INTRODUCTION
SUMMARY
Acute myelogenous leukemia (AML) is the result of a sequence of somatic mutations in a primitive multipotential hematopoietic cell. Exposure to radiation, chronic exposure to high doses of benzene, and chronic inhalation of tobacco smoke increase the incidence of the disease. Obesity has been found to be an endogenous risk factor. A small but increasing proportion of cases develop after a patient with lymphoma, a nonhematologic cancer, or an autoimmune disorder is exposed to intensive chemotherapy, especially with alkylating agents or topoisomerase II inhibitors. The mutant (leukemic) hematopoietic cell acquires the features of a leukemic stem cell capable of self-renewal and desultory differentiation and maturation. It gains a growth and survival advantage in relationship to the normal polyclonal pool of hematopoietic stem cells. As the progeny of this mutant, now leukemic, multipotential cell proliferates to form approximately 10 to 100 billion or more cells, normal hematopoiesis is inhibited, and normal red cell, neutrophil, and platelet blood levels fall. The resultant anemia leads to weakness, exertional limitations, and pallor; the thrombocytopenia to spontaneous hemorrhage, usually in the skin and mucous membranes; and the neutropenia and monocytopenia to poor wound healing and minor infections. Severe infection usually does not occur at diagnosis, but often does if the disease progresses because of lack of treatment or if chemotherapy intensifies the decrease of blood neutrophil and monocyte levels. The diagnosis is made by measurement of blood cell counts and examination of blood and marrow cells and is based on identification of leukemic blast cells in the blood and marrow. The diagnosis of the myelogenous form of acute leukemia is confirmed specifically by identification of myeloperoxidase activity in blast cells or by identifying characteristic cluster of differentiation (CD) antigens on the blast cells (e.g., CD13, CD33). Because the leukemic stem cell is capable of imperfect differentiation and maturation, the clone may contain cells that have the morphologic or immunophenotypic features of erythroblasts, megakaryocytes, monocytes, eosinophils, or, rarely, basophils or mast cells, in addition to myeloblasts or promyelocytes. When one cell line is sufficiently dominant, the leukemia may be referred to by that lineage: for example, acute erythroid, acute megakaryocytic, acute monocytic leukemia, and so on. Certain cytogenetic alterations are more frequent; these abnormalities include t(8;21), t(15;17), inversion 16 or t(16;16), trisomy 8, and deletions of all or part of chromosome 5 or 7. A translocation involving chromosome 17 at the site of the retinoic acid receptor–α (RAR-α) gene is uniquely associated with acute promyelocytic leukemia. AML usually is treated with cytarabine and an anthracycline antibiotic, although other drugs may be added or substituted in poor-prognosis, older, refractory, or relapsed patients. The exception to this approach is the treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and sometimes an anthracycline antibiotic. High-dose chemotherapy and either autologous stem cell infusion or allogeneic hematopoietic stem cell transplantation may be used in an effort to treat relapse or patients at high risk to relapse after chemotherapy treatment. The probability of remission in acute myelogenous leukemia ranges from approximately 80 percent in children to less than 25 percent in octogenarians. The probability for cure decreases from approximately 50 percent in children to virtually zero in octogenarians.
Acronyms and Abbreviations
ALL, acute lymphocytic leukemia; AML, acute myelogenous leukemia; APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; CBF, core binding factor; CD, cluster of differentiation; ceAML, clonally evolved acute myelogenous leukemia; CEBPA, CCAAT-enhancer binding protein A; CML, chronic myelogenous leukemia; CNL, chronic neutrophilic leukemia; DNMT, DNA methyltransferase; FAB, French-American-British classification; FISH, fluorescence in situ hybridization; FLT, Fms-like tyrosine kinase; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-monocyte colony-stimulating factor; GVHD, graft-versus-host disease; HLA, human leukocyte antigen; HSC, hematopoietic stem cell; IDH, isocitrate dehydrogenase; ITD, internal tandem duplication; MDR, multidrug resistance; MDS, myelodysplastic syndrome; NPM1, nucleophosmin-1 mutation; OS, overall survival; PAS, periodic acid–Schiff; PCR, polymerase chain reaction; P-gp, permeability glycoprotein; ppm, parts per million; PTD, partial tandem duplication; RAR, retinoic acid receptor; RT, reverse transcriptase; RUNX, runt-related transcription factor; SAHA, suberoylanilide hydroxamic acid; t, translocation; TdT, terminal deoxynucleotidyl transferase; TET, ten-eleven translocation; TKD, tyrosine kinase domain; TMD, transient myeloproliferative disease; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor; WBC, white blood cell; WHO, World Health Organization; WT, Wilms tumor.
DEFINITION AND HISTORY
Acute myelogenous leukemia (AML) is a clonal, malignant disease of hematopoietic tissues that is characterized by (1) accumulation of abnormal (leukemic) blast cells, principally in the marrow, and (2) impaired production of normal blood cells. Thus, the leukemic cell infiltration in marrow is accompanied, nearly invariably, by anemia and thrombocytopenia. The absolute neutrophil count may be low or normal, depending on the total white cell count.
The first well-documented case of acute leukemia is attributed to Friedreich,1 but Ebstein2 was the first to use the term acute leukämie in 1889. This work led to the general appreciation of the clinical distinctions between AML and chronic myelogenous leukemia (CML).3 In 1878, Neumann,4 who proposed that marrow was the site of blood cell production, first suggested that leukemia originated in the marrow and used the term myelogene (myelogenous) leukemia. The availability of polychromatic stains, as a result of the work of Ehrlich,5 the description of the myeloblast and myelocyte by Naegeli,6 and the earliest appreciation of the common origin of red cells and leukocytes by Hirschfield7 laid the foundation for our current understanding of the disease.
Although Theodor Boveri proposed a critical role for chromosomal abnormalities in the development of cancer in 1914, a series of technical developments in the 1950s was needed to permit informed examination of the chromosomes of human cancer cells. Thereafter, the discovery that a G group chromosome consistently had a foreshortened long arm in the cells of patients with CML (Philadelphia chromosome) supported the concept that chromosome abnormalities may be specifically linked to a cancer phenotype. This finding was followed by the introduction of banding of chromosomes, which enhanced the specific identification of individual chromosomes and the point at which they break in the formation of a translocation, inversion, or deletion. This technologic advance unleashed the power of cancer cytogenetics and initiated an era of leukemia study based not solely on the appearance of cells under the microscope (phenotype) but also by their chromosomal or genetic abnormality (genotype).8 The completion of the human genome project further enhanced the specificity of the identification of gene alterations.9 These advances permitted (1) more precise understanding of the molecular pathology of specific leukemia subtypes, (2) improvement of diagnostic and prognostic methods for the study of AML, and (3) identification of molecular targets for therapy.
The introduction to the clinic by Holland, Ellison, and colleagues10 of arabinosyl cytosine (cytarabine) in the late 1960s as the first potent drug for treatment of AML, followed by their introduction of the combination of 7 days of cytosine arabinoside and 3 days of daunorubicin in the early 1970s (the “7 plus 3 regimen”)11 opened the era of effective therapy for AML. This drug combination or its congeners remains the mainstay of treatment over 4 decades later.12 The description of allogeneic marrow (stem cell) transplantation as a curative therapy for AML by Thomas and colleagues13 in 1977 ushered in the era of hematopoietic stem cell (HSC) transplantation as a modality to cure eligible patients with AML.
ETIOLOGY AND PATHOGENESIS
Table 88–1 lists the major conditions that predispose to development of AML. Only four environmental factors are established causal agents: high-dose radiation exposure,14,15 chronic, high-dose benzene exposure (≥40 parts per million [ppm]-years),16,17,18 chronic tobacco smoking,19 and chemotherapeutic (DNA-damaging) agents.20,21,22 Most patients have not been exposed to an antecedent causative factor. Exposure to high-linear energy transfer radiation from α-emitting radioisotopes such as thorium dioxide increases the risk of AML.23 Case-control studies have sometimes found a relationship between AML and organic solvents, petroleum products, radon exposure, pesticides, and herbicides, but these data have been inconsistent, have shown no association in other studies, and have not reached a level comparable to the strong association that exists for high-dose benzene, high-dose external irradiation, and certain chemotherapeutic agents. There is a significant association between tobacco smoking and AML with a relative risk of about 1.5 to 2.0.24,25 Although formaldehyde has been suspected of being a leukemogen, detailed analysis has not supported this contention.26,27
Environmental factors Acquired diseases
Other hematopoietic disorders Other disorders
Inherited or congenital conditions
|
An endogenous factor that increases risk is obesity. Studies in North America show an increased risk of AML in men and women with elevated body mass index, and this is particularly notable for acute promyelocytic leukemia. The precise mechanisms are still unclear but may be related, in part, to elevated leptin levels, decreased adiponectin levels, shortened telomeres, and as yet unknown factors in obese subjects.28
AML may develop from the progression of other clonal disorders of a multipotential hematopoietic cell, including CML, chronic myelomonocytic leukemia, chronic neutrophilic leukemia (CNL), polycythemia vera, primary myelofibrosis, essential thrombocythemia, and clonal cytopenia or oligoblastic myelogenous leukemia. The latter two are considered forms of myelodysplastic syndrome (MDS) (see Table 88–1). Clonal progression occurs as a result of genomic instability and the acquisition of additional mutations, although with a different probability of occurrence in each chronic myeloid neoplasm (Chap. 83). The frequency of clonal progression to AML is enhanced by radiation or chemotherapy in patients with polycythemia vera (Chap. 84) or essential thrombocythemia (Chap. 85).29 Although some refer to this as secondary AML, it should be called clonally evolved AML (ceAML) to distinguish it from secondary AML that results from radiation or chemotherapy given to patients who do not have a precedent clonal myeloid disease. In the population of patients with preceding clonal myeloid neoplasms, a myeloid leukemic clone already exists and is not induced secondarily. Evolution to AML represents the natural history of the neoplasm, albeit sometimes accelerated by various external mutagens.
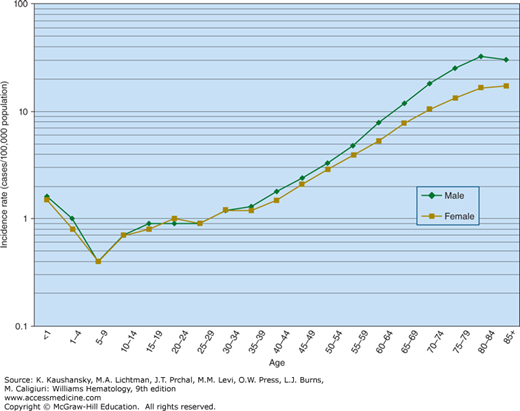
Very low copy number gene mutations characteristic of leukemia or lymphoma have been detected in the blood of healthy individuals. An analysis of blood cell DNA sequence data has identified 77 blood cell–specific mutations in cancer-associated genes, the majority being associated with advanced age. A large majority of these mutations were from 19 leukemia and/or lymphoma-associated genes, and nine were recurrently mutated (DNMT3A, TET2, JAK2, ASXL1, TP53, GNAS, PPM1D, BCORL1, and SF3B1). Additional mutations were found in a very small fraction of blood cells. Comparison of these findings to mutations in hematologic malignancies identified other recurrently mutated genes. The blood cells of more than 2 percent of individuals (5 to 6 percent of people older than 70 years) contain mutations that may represent premalignant events that can cause clonal hematopoietic expansion. These events may, in part, explain the age-dependent incidence of AML (Fig. 88–1).29a
Figure 88–1.
