.143,144,145,146,147,148 and 149 Although there is general agreement that pediatric AML therapy should be based on the use of anthracyclines and cytarabine, pediatric cooperative groups differ in their induction regimens and the use of HSCT transplant in the post-remission period. The major pediatric cooperative groups are also presently evaluating various risk-stratification methods as well as the use of novel agents.
Induction Therapy
The primary goal is to achieve a remission of disease, which is currently defined in the United States as peripheral blood count recovery with a normal or slightly hypocellular marrow with fewer than 5% leukemic blasts and no evidence of extramedullary disease. An increasingly important additional marker is the eradication of detectable minimal residual disease (MRD).
The first significant successful remission-induction regimen for patients with AML included 7 days of continuous infusion Ara-C at 100 mg/m2/day and 3 initial days of an anthracycline, such as daunorubicin, at 45 mg/m2 to 60 mg/m2 per day. This regimen resulted in remission rates of between 60% to 70% in children and young adults.150,151 However, several approaches have been utilized to improve on these remission rates, including altering the schedule and doses of Ara-C and anthracyclines, as well as the introduction of additional agents. Furthermore, supportive care measures, such as the pre-emptive use of broad-spectrum antibiotics and/or blood product transfusions, have proven to reduce significantly remission induction mortality and thus have improved remission rates.
Despite the different approaches taken by the cooperative groups, several important conclusions can be made. First, remission quality, now defined as negative MRD at the end of induction, modifies relapse risk, and more intensive induction regimens may provide deeper (i.e., nondetectable MRD) remissions with lower relapse rates and improved OS. The issue of induction intensification was nicely demonstrated in the Children’s Cancer Group (CCG)-2,891 study, in which the intensively timed DCTER (dexamethasone, cytarabine, thioguanine, etoposide, and rubidomycin) arm had a similar morphologic induction success rate compared to the standard timing regimen, but the relapse rate for the intensively timed arm was lower regardless of post-induction therapy.146 However, more intensive induction regimens also carry greater treatment-related morbidity and mortality that can diminish the net benefit of such intensive therapy. In general, each trial using a very intensive induction regimen has noted an initial toxic death rate of approximately 15%, which decreases to usually less than 5% with acquired treatment experience.152,153
This high induction mortality rate has been reduced in several studies by mandated supportive care guidelines (see the section “Supportive Care”). Although escalating the economic cost of AML therapy, these supportive care guidelines are critical for improved outcomes. In the CCG, these guidelines mandated early initiation of broad-spectrum antibiotics, including vancomycin at the first febrile episode, early initiation of treatment doses of antifungal agents after 3 days of persistent fevers, hospitalization until granulocyte recovery, strict hand washing, and use of high-efficiency particulateair -filtered rooms whenever possible. Institution of these guidelines lowered toxic mortality to ˜5% across cooperative group trials.193
Various trials have also tried to answer additional remission induction questions, including (1) determining the optimal dose and schedule of cytarabine, 2) determining the optimal anthracycline and dose, and (3) determining what agents can be added to the cytarabine and anthracycline backbone to improve outcomes. Although dose intensification of cytarabine has not been demonstrated to improve remission induction rates, higher doses appear to confer lower rates of leukemia relapse in adults in patients with core-binding factor AML.151,154 Becton et al. reported the POG 9421 trial that randomized patients to standard versus high-dose cytarabine in induction therapy. No statistically significant differences in remission induction rate or EFS were observed; patients randomized to high-dose cytarabine had an increased risk of bacteremia/sepsis that approached, but did not reach, statistical significance (28% vs. 22%, P = 0.1).155 A second pediatric study has also demonstrated no benefit for high-dose cytarabine over standard-dose cytarabine when used during induction therapy.148
Debate continues over the optimal anthracycline choice in induction. The Berlin-Frankfurt-Munster (BFM) group showed evidence that idarubicin was superior to daunomycin in induction,156,157 and a meta-analysis by the Medical Research Council (MRC)/Institute for Cancer Research of randomized idarubicin/daunomycin comparisons suggested that idarubicin is superior.158 The CCG-2941 and COG-2961 trials showed that idarubicin was too toxic to be used in sequential courses of intensively timed IdaDCTER therapy.153,159,160