The annual incidence of acute myelogenous leukemia as a function of age. There is a relatively small increase to approximately 1.5 cases per 100,000 persons in the first year of life, representing congenital, neonatal, and infant AML. The incidence falls to a nadir of 0.4 new cases per 100,000 persons over the first 10 years of life and then rises again to 1 case per 100,000 in the second decade of life. From approximately 25 years of age, the incidence increases exponentially (log-linear) to approximately 25 cases per 100,000 population in octogenarians.
Patients who develop AML may have an antecedent predisposing nonmyeloid disease, such as aplastic anemia (poly- or oligoclonal T-cell disorder), myeloma (monoclonal B-cell disorder),30,31 or, rarely, AIDS (HIV-induced polyclonal T-cell disorder).32 An association between Langerhans cell histiocytosis, immune thyroid diseases, and familial polyendocrine disorder and AML has been reported.33,34,35,36 A number of inherited conditions carry an increased risk of AML (see Table 88–1).37–80 In the inherited syndromes, at least several pathogenetic types of gene alterations are represented: (1) DNA repair defects, for example, Fanconi anemia; (2) susceptibility genes favoring a second mutation, for example, familial platelet syndrome; (3) tumor-suppressor defects, for example, dyskeratosis congenita; and (4) unknown mechanisms, for example, ataxia-pancytopenia (See Tables 35-8 and 35-9 in Chap. 35 for further details of each pathogenetic process). There is evidence from central registry studies that any disorder that results in chronic immune stimulation, such as infection or autoimmune diseases may be associated with AML and MDS.81 The prevalence of essential monoclonal gammopathy is not increased in AML patients.82
AML results from a series of somatic mutations in a primitive hematopoietic multipotential progenitor cell or, very occasionally, a more differentiated, more lineage-restricted progenitor cell.83,84 Some cases of monocytic leukemia, promyelocytic leukemia, and AML in younger individuals may arise in a progenitor cell with lineage restrictions (progenitor cell leukemia).85,86,87 Other morphologic phenotypes and older patients likely have a disease that originates in a primitive multipotential cell. In the latter case, all myeloid blood cell lineages can be derived from the leukemic stem cell because it retains the ability for some degree of differentiation and maturation (Chap. 83). Because the T lymphocytes, B lymphocytes, and natural killer cells in cases of AML, often, have not carried a cytogenetic abnormality as did the myeloid cells, claims of origin in the pluripotential lymphohematopoietic cell have been ambiguous. The most compelling data indicate that the bulk of AML cases arise from one of two predominant CD34+ cell populations: CD34+CD45RA+CD38–CD90– (multipotential myeloid progenitor) or CD34+CD38+CD45RA+CD110+ (granulocyte-monocyte progenitor). Both of these cell populations correspond to normal hematopoietic progenitor cells and not the normal pluripotential lymphohematopoietic stem cell.86,88 This finding was confirmed by showing that the two leukemic cell populations were more similar to the corresponding normal progenitor populations than to pluripotential lymphohematopoietic stem cells by microarray gene expression analysis.88 The AML stem cell arises from somatic mutations in one of these populations in most, but not all, cases of AML. Because progenitor cells are not self-renewing, the somatic mutations transform the normal progenitor cell to an AML stem cell capable of sustaining the disease and transplanting it into immunosuppressed (NOD/SCID/IL2Rγ null) mice.
There is, also, experimental evidence that some cases of AML can arise from the accumulation of genetic and epigenetic changes in normal pluripotential HSCs.89 Through single-cell analysis, it has been shown that clonal progression of multiple mutations occurs in the HSC of some AML patients.90 These HSCs have been given the name “preleukemic HSCs” and it is proposed that AML progresses from such cells carrying founder mutations. These are thought to form a reservoir after therapy that can lead to relapse.89 An HSC with DNA methyltransferase 3A (DNMT3A) mutants was found to have multilineage repopulation advantage over nonmutated HSCs in xenografts, establishing their identity as preleukemic HSCs. These cells can be found in remission marrow samples of patients with AML.91 Genes that regulate DNA methylation such as DNMT3A, ten-eleven translocation (TET) 2, and isocitrate dehydrogenase (IDH) 1 and 2 promote self-renewal and block differentiation of stem and progenitor cells. Acquisition of these mutations in an HSC can lead to their clonal expansion resulting in a preleukemia stem cell population.92
Genome sequencing in AML cells shows that most mutations occur at random before acquisition of the initiating driver mutation, giving each clone a mutational history. The founding clone may acquire additional mutations, yielding subclones that contribute to disease progression or relapse.93 When copy number aberrations and copy-neutral loss-of-heterozygosity gene mutation profiles are analyzed in AML cases at diagnosis and at relapse, the relapsed leukemia always reflects reemergence of the founder clone. In persistent AML cases, sometimes two coexisting dominant clones can be seen, one chemotherapy-sensitive and one chemotherapy-resistant, suggesting that refractory or relapsed AML cases represent incomplete eradication of founder clones and not emergence of unrelated clones.94
AML with multiple chromosome aberrations is always characterized by critically short telomeres. Age-related critical telomere shortening may have a role in generating chromosome instability in AML pathogenesis.95 Leukemic cells show variable reduction in length of telomeric DNA, and telomere length in blood cells during remission is greater.96
Somatic mutation results from a chromosomal translocation in a large fraction of patients.97 The translocation results in rearrangement of a critical region of a protooncogene. Fusion of portions of two genes often does not prevent the processes of transcription and translation; thus, the fusion oncogene encodes a fusion protein that, because of its abnormal structure, disrupts a normal cell pathway and predisposes to a malignant transformation of the cell. The mutant protein product often is a transcription factor or an element in the transcription pathway that disrupts the regulatory sequences controlling growth rate or survival of blood cell progenitors and their differentiation and maturation.97,98,99 Examples of genes often mutated are core binding factor (CBF), retinoic acid receptor-α (RAR-α), HOX family, mixed-lineage leukemia (MLL), and others. CBF has two subunits: CBF–β and runt-related transcription factor 1(RUNX1, formerly AML1). Approximately 10 percent of AML cases have translocations involving one or the other of these latter two genes (CBF-β and RUNX1), although the percentage varies depending on the patient’s age at onset. In patients younger than age 50 years, the frequency is approximately 20 percent. In patients older than age 50 years, the frequency is approximately 6 percent. CBF activates genes involved in myeloid and lymphoid differentiation and maturation. These primary mutations are not sufficient to cause AML. Additional activating mutations, for example, in hematopoietic tyrosine kinases Fms-like tyrosine kinase (FLT)3 and KIT or in N-RAS and K-RAS, are required to induce a proliferative advantage in the affected primitive cell. Other protooncogene mutations that occur in leukemic cells involve FES, FOS, GATA-1, JUN B, MPL, MYC, p53, PU.1, RB, WT1 (Wilms tumor 1), WNT, NPM1, CEPBA (CCAAT-enhancer binding protein A), and other genes. Their interaction with loss-of-function mutations in hematopoietic transcription factors probably causes the acute leukemia phenotype characterized by a disorder of proliferation, programmed cell death, differentiation, and maturation. Because the mutant stem or early progenitor cell can proliferate and retains the capability to differentiate, a wide variety of phenotypes can emerge from a leukemic transformation.
AML is a heterogeneous disease, and the extent to which cytogenetic and molecular markers define severity and influence treatment decisions is a rapidly changing arena of investigation as a result of continued refinements in correlating individual or a combination of mutations on disease progression. Using molecular markers to predict disease course in AML is complicated because these are incompletely determined, and they often interact. Several risk scores based on chromosome and molecular markers have integrated factors such as age and white blood cell (WBC) count into the scoring systems.100,101 Others have identified common gene signatures that can be independent predictors of disease progression or therapeutic response and provide a structure for risk stratification. Some of these signatures have 24 genes,102 and some have a seven gene-epigene score.103 Some have relied on genetic proflilng,104 some on expression of a subset of molecular mutations,105 and some have combined epigenetic and genetic markers.106 Prognostic models of AML based solely on molecular markers have been proposed. In one, PML-RARa or CEPBA double mutations were very favorable (overall survival [OS] at 3 years of 83 percent), RUNX1-RUNX1T1, CBFB–MYH11, or NPM1 (nucleophosmin-1 mutation) without FLT3-ITD (OS of 62.6 percent), intermediate with no mutation allowing assignment to other groups (OS of 44 percent), MLL-PTD or RUNX1, or ASXL1 mutation (OS of 22 percent), and very unfavorable, TP53 mutation (OS at 3 years, 0 percent).
In general, those patients with changes involving CBF, that is, t(8/21), inv(16), t(16;16), or t(15;17), a feature of acute promyelocytic leukemia (APL), are considered predictors of a more favorable outcome. Those with complex karyotype, 11q23, t(6;9), abnormalities of chromosome 5 or 7 or inv3 (t3;3) are associated with a poor outcome. The remainder of cytogenetic abnormalities and those patients with a normal karyotype are considered of intermediate risk.107 These are determined by the behavior of the average of very large groups of patients and confidence intervals are not calculated. Patients with favorable cytogenetic patterns may have poor outcomes and those with less favorable patterns may do better than anticipated.
Deletions of all or part of a chromosome (e.g., chromosome 5, 7, or 9) or additional chromosomes (such as trisomy 4, 8, or 13) are common cytogenetic abnormalities (Chap. 11), although the specific causative oncogenes or tumor-suppressor genes in these latter circumstances have not been defined. Deletions in chromosomes 5 and 7 and complex cytogenetic abnormalities are associated with a worse prognosis and are increased in frequency in older patients and cases of AML following cytotoxic therapy compared to de novo cases.108 Because the genes residing on the undeleted homologous segment of chromosome 5 are not mutated, an epigenetic lesion, such as hypermethylation of a gene allelic to one on the deleted segment on chromosome 5, may contribute to the leukemogenic event.