The MRC AML-12 trial randomized daunorubicin, ARAC, and thioguanine (ADE) versus mitoxantrone, ARAC, etoposide (MAE) in combination with cytarabine and etoposide. Although the MAE regimen showed a decrease in the relapse rate compared to the ADE-treated group, the increased risk of treatment-related mortality led to no overall benefit in DFS or OS. These results were similar in children and adults.161,162
Whereas the addition of other agents to the “7 and 3” backbone has helped increase induction rates from 70% to 85%, no randomized trial has demonstrated the superiority of a particular agent, or combination of agents, over any other combination. Specifically, the MRC-10 trial tested 6-thioguanine versus etoposide with daunomycin and cytarabine and found no statistically significant difference between the two induction regimens.145 In an attempt to introduce a novel, noncross-resistant agent into induction therapy, the COG AAML03P1 trial demonstrated that gemtuzumab ozogamicin (GO) can be safely added to a backbone of cytarabine, daunorubicin, and etoposide, resulting in a remission rate of 83.5% after 1 cycle of therapy.147,163 Results of the randomized trial using GO have not been reported at the time of this writing. However, the randomized study from the MRC has demonstrated no change in remission rates with the addition of GO, but an OS advantage particularly for patients with core-binding factor AML.164 No advantage was observed in a Nordic Society of Pediatric Hematology and Oncology (NOPHO) trial randomizing GO in the post-remission setting.165
In summary, current pediatric AML induction regimens successfully induce remission in approximately 85% of patients using a variety of induction strategies. The improvements in induction remission rates have come primarily from intensification of therapy, either by adding additional agents to the “7 and 3” backbone or from dose intensification. Although successful, it appears unlikely that further dose escalation or intensification will significantly improve remission rates. Thus, a central remission induction question now centers on the selection and safe integration of other novel agents aside from GO, in order to increase antileukemic activ ity with less toxicity than conventional chemotherapy.
Post-remission Therapy
Once a complete remission has been achieved, including negative MRD status, additional therapy is required to avoid disease relapse. Various combinations and numbers of courses of therapy have been tested.
Dose and Duration
Although agreement exists on the role of cytarabine-based intensification therapy, especially for core-binding factor myeloid leukemias in which additional courses of cytarabine appear to decrease relapse risk significantly, the optimal number of cycles of intensification chemotherapy is not known.146,166,167 The published pediatric AML trials have used either 2 or 3 courses of consolidation therapy, for a total of 4 or 5 courses of therapy.168,169 and 170 The MRC AML-10 trial randomized patients to either auto-HSCT transplant or no further therapy and demonstrated a decreased relapse risk in patients receiving auto-HSCT. However, the addition of auto-HSCT was associated with significant morbidity and mortality, thus abrogating any OS advantage.169 The CCG-2961 trial gave patients a total of 3 courses of chemotherapy with a resulting overall survival of 57% following changes in supportive care recommendations.171 The MRC AML-12 trial randomized patients to a total of 4 versus 5 courses of chemotherapy with an OS of 81% versus 78%, respectively, at 5 years (P = 0.5). However, the survival for patients with very high risk AML was significantly less than for patients with intermediate and favorable risk AML; thus, any attempts to test 4 versus 3 courses of therapy should likely be done in the latter groups.162
With the exception of patients with APL, the use of maintenance therapy with relatively low dose chemotherapy has been currently abandoned with the exception of BFM studies, which in part base this choice on the results of BFM-87, in which a maintenance phase was beneficial to a low-risk group of patients who did not receive HSCT. However, when only randomized patients were analyzed, no significant difference in outcome was observed, and no significant difference was observed.172 Other cooperative groups, however, have shown that maintenance therapy is associated with a decrease in both EFS and OS when compared to shorter, more intensive regimens, with relapsed disease being more resistant to subsequent therapies.173,174,175
Hematopoietic Stem Cell Transplantation
Sustained improvements in chemotherapeutic regimens and supportive care have continued to reduce the need for patients to receive allogeneic HSCT. Furthermore, several studies have shown equivalent overall survival when compared to chemotherapy; most co-operative groups have thus omitted autologous HSCT from consideration in order potentially to avoid greater short- and long-term toxicities.145,168,176,177 However, with equivalency of overall outcome between autologous HSCT and chemotherapy, one might also conclude that either approach would be a reasonable, evidenced-based recommendation.178 In addition, alternative approaches to transplantation, such as the use of nonablative or intensity-reduced allogeneic or haplo-identical HSCT, remain experimental and need prospective testing in comparison to more conventional approaches.
Pediatric cooperative groups generally agree that intensive cytarabine-based post-remission induction therapy is required to minimize relapse risk. The role of allogeneic HSCT has continued to evolve. This has in large part been due to the advances in chemotherapy-based treatments for patients who might have previously been considered candidates for allogeneic HSCT. Key questions are whether allogeneic HSCT provides an overall improvement in survival and quality of life compared to chemotherapy-only treatment approaches. In order to answer these questions, it has been necessary to define risk groups more carefully in terms of outcome and potential for benefit from HSCT. In addition, whereas most trials have analyzed outcomes on whether patients did or did not have an HLA-matched donor, the improving success being obtained using matched unrelated donor HSCT has led to outcome analysis based on availability of the best HLA-matched donor.162
Many trials have shown that allogeneic HSCT results in an improved disease-free survival compared to chemotherapy or autologous HSCT.162,168,177,179,180,181 However, HSCT has not usually resulted in an improved event-free or overall survival, reflecting the associated increased treatment-related mortality. Such results suggest that HSCT may benefit some groups more than others. Several studies have attempted to define such subgroups more precisely.
The availability of an HLA-matched donor was used to analyze outcome in the MRC AML 10 trial.182 There was no statistically significant difference in overall survival between those children with (68%) or without (59%) a donor at 10 years, even those there was a significant difference in relapse rate (30% with donors vs. 45% without donors). Based on the MRC cytogenetic and response-based risk stratification, overall survival at 5 years from the time of relapse was 57%, 14%, and 8% for good, standard, and poor risk groups, respectively; this led to the conclusion that allogeneic HSCT should be done in good risk AML in CR2. Although outcomes from MRC AML 10 also suggested that allo-HSCT in first remission as improved for patients with intermediate- and high-risk AML, combined data from the MRC AML 10 and 12 trials showed no statistically significant benefit for these groups of patients compared to chemotherapy alone.182 BFM trials and POG 8,821 and 9,421 studies showed comparable results although in the POG trials did not do a detailed subgroup stratification.155,183,184
In contrast, analysis of post-remission treatment of 1,464 children less than age 21 years on 5 consecutive CCG trials from 1979 to 1996 has shown an advantage to those patients assigned a HSCT in terms of overall survival (P = 0.026), disease-free survival (P = 0.005), and relapse rate (P < 0.001).185 Subgroup analysis demonstrated that HSCT was associated with improved survival for patients with WBC greater than 50,000/µL and for those with normal karyotype, but was not beneficial for patients with AML characterized by good -risk cryogenics, such as inv(16) or t(8;21). In support of such data, a report from the CCG 2961 trial showed no statistically significant advantage of having a HSCT donor in terms of OS or DFS in the subgroup of patients with inv(16) or t(8;21) chromosomal translocations.171 A detailed analysis of the effect of donor availability on patients with standard and poor risk features was not reported although no advantage of having a HSCT matched family member donor was observed compared to chemotherapy for the entire study population. Overall, these results appear to be consistent with those reported from pediatric MRC AML trials.
A more detailed analysis of the MRC AML 12 outcomes has reported no advantage of HSCT for patients in the good and intermediate groups, but a statistically significant advantage for relapse-free survival and overall survival for a subset of patients with high-risk AML; for example, in the 12% of patients defined as having poor-risk AML, HSCT was associated with an overall survival of 41% compared to 10% for those who received only chemotherapy (P = 0.001).162
Whether patients with other subtypes of AML might also benefit from receiving an allogeneic HSCT remains controversial. A high FLT3-ITD mutant to normal allele frequency has been uniformly associated with a poor prognosis when patients are treated with standard chemotherapeutic regimens alone.186,187,188 A significant question is whether more allogeneic HSCT is able to improve outcomes in patients with a high allelic ratio FLT3-ITD AML.