In APL, PML-RAR-α fusion protein represses retinoic acid-inducible genes, which prevent appropriate maturation of promyelocytes. The induced disruption, which involves corepressor–histone deacetylase complexes, results in the leukemic phenotype (see “Acute Promyelocytic Leukemia” below).109,110
Patients with CBF leukemias are younger on average and in addition to t(8;21) or inv(16)/t(16;16) may have RUNX1/RUNX1T1 and CBFB/MYH11 oncogenes.111 The cure rate in these so-called good-risk patients is only approximately 55 percent, however. Patients with CBF leukemias expressing KIT have a worse prognosis.112 In the case of inv(16)/t(16;16), different fusion transcripts can be formed, and these may have associated with KIT mutations and other abnormal chromosomal associations with differing prognosis, possibly from activation of caspase activity.113 Secondary genetic changes in inv(16) or t(16;16) cases may have an impact on prognosis. RAS, KIT, FLT3-internal tandem duplication (ITD), and FLT3-TKD each affect prognosis. FLT3-TKD, trisomy 8, age, and therapy-related AML were associated with worse prognosis.114 In t(8;21) leukemias, epigenetic silencing of microRNA-193a activates the PTEN/PI3K signaling pathway,115 and wild-type RUNX1 can attenuate nuclear factor-kappaB (NF-κB) signaling, events not present in the t(8;21) translocation leukemias.116
3q Abnormalities EVI1 and MDS1/EVI1 expression in AML is associated with poor prognosis and is a distinct entity. These chromosome 3 abnormalities are found in only approximately 4 percent of AML cases. These include inv(3) or t(3;3), t(3q26), t(3q21), and other miscellaneous 3q abnormalities.117 These generally have an unfavorable prognosis.118
Monosomal Karyotype A monosomy has been associated with decreased chance of achieving remission or of survival, especially when combined with TP53 mutations.119,120
Approximately 45 percent of AML cases have a normal karyotype. Sequencing has shown that mutations in NPM1, DNMT1, FLT3, KIT, CEBPA, TET2, and others may have diagnostic and prognostic implications. When genomes of APL with a known founder event (PML-RARa) are sequenced and compared with normal karyotype AML and exomes of HSCs from normal donors, most mutations in AML genomes are random events that occurred in HSC before the initiating mutation occurred. As the clone expands, one or two additional, cooperating mutations may result in development of a leukemia, and these clones may acquire additional mutations, leading to subclones.121 DNA sequences of leukemia cell and normal skin cell genomes of a patient with AML showed 12 acquired mutations within coding sequences of genes and 532 somatic point mutations in conserved or regulatory portions of the genome.122 When whole-genome or whole-exome sequencing was performed in 200 AML cases, it was found that an average of only 13 mutations occurred in the genes. Only a total of 23 genes were mutated. There were nine categories of genes thought relevant for pathogenesis: (1) transcription-factor fusions, (2) nucleophosmin, tumor-suppressor, (3) DNA methylation-related, (4) signaling, chromatin-modifying, (5) transcription-factor, (6) cohesion-complex, and (7) spliceosome-complex genes. Many of these genes had patterns of cooperation and mutual exclusivity.123 Table 88–2 lists commonly mutated genes in cytogenetically normal AML in order of decreasing frequency.
| Mutated Gene | Approximate Frequency in AML with Normal Karyotype (%) | Implication | Comments | References |
|---|---|---|---|---|
| NPM1 | 50 | More-favorable outcomes | Most frequently mutated gene in AML. Allogenic transplantation not needed in first remission if this mutation occurs in absence of mutated FLT3-ITD | 124,125,126,127,128,129 |
| FLT3 ITD | 40 | Less-favorable outcomes | 124,125,130,131,132,133,134,135,136,137 | |
| DNMT3A | 20 | Less-favorable outcomes | Seen more often in AML patients with normal cytogenetics. Mutant NPM1, FLT3-ITD, and IDH1 have been found more frequently in AML patients with DNMT3A mutations compared to those with wild-type DNMT3A | 137,138,139,140,141,142,143 |
| RUNX1 | 15 | Less-favorable outcomes | 144,145,146,147,148,149 | |
| TET2 | 15 | Less-favorable outcomes | Coincidence of mutated TET2 with NPM1 mutation in the absence of FLT3-ITD mutation predicts a less-favorable outcome | 150,151,152,153 |
| CEBPA | 15 | More-favorable outcomes | Only cases with double mutations associated with favorable outcomes | 124,154,155,156,157 |
| NRAS | 10 | Little effect on prognosis | 144 | |
| IDH1 or IDH2 | 10 | Little effect on outcomes | More frequent in AML patients with normal cytogenetics. Frequently associated with NPM1. Adverse prognostic factor if present with mutated NPM1 without FLT3-ITD. Serum 2-hydroxyglutarate levels indicate high probability of IDH mutation | 138,158,159,160,161,162,163,164 |
| MLL-PTD | 8 | Less-favorable outcomes | 144 | |
| WT1 | 6 | Less-favorable outcomes | More frequent in females than in males (6.6 vs. 4.7%; P = 0.014) and in patients <60 than in patients >60 years (P <0.001) | 166,167 |
| FLT3-TKD | 6 | Little effect on outcomes | May appear after use of FLT3-ITD inhibitor | 132,136 |
Nucleophosmin-1 Mutations NPM1 mutations are the most frequent genetic alterations in AML, found in approximately half of patients with a normal karyotype.124,125 The mutation in exon 12 results in loss of the residue that requires its binding to nucleoli such that the NPM1 protein is abnormally localized to the cytoplasm.126 Studies show that mutant NPM1 without FLT3-ITD represents a favorable prognostic marker.127 NPM1 mutations also have a favorable prognostic impact in older patients.128 Mutated regions of NPM1 elicit T-cell responses which might indicate that immunotherapy could have a role in these mutated cases.129
FLT3 Mutations FLT3 encodes a tyrosine kinase receptor in normal myeloid and lymphoid progenitors. ITD of FLT3 on chromosome 13 occurs in approximately 25 percent of adult AML cases, but occurs more frequently in cases of AML with normal cytogenetic patterns, monocytic phenotype, and PML-RAR-α or DEK-CAN translocations.124,125,130 The FLT3-ITD mutation confers a poor prognosis if the ratio of mutant to wild-type expression is high.130,131,132 FLT3-ITD expression is often higher at relapse.133 FLT3-ITD upregulates MCL-1 to promote survival of AML stem cells through signal transducer and activator of transcription (STAT) 5 activation.134 FLT3-ITD adversely affects the outcome of an allogeneic stem cell transplant, but more than half of patients harboring this mutation who receive transplants can survive leukemia free for 2 or more years.135 Point mutations in the tyrosine kinase domain (TKD) of FLT3 (FLT3-TKD) mutations occur in approximately 6 percent of AML cases and have little impact on outcomes.136
DNMT3A Mutations The DNMT3A gene encodes a DNA methyltransferase isoform. The process of DNA methylation involves the addition of a methyl group on a cytosine residue at a C-G site. If this methylation happens in the promoter region of a coding gene, the gene will be silenced. The DNMT enzymes contribute to leukemogenesis by mediating tumor suppressor gene silencing.137 DNMT3A mutations have been found in approximately 20 percent of AML patients with normal cytogenetic patterns.138 These cases more frequently had mutations in NPM1, FLT3, and IDH1 genes as well.139 DNMT3A mutations are associated with a poorer prognosis139,140,141,142 and their significance appears to be age-dependent.143 The R882 mutation was associated with adverse prognosis in older patients, and non-R882 mutations with adverse prognosis in younger patients.
RUNX1 Mutations The RUNX1 gene is located on chromosome 21q22 and is involved in hematopoiesis at all stages through its interaction with CBFβ. It acts as an activator or repressor of numerous genes, including transcription factors.144,145 In de novo AML, RUNX1 mutations were found with normal and noncomplex karyotypes. They were sometimes associated with MLL-PTD (partial tandem duplication [PTD]) and FLT3-ITD, and they were associated with a poor prognosis independent of other molecular mutations.146 Another group found these mutations in 5.6 percent of cases, associated with cytogenetically normal AML and an association with MLL-PTD mutations, refractory disease, and, as an independent risk factor, an inferior relapse-free survival and overall survival. The use of allogeneic HSC transplant did have a favorable impact in such cases.147 Another series found RUNX1 mutations to be twice as common in older than younger patients with normal cytogenetics, and to have an adverse outcome effect in both age groups. Mutated blasts had molecular signatures suggesting origin in a primitive hematopoietic cell.148 RUNX1 mutations have been found to cooperate with granulocyte colony-stimulating factor receptor (CSFR) mutations in congenital neutropenia to lead to acute leukemia or MDS.149
TET2 Mutations The TET2 protein inactivation may occur through a loss of function mutation, deletion, or through IDH1/2 mutations. It is a member of a family of dioxygenases that catalyze conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine and promote DNA demethylation. TET2 has many roles in normal hematopoiesis, and knockout mice show that it is a tumor suppressor, which haploinsufficiency initiates myeloid transformations.150 TET2 mutations are found in approximately 25 percent of patients and in those who have mutated CEBPα and/or mutated NPM1 without a FLT3/ITD mutation.151,152 Patients with AML and a TET2 mutation had a shorter event-free and overall survival compared with patients who were TET2 wild-type. They did not predict for outcomes in those with cytogenetically normal AML and with wild-type CEBPα, NPM1, and/or FLT3/ITD.153 Whether these patients would benefit from alternate therapies, such as hypomethylating agents or HSC transplantation, has not been determined.
CEBPα Mutation CEBPα is a leucine zipper transcription factor involved in myeloid differentiation. Mutations have been described in approximately 10 percent of AML patients.124 Single or double mutations can occur, and these rarely are associated with FLT3/ITD or with NPM1 mutations. CEBPα-double, but not CEBPα-single, mutation patients had a significantly better overall survival at 8 years than wild-type, CEBPα-single, or CEBPα-double and FLT3/ITD-positive patients.154 A multivariate analysis found that only double-mutant CEBPα was associated with a favorable event-free, relapse-free, and overall survival. Double-mutant cases were also associated with a unique gene signature as compared with single-mutant cases.155,156 Some AML patients with CEBPα-double mutations harbor TET2 and GATA2 mutations, which can affect prognostic outlook unfavorably with TET2 or favorably with GATA2 mutations.157
IDH1 and IDH2 Mutations The IDHs catalyze oxidative decarboxylation of isocitrate into α-hemoglutarate. The nicotinamide adenine dinucleotide phosphate–dependent IDH1 enzyme is encoded by the IDH1 gene on chromosome 2q33.3, and the nicotinamide adenine dinucleotide phosphate–dependent-dependent IDH2 enzyme is encoded by the IDH2 gene on chromosome 15q26.1.158 Mutations in IDH1 (R132) or IDH2 (R172) occur in 10 percent of AML patients.158,159 Both were found to adversely impact relapse-free survival and overall survival. Multivariate analysis showed that IDH mutation conferred an adverse impact in those patients with an NPM1 mutation without FLT3-ITD. Favorable genotype cytogenetically normal AML is therefore defined as NPM1 or CEBPα mutation with neither a FLT3-ITD nor an IDH1 mutation. An IDH1 mutation was also associated with a higher relapse rate and shorter overall survival.160 Another group found a higher frequency of IDH1 and IDH2 mutations in cytogenetically normal AML. Both were found to have an unfavorable impact on outcome.161 IDH1 was exclusive of other mutations. Serum 2-hydroxyglutarate production has been found to predict for the presence of IDH1/2 mutations.162,163 A level of 700 mg/mL was found to discriminate mutated from nonmutated cases, and those with levels greater than 20 ng/mL at the time of remission had shorter overall survival.163 Mutant IDH1 has been found to accelerate cell-cycle transition and to activate mitogen-activated protein kinase signaling. Mutant IDH1 can be inhibited, suggesting this may be a therapeutic target.164
WT1 Mutations Mutations of the WT1 gene have been reported in approximately 5 to 10 percent of cytogenetically normal patients with AML.165 Some studies suggest association with a poor prognosis, but others have not. WT1 SNP rs 16754 was associated with a favorable risk, but acquired mutations did not affect the development of complete remission, relapse-free survival, or overall survival.166 A study of WT1 mutations in older patients with cytogenetically normal AML also showed poor treatment response across all age-groups and association with a distinct gene expression signature.167
Other methodologies to evaluate genomic aberrations have been reported to have prognostic importance beyond the impact of the individual mutations described above and in Table 88–2. Abnormal genome-wide single nucleotide polymorphisms have adverse prognosis in patients with AML and a normal karyotype.168 Expression signatures of cytokines and chemokines have an independent prognostic impact in AML.169 Profiling transcriptional pathways may have prognostic importance in AML as well.170
There is also interplay among molecular aberrancies in AML. These include: (1) gene interaction with a microRNA; for example, BAALC and miR-3151 in cytogenetically normal AML,171 (2) distinct patterns of dual or multiple gene mutation patterns that have prognostic impact,172 and (3) concurrence of somatic mutations and transcriptional regulators such as interaction between ERG expression and a heptad of transcriptional factors173 that maintains a stem cell-like signature. Furthermore, interactions between genetic and epigenetic changes (DNA methylation, histone acetylation, histone methylation, and others) are anticipated to have prognostic impact.174,175
The mutations in AML result in deregulation of any of several signal transduction pathways, which disrupt pathways that ensure the normal behavior of (1) differentiation and maturation, (2) proliferation, and (3) survival signals in hematopoietic cells. The pathways involved are myriad, but several represent the majority of cases such as the (1) PI3K-AKT, (2) RAS-RAF-MEK-ERK, and (3) STAT3 signaling sequences.176 The expectation is that a relative small number of downstream signaling pathways mediate the leukemogenic effect of gene mutations, making the potential targets for therapy less diffuse than suggested by the number of gene mutations involved in AML.
In most cases, little evidence is seen for a strong influence of inherited factors. The identical twin of a child with acute leukemia has a heightened risk of developing the disease. However, the risk appears to be related to intraplacental metastasis and thus falls to the risk of a nonidentical sibling after the first few years of life.177,178 The risk of AML in a nonidentical sibling in the United States is elevated, perhaps twofold to threefold, compared to the risk of AML in unrelated American children of European descent younger than age 15 years.177,179 A registry study in Sweden showed no significant aggregation in relatives of patients with AML. An increased risk of AML/MDS was found among relatives of patients diagnosed at younger than age 21 years (relative risk 6.5).180 Clusters of AML cases in families have been documented, but their frequency is low.58 Clusters of AML in unrelated persons in a community are uncommon and, when investigated, usually prove to be a chance occurrence. Heritable GATA2 mutations may be associated with familial MDS and AML,181 and loss-of-function germline GATA2 mutations (the MonoMAC [monocytopenia and mycobacterial infections] syndrome) may be associated with primary lymphedema and a predisposition to AML (Emberger syndrome).182,183,184 Mutations of CEBPα have been found in familial AML.185 In one study of 27 families with familial MDS/AML, genetic characterization could be shown in 10 (four with GATA2 mutations, five with telomerase mutations, and one with mutated RUNX1).186 Mutations in telomerase RNA (TERC) or telomerase reverse transcriptase component (TERT) are also associated with familial AML.185,187
AML is the predominant form of leukemia during the neonatal period but represents a small proportion of cases during childhood and adolescence. Approximately 20,000 new cases of AML occur annually, representing approximately 35 percent of the new cases of leukemia in the United States each year. Approximately 12,000 patients with AML in the United States die each year as a result of the disease. The incidence rate of AML is approximately 1.5 per 100,000 in infants younger than 1 year of age, decreases to approximately 0.4 per 100,000 children ages 5 to 9 years, increases gradually to approximately 1.0 persons per 100,000 population until age 25 years, and thereafter increases exponentially until the rate reaches approximately 25 per 100,000 persons in octogenarians (see Fig. 88–1). The exception to this exponential age-related increase in incidence is APL, which does not change greatly in incidence with age.188
AML accounts for 15 to 20 percent of the acute leukemias in children and 80 percent of the acute leukemias in adults. It is slightly more common in males. Little difference in incidence is seen between individuals of African or European descent at any age. A somewhat lower incidence is seen in persons of Asian descent.189 An increase in the frequency of AML is seen in Jews, especially those of Eastern European descent. The acute promyelocytic variant of AML is somewhat more common in Latinos.190,191 In a large population study of 426,068 patients treated with chemotherapy for malignancy, 301 AML cases occurred, 4.7 times the number expected. Over time (1975 to 2008), the risks increased for non-Hodgkin lymphoma, declined for ovarian cancer and myeloma, and were heterogeneous for breast and Hodgkin lymphoma, reflecting changing treatment patterns.192
CLASSIFICATION
Variants of AML can be identified by morphologic features of blood films using polychromatic stains and histochemical reactions,193 monoclonal antibodies against surface markers,194 or by the presence of specific chromosome translocations or other molecular changes as discussed above.104,105 The epitopes on the progenitor cells of several phenotypic variants overlap, and several monoclonal antibodies are required to make specific distinctions among cell types (Table 88–3; see also “Morphologic Variants of Acute Myelogenous Leukemia” below). Correlation between morphologic and immunologic phenotyping of AML is poor. However, poor correlation is expected because morphologic phenotyping is more subjective, given to observer variation, and is based on qualitative factors, whereas the immunologic phenotyping, which characterizes surface molecular features, is more accurate and reproducible. The correlation is improved only somewhat if morphology and histochemistry are coupled.195 Gene-expression profiling is early in its use as a classification technique for AML but will be more specific and informative than current methods.104,105 The outcome will depend on the simplification and automation of such techniques, and the availability of drugs that make such distinctions in the prognostic category of practical utility. Chapter 83 contains the classification of morphologic variants of AML (see Chap. 83, Table 83–1 and Fig. 83–3). A cogent argument has been made that, for practical purposes, a classification that initially considers morphologic phenotype and immunophenotype is advisable. Cytogenetics, molecular genetics, gene-expression profiling, and other considerations can, and should, be layered on as available and useful in influencing therapy, and these features are starting to be incorporated into the World Health Organization (WHO) Classification of AML.196 It is anticipated that molecular classifications will continue to evolve and dominate clinical decision making in the future.105
| Phenotype | Usually Positive |
|---|---|
| Myeloblastic | CD11b, CD13, CD15, CD33, CD117, HLA-DR |
| Myelomonocytic | CD11b, CD13, CD14, CD15, CD32, CD33, HLA-DR |
| Erythroid | Glycophorin, spectrin, ABH antigens, carbonic anhydrase I, HLA-DR, CD71 (transferrin receptor) |
| Promyelocytic | CD13, CD33 |
| Monocytic | CD11b, 11c, CD13, CD14, CD33, CD65, HLA-DR |
| Megakaryoblastic | CD34, CD41, CD42, CD61, anti–von Willebrand factor |
| Basophilic | CD11b, CD13, CD33, CD123, CD203c |
| Mast cell | CD13, CD33, CD117 |
CLINICAL FEATURES
Signs and symptoms that signal the onset of AML include pallor, fatigue, weakness, palpitations, and dyspnea on exertion. The signs and symptoms reflect the development of anemia; however, weakness, loss of sense of well-being, and fatigue on exertion can be disproportionate to the severity of anemia.197,198,199,200,201
Easy bruising, petechiae, epistaxis, gingival bleeding, conjunctival hemorrhages, and prolonged bleeding from skin injuries reflect thrombocytopenia and are frequent early manifestations of the disease. Very infrequently, gastrointestinal, genitourinary, bronchopulmonary, or CNS bleeding occurs at the onset of disease.
Pustules or other minor pyogenic infections of the skin and of minor cuts or wounds are most common. Major infections, such as sinusitis, pneumonia, pyelonephritis, and meningitis, are uncommon presenting features of the disease, partly because absolute neutrophil counts less than 0.5 × 109/L are uncommon until chemotherapy starts. With intensification of neutropenia and monocytopenia after chemotherapy, major bacterial, fungal, or viral infections become more frequent. Anorexia and weight loss are frequent findings. Fever is present in many patients at the time of diagnosis.200,202,203,204 Palpable splenomegaly or hepatomegaly occurs in approximately one-quarter of patients.197,198,201 Lymphadenopathy is extremely uncommon,201,205,206 except in the monocytic variant of AML.207
Leukemic blast cells circulate and enter most tissues in small numbers. Occasionally, biopsy (or autopsy) uncovers marked aggregates or infiltrates of leukemic cells. Collections of such cells may cause functional disturbances. Extramedullary involvement is most common in monocytic or myelomonocytic leukemia.208,209
Skin involvement may be of three types: nonspecific lesions, leukemia cutis, or granulocytic (myeloid) sarcoma of skin and subcutis.210,211,212,213 Nonspecific lesions include macules, papules, vesicles, pyoderma gangrenosum, vasculitis,214,215,216 neutrophilic dermatitis (Sweet syndrome),217 cutis vertices gyrata,218 and erythema multiforme or nodosum.211,212 Skin involvement preceding marrow and blood involvement or relapse occurs, but is rare.219,220,221,222
Sensory organ involvement is very unusual, but retinal, choroidal, iridial, and optic nerve infiltration can occur.223 Otitis externa and interna, inner ear hemorrhage, and mastoid tumors with seventh nerve involvement may be presenting signs.224,225,226
The gastrointestinal tract may be involved at any point, but functional disturbances are unusual.227,228 The mouth, colon, and anal canal are sites of involvement that most commonly lead to symptoms. Oral manifestations may prompt the patient to visit the dentist. Gingival or periodontal infiltration and dental abscesses may lead to an extraction, followed by prolonged bleeding of an infected tooth socket.229 Ileotyphlitis (enterocolitis), a necrotizing inflammatory lesion involving the terminal ileum, cecum, and ascending colon, can be a presenting syndrome or occur during treatment.230,231,232,233 Fever, abdominal pain, bloody diarrhea, or ileus may be present and occasionally mimic appendicitis. Intestinal perforation, an inflammatory mass, and associated infection with enteric gram-negative bacilli or clostridial species often are associated with a fatal outcome. Isolated involvement of the gastrointestinal tract is rare.234,235 Proctitis, especially common in the monocytic variant of AML, can be a presenting sign or a vexing problem during periods of severe granulocytopenia and diarrhea.227
The respiratory tract can be involved by infiltrates or tumors, leading to laryngeal obstruction, parenchymal infiltrates, alveolar septal infiltration, or pleural seeding. Each of these events can result in severe symptoms and radiologic findings.236,237,238,239,240
Cardiac involvement is frequent but rarely causes symptoms. Symptomatic pericardial infiltrates, transmural ventricular infiltrates with hemorrhage, and endocardial foci with associated intracavitary thrombi can occasionally cause heart failure, arrhythmia, and death.241 Infiltration of the conducting system or valve leaflets or myocardial infarction has occurred.242
The urogenital system can be affected. The kidneys are infiltrated with leukemic cells in a high proportion of cases, but functional abnormalities are rare. Hemorrhage in the pelvis or collecting system is frequent.243,244 Cases of vulvar, bladder neck, prostatic, and testicular involvement have been described.245,246,247
Osteoarticular symptoms may occur. Bone pain, joint pain, and bone necrosis can occur, and, rarely, arthritis with effusion is present.248 Crystal-induced arthritis of either calcium pyrophosphate dihydrate (pseudogout) or monosodium urate (gout) may be responsible for the synovitis in some cases.249
Central or peripheral nervous system involvement by infiltration of leukemic cells is very uncommon, although meningeal involvement is an important consideration in the treatment of the monocytic type of AML.250,251 An association of CNS involvement and diabetes insipidus in AML with monosomy 7252 and inversion of chromosome 16253,254 has been reported.