Some studies have strongly suggested an advantage of HSCT for patients with a high mutant allele frequency of FLT3-ITD mutations.188,189,190 and 191 Data from the CCG 2941 and 2961 trials showed a borderline significant difference in relapse for patients with FLT3-ITD positive AML who received a allogeneic matched sibling donor HSCT (27% ± 27%) compared to those treated with only chemotherapy (65% ± 15%, p = 0.05). However, overall survival at 4 years from the end of the second course of treatment was not significantly different (64% ± 29% for those with FLT3-ITD AML who received an allogeneic HSCT and 48% ± 17% for those treated with chemotherapy [p = 0.4]).188,192 The MRC AML 10 and 12 trials concluded that there was no strong evidence that FLT3 status should be considered as to whether to perform a HSCT based on their analysis of 1,135 young adult patients.193 Because OS was not significantly improved by having a donor in patients with FLT3-ITD positive or negative AML, it was concluded that allogeneic HSCT is not able to overcome the intrinsic chemoresistance or radiation resistance of FLT3-ITD positive AML. Thus, whether allogeneic HSCT can improve outcomes for patients with FLT3-ITD remains an open question. Nevertheless, most ongoing clinical trials for children and young adults assign allogeneic HSCT for patients with AML characterized by a high mutant FLT3-ITD to normal allelic ratio, often in the context of additional targeted therapy directed toward inhibition of the mutant receptor.
Current strategies for determining which patients should receive matched or single mismatched family donor or alternative donor HSCT are thus based on risk assessment and stratification. Patients with AML characterized by alternations in core binding or transcription factors (e.g., t(8;21), inv(16), biallelic CEBPA mutations) have an approximately 80% overall survival with chemotherapy alone and, thus, HSCT is recommended only in CR2. Similarly, children and young adults with APL have an overall survival of 75% to 90%, depending on risk group, with chemotherapy plus all-trans-retinoic acid (ATRA) and, more recently, arsenic.194,195 Thus, HSCT is not usually recommended in CR1 for these patients, but instead following CR2, in which case allogeneic, and in some instances autologous, HSCT result in an approximately 70% overall survival.195,196 and 197
MLL-rearranged AML represents an extremely heterogeneous group of leukemias associated with variable outcomes. For example, an international trial has reported that survival is 100%, 63%, 27%, and 22% for patients with the t(1;11), the t(9;11), the t(4;11), and the t(6;11), respectively.198 Because of the wide variability of outcomes along with small numbers of patients with MLL subtypes as well as no prospective definitive data that demonstrate HSCG improves outcome in this group of patients, most cooperative group clinical trials have not used allogeneic HSCT in CR1.178 An intention-to-treat analysis of the AML-BFM 98 study has suggested an improved OS with allogeneic MSD HSCT for patients with 11q23 (MLL) rearrangements.199
AML characterized by a normal karyotype represents a large percentage of cases, however, it is also proving to be a molecularly heterogeneous group. For example, nucleophosmin member 1 (NPM1) mutations are associated with an improved outcome, although not necessarily in the presence of high mutant FLT3-ITD to normal allelic ratio is somewhat controversial.200,201 AML with CEBPA mutations is usually associated with normal karyotype AML and improved overall survival, thus making HSCT undesirable in CR1.50 Point mutations involving KIT, RAS, or WT1 (Wilms tumor 1) have not yet been definitively shown to improve outcome although some data exist linking them to a poorer prognosis.202,203 Furthermore, patients with AML having high-risk cytogenetics, such as monosomy 7 or del(5q)- and -5, are recommended to have HSCT in CR1 (Table 77.2).
The Children’s Oncology Group (COG) frontline AML clinical trial AAML1031 uses allogeneic HSCT in first remission only for patients with predicted high risk of treatment failure based on unfavorable cytogenetic, molecular characteristics and elevated end-of-induction MRD levels.204 In contrast, the AML-BFM 2004 clinical trial restricted allogeneic HSCT to patients in second CR and to refractory AML, based on results from their AML-BFM 98 study showing no improvement in DFS or OS for high-risk patients receiving allogeneic HSCT.205 Although the optimal timing for allogeneic HSCT has not been determined, most cooperative groups recommend doing HSCT following the second or third course of chemotherapy, based in part due to the time involved in obtaining HLA typing. The use of matched unrelated donors (MUD), haploidentical donors, single- or double-cord blood donor, or nonablative approaches for HSCT in CR1 are not as clearly established as MSD HSCT, but are increasingly used to provide potential curative treatment for patients with recurrent AML (Table 77.3).206,207
Only gold members can continue reading. Log In or Register to continue
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