Myeloid sarcoma (synonyms: granulocytic sarcoma, chloroma, myeloblastoma, monocytoma) is a tumor composed of myeloblasts, monoblasts, or megakaryocyes.255,256,257,258,259,260 The tumor may occur as an extramedullary mass without evidence of leukemia in blood or marrow, so-called nonleukemic myeloid sarcomas, or in association with AML. When the tumor appears as an isolated lesion, it initially may be misdiagnosed as extranodal lymphoma because they look like lymphoid cells on biopsy.257 They may be found in virtually any location, including the skin; orbit; paranasal sinuses; bone; chest wall; breast; heart; gastrointestinal, respiratory, or genitourinary tract; central or peripheral nervous system; or lymph nodes and spleen. The tumors originally were called chloromas because of the green color imparted by the high concentration of the enzyme myeloperoxidase present in myelogenous leukemic cells. Biopsy specimens are positive for chloracetate esterase, lysozyme, myeloperoxidase, and cluster of differentiation (CD) markers of myeloid cells. When myeloid sarcomas are the initial manifestation of AML, the appearance of the disease in the blood and marrow may follow weeks or months later. Abnormalities in chromosome 8 are the most frequent cytogenetic disturbance in myeloid sarcomas.258 Systemic chemotherapy, rather than local therapy, should be used for treatment, although the long-term outcome in such cases usually is poor.260,261,262 Patients having AML with t(8;21) or inv16 have a propensity to develop extramedullary leukemia,263,264,265,266 and such patients with myeloid sarcomas have a poorer outcome after treatment than those who do not have extramedullary lesions.263,265
LABORATORY FEATURES
Anemia is an almost constant feature.197,198,199,200,201 Red cell life span may be mildly shortened, but the principal cause of anemia is inadequate production of red cells. The reticulocyte count usually is between 0.5 and 2.0 percent. Occasionally patients have rapid destruction of autologous and transfused red cells as a result of an unknown mechanism, referred to as milieu hemolysis. The presence of red cell autoantibodies (positive direct antiglobulin test) is very uncommon and may be nonspecific (anti-C3), perhaps related to circulating immune complexes. Red cell morphology is mildly abnormal, with exaggerated variation in cell size and occasional poikilocytes. Nucleated red cells or stippled erythrocytes may be present. Less often, extreme abnormalities of red cell size, shape, and hemoglobin content occur (AML with trilineage dysmorphia), but these changes are seen more often in oligoblastic myelogenous leukemia (Chap. 87).
Thrombocytopenia is nearly always present at the time of diagnosis. The mechanism of thrombocytopenia is a combination of inadequate production and decreased survival of platelets. More than half of patients have a platelet count less than 50 × 109/L at the time of diagnosis.267 Giant platelets and poorly granulated platelets with functional abnormalities can occur.268 Defects in platelet aggregation and 5-hydroxytryptamine release are frequent.268
The total leukocyte count is less than 5 × 109/L in approximately half of patients at the time of diagnosis.197,198,199,200,201 The absolute neutrophil count is less than 1 × 109/L in more than half of cases at diagnosis.97–201 Patients with very elevated total leukocyte counts have a low proportion of mature neutrophils but may have a normal absolute neutrophil count. Hypersegmented, hyposegmented, and hypogranular mature neutrophils may be present. Cytochemical abnormalities of blood neutrophils include low or absent myeloperoxidase or low alkaline phosphatase activity.269 Defects in phagocytosis or microbial killing are common.270,270A
Myeloblasts almost always are present in the blood but may be infrequent in severely leukopenic patients. Diligent search may uncover the myeloblasts, or examination of a white cell concentrate (buffy coat) may permit their identification. Classic leukemic blast cells are agranular, but mixtures of immature cells, including agranular and slightly granular cells ranging up to overt progranulocytes, can occur. Auer rods are elliptical cytoplasmic inclusions approximately 1.0 to 1.5 μm long and 0.5 μm wide that derive from azurophilic granules (Fig. 88–2B). The inclusions are present in the blast cells of approximately 15 percent of cases. When present, the inclusions are found in only a small percentage of blast cells when examined with polychrome stains.193 An exception is APL, in which a higher proportion of cells have Auer rods and some have multiple (bundles) of rods (faggot cells). This finding can be dramatic if peroxidase stain is used to highlight the Auer rods.
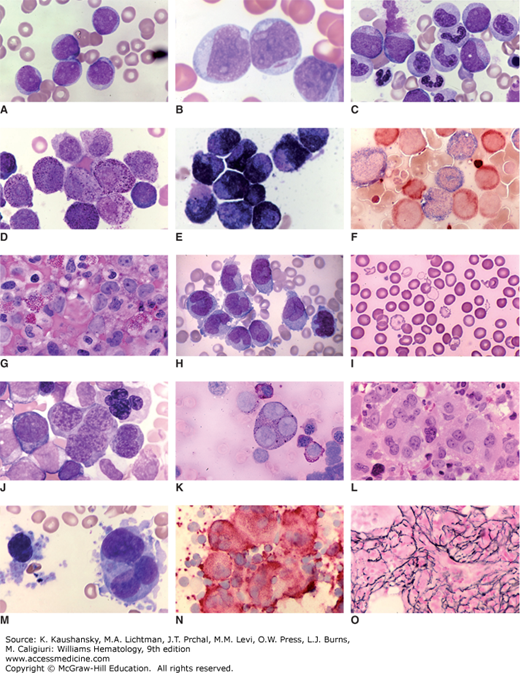
Figure 88–2.
Blood and marrow images of major subtypes of acute myelogenous leukemia. A. Blood film of acute myelogenous leukemia (AML) without maturation (acute myeloblastic leukemia). Five myeloblasts are evident. High nuclear-to-cytoplasmic ratio. Agranular cells. Nucleoli in each cell. B. Blood film. AML without maturation (acute myeloblastic leukemia). Three myeloblasts, one containing an Auer rod. C. Marrow film. AML with maturation. Three leukemic myeloblasts admixed with myelocytes, bands, and segmented neutrophils. D. Blood film. Acute promyelocytic leukemia. Majority of cells are heavily granulated leukemic promyelocytes. E. Blood film. Acute promyelocytic leukemia. Myeloperoxidase stain. Intensely positive. Numerous stained (black) granules in cytoplasm of leukemic progranulocytes. F. Blood film. Acute myelomonocytic leukemia. Double esterase stain. Leukemic monocytic cells stained dark blue and leukemic neutrophil precursors stained reddish-brown. G. Marrow film. AML with inv16. Note high proportion of eosinophils in field. Note myeloblasts with very large nucleoli at upper right. Also, intermediate leukemic granulocytic forms. H. Blood film. Acute monocytic leukemia. Leukemic cells have characteristics of monocytes with agranular gray cytoplasm and reniform or folded nuclei with characteristic chromatin staining. This case had hyperleukocytosis as evident by leukemic monocyte frequency in the blood film. I. Blood film. Acute erythroid leukemia. Note population of extremely hypochromic cells with scattered bizarre-shaped poikilocytes admixed with normal-appearing red cells. J. Marrow film. Acute erythroid leukemia. Giant erythroblasts with multilobulated nuclei. K. Marrow film. Acute erythroid leukemia. Note giant trinucleate erythroblast and other leukemic erythroblasts with periodic acid–Schiff–positive cytoplasmic staining (reddish granules). L. Marrow section. Acute megakaryoblastic leukemia. Marrow replaced with atypical two- and three-lobed leukemic megakaryocytes with bold nucleoli. M. Marrow film. Acute megakaryoblastic leukemia. Marrow replaced with atypical megakaryocytes and megakaryoblasts with cytoplasmic disorganization, fragmentation, and budding. N. Marrow film. Acute megakaryoblastic leukemia. Marrow replaced with atypical megakaryocytes and megakaryoblasts staining for platelet glycoprotein IIIA (reddish-brown). Platelets in background also stained. O. Marrow section. Acute megakaryoblastic leukemia. Argentophilic (silver) stain shows marked increase in collagen, type III fibrils (marrow reticulin fibrosis), characteristic of this AML subtype. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
The marrow always contains leukemic blast cells. From 3 to 95 percent of marrow cells are blasts at the time of diagnosis or relapse (see Fig. 88–2A). The WHO has invoked an arbitrary threshold of 20 percent of marrow nucleated cells being blast cells to distinguish polyblastic AML (≥20 percent blasts) from oligoblastic myelogenous leukemia (<20 percent blasts).197,198,199,201 The latter situation is referred to as refractory anemia with excess blasts, a MDS (Chap. 87). The WHO choice of ≥20 percent blasts is an arbitrary standard as acute monocytic leukemia, APL, acute erythroid leukemia, and other variants often have less than 20 percent blast cells at the time of diagnosis,271 and if any blasts are found with a case of AML in which the cells have a t(8;21) or other CBF inversions or translocations, AML is the diagnosis. Moreover, relapse of AML can be identified at any increase in blast count >2 percent. In addition, patients with oligoblastic leukemia with 10 to 19 percent marrow leukemic blast cells are identical in all other phenotypic findings and survival to those with 20 to 29 percent marrow blast cells. Any distinctions between the two groups in survival are a function of age, cytogenetic risk category, and molecular features, not the blast count.272 This arbitrary boundary prevents patients, otherwise suitable, to enter clinical trials.
Myeloblasts are distinguished from lymphoblasts by any of three pathognomonic features: reactivity with specific histochemical stains; Auer rods in the cells (see Fig. 88–2B); or reactivity with a panel of monoclonal antibodies against epitopes present on myeloblasts (e.g., CD13, CD33, CD117) (see Table 88–3). Leukemic myeloblasts give positive histochemical reactions for myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate esterase stains. Auer rods can be found in the marrow blast cells in approximately one-sixth of cases. Blast cells may express granulocytic (CD15, CD65) or monocytic (CD11b, CD11c, CD14, CD64) surface antigens. They typically do not express either lymphoid surface markers or membrane or cytoplasmic immunoglobulin. No immunoglobulin gene rearrangement or T-lymphocyte receptor gene rearrangement is evident with molecular probes (see “Hybrid and Mixed Leukemias” below). In a proportion of otherwise typical cases of AML, the cells may contain terminal deoxynucleotidyl transferase (TdT).273,274 Variations in marrow findings are discussed below in “Morphologic Variants of Acute Myelogenous Leukemia.” Normal erythropoiesis, megakaryocytopoiesis, and granulopoiesis are decreased or absent in the marrow aspirate. The biopsy may contain residual islands of erythroblasts or megakaryocytes. Dysmorphic changes in hematopoietic cells, including very small or large erythroblasts with nuclear fragmentation or binucleation or delayed nuclear condensation; small or monolobed megakaryocytes; or hypogranulated, bilobed, or monolobed neutrophils, may occur in 30 to 50 percent of patients with de novo AML.275 Marrow reticulin fibrosis is common but usually is slight to moderate except in cases of megakaryoblastic leukemia, in which intense fibrosis is the rule.276 Increased blood vessel density (angiogenesis) is present in the marrow of patients with AML compared to normal subjects.277,278 Various angiogenic factors, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor, angiogenin, and angiopoietin-1, are increased. VEGF detected histochemically in human marrow is closely correlated with the prevalence of leukemic myeloblasts in the various AML subtypes.279 AML cytogenetic variants may result in marrow basophilia (usually t(6;9))280 or marrow eosinophilia (usually inv16 or t(16;16)).281
An abnormal number (aneuploidy) or structure (pseudodiploidy) of chromosomes or both are evident in approximately 55 percent of cases.282,283,284,285 The most prevalent abnormalities are trisomy 8, monosomy 7, monosomy 21, trisomy 21, and loss of an X or Y chromosome. However, any chromosome can be rearranged, added, or lost (Chap. 13). In cases of AML following chemotherapy or radiotherapy, loss of part or all of chromosomes 5 and 7 are a common features,286,287,288 as are the cytogenetic findings noted above for AML, occurring de novo. Table 88–4 lists the most frequent abnormalities and translocations seen in AML.282,283,286–308 The t(8;21) and inv(16) confer a more favorable outcome on average. t(15;17) confers a highly favorable prognosis. Deletion of all or part of chromosomes 5 and 7 or the presence of complex changes (greater than 3 abnormalities) confers an unfavorable prognosis. Other findings (e.g., normal karyotype, +8, 11q23) generally confer an intermediate prognosis (Chap. 13 has further details and discussion regarding impact of specific translocations).282,283,284
| Chromosome Abnormality | Genes Affected | Clinical Correlation |
|---|---|---|
Loss or gain of chromosome | ||
| Deletions of part or all of chromosome 5 or 7 | Not defined | Frequent in patients with acute myelogenous leukemia (AML) occurring de novo and in patients with history of chemical, drug, or radiation exposure and/or previous hematologic disease.282,283,286,287 |
| Trisomy 8 | Not defined | Very common abnormality in acute myeloblastic leukemia. Poor prognosis, often a secondary change.283,289 |
| Translocation | ||
| t(8;21) (q22;q22) | RUNX1(AML1)–RUNX1T1(ETO) | Present in ~8% of patients <50 years old and in 3% of patients >50 years old with AML.288 Approximately 75% of cases have additional cytogenetic abnormalities, including loss of Y in males or X in females. Secondary cooperative mutations of KRAS, NRAS, KIT common. Present in ~40% of myelomonocytic phenotype. Higher frequency of myeloid sarcomas.263,264,265,266 |
| t(15;17) (q31; q22) | PML-RAR-α | Represents ~6% of cases of AML.288 Translocation involving chromosome 17, t(15;17), t(11;17), or t(5;17) is present in most cases of promyelocytic leukemia.290,291 |
| t(9;11); (p22; q23) | MLL (especially MLLT3) | Present in ~7% of cases of AML. Associated with monocytic leukemia.292,293 11q23 translocations in 60% of infants with AML and carries poor prognosis. Rearranges MLL gene.292,293,294,295,296 Many translocation partners for 11q23 translocation.295,296,297,298 MLL1, MLL4, MLL10 may also result in AML phenotype. |
| t(9;22) (q34; q22) | BCR-ABL1 | Present in ~2% of patients with AML.299,300 |
| t(1;22)(p13;q13) | RBMIS-MKL1 | <1% of cases of AML. Admixture of myeloblasts, megakaryoblasts, micromegakaryocytes with cytoplasmic blebbing, dysmorphic megakaryocytes. Reticulin fibrosis common.301 |
| t(10;11)(p12-13;q14-21) | PICALM-MLLT10 | Outcome similar to that of intermediate prognosis group; more extramedullary disease and CD7 expression.302 |
| Inversion | ||
| Inv(16) (p13.1;q22) or t(16;16) (p13.1;q22) | CBF-β MYH11 | Present in ~8% of patients <50 years of age and in ~3% of patients >50 years of age with AML288; often acute myelomonocytic phenotype; associated with increased marrow eosinophils; predisposition to cervical lymphadenopathy,303 better response to therapy.304,305,306,307 Predisposed to myeloid sarcoma. |
| Inv(3) (q21q26.2) | RPN1-EVI1 | ~1% of cases of AML. Approximately 85% of cases with normal or increased platelet count. Marrow has increased dysmorphic, hypolobulated megakaryocytes. Hepatosplenomegaly more frequent than usual in AML.308 |
Approximately 45 percent of cases of AML contain cells that are cytogenetically normal. When five genes—NPM1, FLT3, CEPBA, MLL, and NRAS—were examined in 872 adults who were younger than 60 years of age with a normal karyotype, approximately 85 percent had a mutation in at least one of these genes. Mutations in NPM1 or CEPBA were associated with more favorable outcomes, analogous to the category of favorable cytogenetics noted above. The microarray expression signature in patients with AML younger than age 60 years who have cytogenetically normal cells but high-risk molecular features, especially FLT3-ITD and/or wild-type NMP1 expression, is correlated with outcome of therapy (see “Effect of Molecular and Cytogenetic Markers on Disease Progression and Therapeutic Responsiveness” above and Table 88–2). MicroRNAs regulate gene expression and the downregulation of the microRNA-181 family predicts a poor outcome. The microRNAs studied also revealed several important gene families that appear to be involved in the pathogenesis of AML, including genes involved in innate immunity (e.g., toll-like receptors and interleukin-1β expression and regulation).309 (Chapters 13 and 83 provide further discussion of gene-array profiling and microRNA analysis and the section “Other Acquired Mutations” on molecular pathogenesis has a more detailed discussion of specific molecular markers.) Microarray-based gene-expression profiling is anticipated to become more important in precise diagnosis and subclassification of AML in the future.310
Prior to treatment, mild to moderate increases in serum uric acid and lactic dehydrogenase levels are frequent. Both levels are higher in myelomonocytic and monocytic AML than in other AML phenotypes.200,201 Occasional patients have very elevated uric acid levels, which usually occur after chemotherapy if proper precautions are not taken (e.g., hypouricemic agents and hydration therapy).311 Abnormalities of sodium, potassium, calcium, or hydrogen ion concentration are infrequent and usually mild.312,313 Severe hyponatremia associated with inappropriate antidiuretic hormone secretion has occurred at presentation.312,313 Severe hypernatremia as a consequence of diabetes insipidus can be an initial event.314 Hypokalemia is a more frequent finding at presentation and is related to kaliuresis, although the reason for the proximal renal tubular dysfunction is unclear.312,313,315 The hypokalemia can be severe and often is worsened by the effects of treatment, especially use of kaliuretic antibiotics.315 Factitious elevations in serum potassium levels have been reported in patients with hyperleukocytosis as a result of leakage from white cells in vitro.316,317 Factitious hypoglycemia and spurious hypoxia from the effects of high blast cell counts in blood can occur.314,318
The presence of hypercalcemia is multifactorial,319 but cases with increased ectopic parathormone-like activity in the plasma have been described.320 Severe lactic acidosis prior to treatment has been reported.312,321,322 Hypophosphatemia as a result of phosphate uptake by leukemic cells can occur.323 Ectopic adrenocorticotropic hormone secretion,324 circulating immune complexes,325 and abnormal concentrations of coagulation factors or their inhibitors326 may be present.
Although prothrombin and partial thromboplastin times usually are normal or near normal, abnormal concentrations of coagulation factors are frequent. Elevations of platelet factor 4 and thromboxane B2 occur often.327 Decreases in α2-antiplasmin, protein C, and antithrombin III levels are frequent327 and may be associated with venous thrombosis.328 APL and acute monocytic leukemia are associated with hypofibrinogenemia and other indicators of activation of coagulation or fibrinolysis (see “Morphologic Variants of Acute Myelogenous Leukemia” below).329
Leukocyte count is an independent prognostic factor in the outcome of AML treatment.330 Approximately 5 percent of patients with AML develop signs or symptoms attributable to a markedly elevated blood blast cell count, usually greater than 100 × 109/L (Chap. 83).331 Several subsets of AML are associated with a greater likelihood of presenting with hyperleukocytosis. These include acute myelomonocytic, acute monocytic, the microgranular variant of APL, and AML with inv16,11q23 rearrangements, or FLT3-ITD. The circulations of the CNS, lungs, and penis are most sensitive to the effects of leukostasis. Intracerebral hemorrhage from vascular occlusion, invasion, and disruption, sometimes complicated by thrombocytopenia and vascular insufficiency are the most virulent manifestations of the syndrome.332,333,334,335,336 Dizziness, stupor, dyspnea, and priapism may occur. Diabetes insipidus is another association.337,338 Other severe organ involvement also may occur infrequently. A high early mortality in patients with AML correlates with hyperleukocytosis greater than 100 × 109/L.334,335,339,340 Chemotherapy in hyperleukocytic patients may lead to a pulmonary leukostatic syndrome, presumably from the effects of rigid, effete blast cells, or the discharge of large amounts of cell contents and resultant cell aggregation or other effects.341,342,343 Larger-vessel vascular occlusion as a result of white thrombi or masses of leukemic cells is rare.344,348 The upregulation of endothelial cell intercellular adhesion molecule-1 and of leukemic blast cell lymphocyte function-associated antigen-1 may mediate the vessel wall interaction contributing to leukostasis.349
Approximately 10 percent of patients with AML present with a syndrome that includes pancytopenia, often with inapparent blood blast cells, and absence of hepatic, splenic, or lymph nodal enlargement.350,351,352 If one corrects for the decrease in marrow cellularity with age, hypoplastic AML occurs in approximately 2 percent of cases.353 Approximately 75 percent of these patients are men older than 50 years of age. Marrow biopsy is hypocellular, which is the unusual feature of the syndrome, but leukemic blast cells are evident and present in a proportion of 10 to 90 percent of marrow cells. Response to intensive chemotherapeutic treatment, often with low-dose cytarabine because of the patients’ very advanced age, has been relatively good, and 3-year survival rates are approximately the same as the rates of other age-matched patients.354
Not infrequently, usually in patients older than 50 years of age, myelogenous leukemia is manifested by anemia and often thrombocytopenia. The leukocyte count may be low, normal, or increased, and a small proportion of blast cells are present in the blood (0 to 15 percent) and marrow (3 to 19 percent). Such cases have been referred to as oligoblastic myelogenous leukemia, subacute, or smoldering leukemia,355 or classified as a MDS, particularly refractory anemia with excess blasts. The clinical course of the untreated disease can be protracted. The disease has a high morbidity and mortality from infection and hemorrhage and can evolve into overt (polyblastic) AML. The smoldering or oligoblastic leukemias (refractory anemia with excess blasts) historically have been grouped along with the clonal cytopenias as composing MDS; and, the diagnosis and treatment of these variants are discussed in Chap. 87. Biologically and clinically, the disorders in this subset of the MDS with blast cell proportions in the marrow above normal are leukemias, not dysplasias, but they have a slower rate of progression than polyblastic myelogenous leukemia. Dysmorphogenesis of red cells, neutrophils, and platelets is more frequent and more striking than in the average case of polyblastic AML (Chap. 87), but such dysmorphogenesis also occurs in polyblastic leukemia, so-called AML with trilineage dysmorphia.275 A discussion of the spectrum of myelogenous leukemias, ranging from minimal to severe deviation neoplasms, can be found in Chap. 83.
Approximately 2 percent of patients with AML have the Philadelphia (Ph) chromosome t(9;22)(q34;q11) in a significant proportion (10 to 100 percent) of leukemic blast cells.356,357,358 The blast cells have surface antigens, such as CD13 and CD33, characteristic of myeloid leukemias.359,360 One interpretation of the concurrence of AML with t(9;22) is that it represents CML presenting in myeloid blast crisis.361,362,363 The arguments in favor of this proposal are as follows: (1) Blast crisis may occur within days after diagnosis of Ph chromosome–positive CML. (2) Cases can present with additional cytogenetic changes comparable to CML in blast crisis.361,363 (3) Marked hepatosplenomegaly, uncharacteristic of AML, may be present.362,363 (4) Platelet counts may be normal, and basophils can be increased.361,363 (5) A long prodromal period of weakness and weight loss may occur, and some features of CML, such as granulocytosis, can appear after treatment with chemotherapy.364 (6) Ph chromosome–positive AML has a poor prognosis, as in myeloid blast crisis of CML. (7) The breakpoint on chromosome 22 in the M-bcr may be typical of CML, and the product of the fusion BCR-ABL gene is a p210 tyrosine kinase identical to that of classic CML.360,363,364,365,366,367,368 (8) Occasional cases express p210 and p190 tyrosine kinases, now known to be features of CML.368 (9) Some patients enter a remission by converting to a phenotype analogous to chronic phase CML. An alternative view has been promulgated because (1) cases of Ph chromosome–positive AML can be a mosaic (normal and abnormal karyotypes)360; (2) the Ph chromosome may appear later in the course of the disease369; (3) additional chromosomal abnormalities often are different from those seen in the myeloblastic crisis of CML360,370,371; and (4) in some cases, the BCR-ABL gene is not encoding a p210 but a p190 mutant tyrosine kinase,357,365,368,372 the former being most characteristic of CML. Moreover, Ph chromosome–positive AML has developed following Ph chromosome–negative oligoblastic myelogenous leukemia.357,373,374 Many cases of Ph chromosome–positive acute leukemia are myeloid-lymphoid hybrids.364,368,370,375 Thus, Ph chromosome–positive AML comes in two varieties: one with a break in M-BCR of chromosome 22 with a p210 product, which could be considered analogous to acute blast crisis of CML, and one with a molecular pathology resulting in the oncogene product being a p190 protein (m-BCR) that could be considered a de novo case.
Necrosis of the marrow is an uncommon event and can be seen in a wide variety of malignant and nonmalignant clinical disorders, but approximately two-thirds of cases are associated with lymphoid or myeloid malignancies and about one-quarter of cases occur in patients with AML.376 Bone pain (approximately 80 percent of patients) and fever (approximately 70 percent of patients) are the two most common symptoms or signs. Anemia and thrombocytopenia, if not already present, results. White cell counts may be low or high. The blood may contain nucleated red cells and myeloid immaturity (approximately 50 percent of cases). Lactic dehydrogenase and alkaline phosphatase are elevated in approximately 50 percent of cases. The marrow aspirate is often watery and serosanguineous. An amorphous extracellular eosinophilic background with disintegrating cells that have lost their staining characteristics with indistinct margins and varying degrees of pyknosis and karyorrhexis is characteristic. Rare cases have been described in which the marrow contained Charcot-Leyden crystals without an increase in eosinophils or basophils.377 Bony spicules may also show evidence of necrosis. Destruction of spicule architecture with loss of osteocytes, osteoblasts, and osteocytes may be seen. It is important not to identify these changes as artifact. Usually more than 50 percent of the biopsy is involved. Careful search may identify the underlying hematologic disorder in small islands of intact cells. Technetium-99m sulfur colloid scans show little or no uptake. Magnetic resonance imaging (MRI) may not be diagnostic but can show the extent of the necrosis by changes in signal intensity signifying an increase in water content in relation to fat. Both technetium scanning and MRI can point to areas of intact marrow that may be used to make a diagnosis of the underlying disease, if it is unknown. The pathophysiology is uncertain but is thought to be related to marrow vascular injury and or thrombosis secondary to inflammatory or immune factors and cytokines. The prognosis of marrow necrosis is largely related to the underlying disease. Repair of marrow can occur, if the patient enters remission.
Four myeloproliferative syndromes related to AML have been identified in the neonate: transient myeloproliferative disorder, transient leukemia, congenital leukemia, and neonatal leukemia. Transient myeloproliferative disorder and transient leukemia are considered to represent the same phenomenon.
Transient myeloproliferative disease (TMD) can be present at birth or occur shortly thereafter in approximately 10 percent of infants with Down syndrome.378,379,380,381,382,383,384 The leukocyte count is markedly elevated, blast cells are present in the blood and marrow, and anemia and thrombocytopenia may be present, but the latter are not constant findings. The liver and spleen may be enlarged. Results of cytogenetic studies and marrow cell culture studies are normal, except for trisomy 21, which is characteristic of Down syndrome. The blast cells usually have the immunophenotype of megakaryocytes. In contrast to congenital leukemia, the elevated white cell and blast cell counts disappear in most patients (approximately 80 percent) over a period of weeks to months. In approximately 20 percent of patients, severe and potentially lethal complications of hydrops fetalis, hepatic fibrosis, or cardiorespiratory failure may occur.
In some cases, an additional cytogenetic abnormality is present, which disappears after regression of the myeloproliferative syndrome, suggesting a reversible clonal disorder (transient leukemia) that is replaced by normal hematopoiesis. The presence of a trisomy of chromosome 21 is essential for the disease as judged by three observations: the trisomy occurs in (1) the TMD clone of patients with constitutional trisomy 21, (2) the TMD clone in patients with Down syndrome with a cell mosaic pattern of trisomy 21, and (3) in the TMD clone of phenotypically normal infants without a constitutional trisomy 21, but with TMD. In the last case, trisomy 21 disappears with resolution of the myeloproliferation.385 Candidate oncogenes on chromosome 21 responsible for the phenomenon include FPDMM, RUNX1 (CBF-β), and IFNAR, among others.385 GATA-1 mutations have been found in nearly all patients with TMD and in acute megakaryocytic leukemia in Down patients.386 The TMD syndrome may disappear, only to be followed shortly thereafter by acute leukemia, predominantly AML, but occasionally acute lymphocytic leukemia (ALL).
One hypothesis for TMD is that the disorder originates in a primitive cell of fetal hepatic hematopoiesis. The cell involutes and is replaced with marrow stem cells. Approximately 25 percent of newborns with Down syndrome and transient leukemia develop acute megakaryocytic leukemia in the first 4 years of life.387,388,389
Very-low-dose cytarabine has been suggested for those patients with severe hepatic fibrosis, very high white cell counts, or hydrops fetalis.385 TMD cells in these infants are very sensitive to cytarabine.390,391
Children with Down syndrome have a 150-fold risk of AML and about a 40-fold risk of ALL by age 5 years. A slightly increased risk of acute leukemia persists into older age. Myelogenous leukemia in patients with Down syndrome often has a megakaryoblastic or erythroid phenotype and may have an interstitial deletion of chromosome 21.380,381,392,393,394,395 This requires mutation in the GATA1 gene in addition to trisomy 21, and sequential epigenetic changes occur as well in the evolution of acute megakaryoblastic leukemia.395,396 The response rate of infants with Down syndrome and AML to chemotherapy is very high over prolonged followup and better than the response of patients without Down syndrome.387,391,397,398 The response to adjusted-dose anthracycline antibiotic and cytarabine in Down syndrome children with AML is approximately 90 percent and the event-free 5-year survival is approximately 80 percent.394 In those cases with relapsed or refractory disease, outcomes are poor even with allogeneic HSC transplantation.399 ALL may occur, and the response to therapy is similar to the response of patients without Down syndrome of the same age. Most solid tumors occur less frequently in Down syndrome patients.390
Congenital or neonatal leukemia, a rare syndrome, occurs 10 times more frequently in newborns with Down syndrome than in newborns without trisomy 21.392,393 Leukocytosis, blood and marrow blast cells, hepatosplenomegaly, thrombocytopenia, purpura, anemia, and skin infiltrates are usual. The disease has been diagnosed prenatally. Cytogenetic abnormalities can occur and mark the leukemic clone.393,400,401 Monocytic leukemia and t(4;11) are the most common phenotype and karyotype.401,402,403 A case of vertical (transplacental) transmission of acute monocytic leukemia from mother to son has been reported.404
Infants who are normal at birth but develop AML in the first few weeks of life (neonatal leukemia) often display pallor, inadequate food intake, insufficient weight gain, diarrhea, and lethargy. The presence of a cytogenetic abnormality on band q23 of chromosome 11 is a very poor prognostic sign. Most infants with congenital or neonatal leukemia do not survive for more than a few weeks or months. Because treatment has been largely ineffective, observation to ascertain if TMD or a transient leukemia is present has been recommended if the clinical picture is unclear.405
Although coincidental myeloid and lymphoid clonal diseases have been reported for more than 60 years, the availability of techniques to identify surface antigens with monoclonal antibodies, immunoglobulin gene, and T-lymphocyte receptor gene rearrangements with molecular methods, and chromosome translocations by chromosome banding cytogenetic techniques has led to the appreciation of several types of hybrid acute leukemia.406,407,408,409,410,411
In bilineal (interlineal) acute leukemias, a proportion of cells (>10 percent) have lymphoid and myeloid markers; interlineal here refers to lymphopoietic and myeloid gene expression. Bilineal (biphenotypic) leukemias are heterogeneous. Some patients have cells with both lymphoid and myeloid markers (chimeric), whereas other patients have cells with either lymphoid or myeloid markers but evidence that all the cells are part of the same malignant clone (mosaic). The bilineal leukemias may be synchronous (lymphoid and myeloid cells are present simultaneously) or asynchronous (in which lymphoid cells are succeeded by myeloid cells or vice versa), but evidence exists for their origin from the same clone.
Cases of biphenotypic leukemia that are morphologically or cytochemically indicative of myelogenous leukemia have been referred to as LY+AML; the cases that are more indicative of lymphocytic leukemia are referred to as MY+ALL. As a group, interlineal hybrid leukemias treated with current regimens respond to therapy at approximately the same rate as AML cases without lymphoid markers.406 Some observers suggest altering drug regimens, depending on the balance between lymphoid and myeloid biochemical (drug-response) patterns.412
Acute leukemias may be intralineal hybrids in that the blast cells have markers for two or more myeloid lineages (e.g., erythroid, granulocytic, and megakaryocytic) or, in the case of lymphocytic leukemias, both immunoglobulin gene rearrangement (B-lymphocyte type) and T-cell receptor gene rearrangement (T-lymphocyte type).
Although most hybrid leukemias share myeloid and either B- or T-lymphocyte markers, two notable syndromes are associated with hybrid leukemias: (1) the myeloid leukemia and natural killer cell hybrid (CD56+, CD7+, CD13+, CD33+)413,414,415,416,417,418,419 and (2) the lymphoma, eosinophilia, and t(8;13) myeloid leukemia hybrid.420,421 Signs of lymphoma, such as mediastinal or other lymphadenopathy and extranodal lymphoid tumor, are mixed with findings compatible with AML in both syndromes. The morphology of the myeloid–natural killer cell leukemia often simulates APL, with hypergranular cytoplasm present but abnormality of chromosome 17 absent. The hybrids can appear de novo or after relapse of a lymphoma, T-cell leukemia, or blast crisis of CML. The hybrid leukemias usually have a poor prognosis. Myeloid antigens may not be evident at diagnosis in the natural killer cell hybrid but appear later in the course.422 Hematopoietic stem cell transplantation should be considered in an eligible patient.423
Hybrid leukemias may result from either lineage infidelity caused by genetic misprogramming413 or promiscuous gene expression, which occurs transiently in the differentiation of normal pluripotential HSCs. In the case of promiscuity, persistence of the transient normal event is thought to be present because of the block in differentiation that occurs.408 Genetic misprogramming (infidelity) could result from rearrangements of the DNA sequences that control the transcription of genes designating differentiation antigens.424
In these cases, lymphoid and myeloid cells are present simultaneously but are derived from separate clones, or sequential myeloid and lymphoid leukemia are present but the two lineages are derived from separate clones.
An unusual but significant concordance has been reported between nonseminomatous mediastinal germ cell tumors and AML, especially the megakaryoblastic variant.425,426,427,428,429,430 Mediastinal tumors are rare variants of germ cell tumors. The latter ordinarily occur as testicular teratomas and seminomas in men or as ovarian teratomas in women. They are thought to be derived from yolk sac cells that failed to migrate.428,429 AML is a HSC tumor derived from a cell type that is present in the yolk sac. Cytogenetic studies are compatible with a clonal relationship (identity) of mediastinal germ cells and myelogenous leukemia cells.426,427 Apparently, hematopoietic lineage genes are predisposed to expression in extragonadal (mediastinal) germ cell tumors. Use of etoposide, platinum, and related cytotoxic drugs for treatment of mediastinal germ cell tumors may induce secondary AML in a predisposed cell population.431
A study of 1892 patients with KIT-positive mesenchymal gastrointestinal stromal tumors found a significant subsequent incidence of AML (nine patients). The standardized incidence ratio was approximately 3.0 (confidence interval: 1.1 to 5.8). The patients had not received prior chemotherapy or radiotherapy and the median duration of gastrointestinal stromal tumors before onset of AML was 6 years.432
MORPHOLOGIC VARIANTS OF ACUTE MYELOGENOUS LEUKEMIA
Morphologic variants of AML (Table 88–5) may occur de novo or may be the manifestation of clonal evolution from essential thrombocythemia, idiopathic myelofibrosis, CML, or other chronic clonal myeloid disorders. For example, every phenotypic variant of AML can occur as the blast crisis of CML (Chap. 89).
| Variant | Cytologic Features | Special Clinical Features | Special Laboratory Features |
|---|---|---|---|
| Acute myeloblastic leukemia (M0, M1, M2) |
|
|
|
| Acute promyelocytic leukemia (M3, M3v) |
|
|
|
| Acute myelomonocytic leukemia (M4, M4Eo) |
|
|
|
| Acute monocytic leukemia (M5) |
|
|
|
| Acute erythroid leukemia (M6) |
|
|
|
| Acute megakaryocytic leukemia (M7) |
|
|
|
| Acute eosinophilic leukemia |
|
|
|
| Acute basophilic leukemia | Mixture of blast cells and cells with basophilic granules in blood and marrow. |
|
|
| Acute mast cell leukemia |
|
|
|
The designation acute myeloblastic leukemia came into existence in the second decade of the 20th century,4 following the specific description of the myeloblast.6 Approximately 25 percent of AML cases have the features of acute myeloblastic leukemia, a variant in which the leukemic myeloblast is the predominant cell in the marrow. Acute myeloblastic leukemia was divided into two forms, designated M0 and M1 in the French-American-British (FAB) Classification. In either type, little evidence of maturation of myeloblasts exists, and the marrow is replaced by a monotonous population of blasts. In acute myeloblastic leukemia (M0), the patient’s age distribution, presenting white cell count, and cytogenetic abnormalities are not distinctive. The blasts are nonreactive when stained for myeloperoxidase activity, and Auer rods are not seen. The blasts react with antibodies to myeloperoxidase and antibodies to CD13, CD33, and CD34. Human leukocyte antigen (HLA)-DR is positive in most patients. Occasional cases require in situ hybridization to identify the myeloperoxidase gene433 or genomic profiling for early myeloid-associated genes.434 Abnormal and unfavorable karyotypes (e.g., 5q–,7q–) and expression of the multidrug resistance (MDR) glycoprotein (p170) are more frequent. This phenotypic variant has a poor prognosis.435,436,437,438 In the other type of myeloblastic leukemia, designated M1, myeloblasts are present in the blood and make up more than 70 percent of marrow cells. Less than 15 percent of marrow cells are promyelocytes and myelocytes. Auer rods may be present in occasional blasts, but azurophilic granules are not evident in the blasts by light microscopy. At least 3 percent, but usually a much higher percentage, of the blast cells, have a positive reaction when stained for peroxidase or with Sudan black or react with monoclonal antibodies specific to myeloblasts, such as CD33. This morphologic subtype is denoted as M1 in the FAB classification. The WHO has divided acute myeloblastic leukemia into three types: AML without differentiation, AML without maturation, and AML with maturation. There is no evidence of a clinical distinction in response to therapy or in prognosis within these rarified designations.
In many cases of myeloblastic leukemia, more prominent granulocytic maturation is evident (FAB type M2 or WHO designation AML with maturation). This variant is present in approximately 15 percent of AML cases; thus, approximately 45 percent of cases of AML are myeloblastic leukemia with or without maturation. Blasts usually constitute at least 20 percent of the marrow cells. Auer rods may be present in blast cells. Promyelocytes, myelocytes, and segmented neutrophils, the latter often with the acquired Pelger-Huët anomaly, may constitute 20 to 60 percent of marrow granulocytes. The anomaly is reflected in bilobed or monolobed neutrophils. Histochemical and surface markers of blast cells are typical of myeloblastic leukemia, and monocytic markers are absent or infrequent. Monocytes represent less than 10 percent of cells. A translocation between chromosomes 8 and 21 t(8;21)(q22; q22), often accompanied by loss of the Y chromosome in men or loss of an X chromosome in women, is associated with the phenotype and occurs in younger patients (average age approximately 30 years).439,440,441 Patients whose cells contain t(8;21) are more prone to develop myeloid sarcoma.263,266
The ability of AML to express cells of the monocytic and granulocytic lineages was first highlighted in the early 1900s by Naegeli. Later, Hal Downey, a leading hematologist of the day, proposed the eponym Naegeli type for myelomonocytic leukemia.442 Approximately 15 percent of patients with AML present with this variant, and they are more likely to have extramedullary infiltrates in gingiva, skin, or CNS than are patients with acute myeloblastic leukemia (see “Myeloid [Granulocytic] Sarcoma” above).443 A mixture of myeloblasts and monoblasts is found in the blood and marrow. More than 30 percent of marrow cells are a mixed population of myeloblasts, which react with peroxidase or chloracetate esterase, and monoblasts or promonocytes, which react with fluoride-inhibitable nonspecific esterase (see Fig. 88–2F). More than 20 percent of cells are monoblasts or promonocytes in blood and marrow. In some cases, individual cells react with monocytic and granulocytic histochemical stains.444 Serum and urinary lysozyme levels are increased in most cases. This variant of AML is referred to as M4 in the FAB classification and as acute myelomonocytic leukemia in the WHO classification. Translocations involving chromosome 3 are associated with this phenotype.445
The proportion of marrow eosinophils446 or basophils447 may be increased. A particular variant of myelomonocytic leukemia has increased numbers of marrow eosinophils (10 to 50 percent), Auer rods in blast cells, and inversion or rearrangement of chromosome 16 (see Fig. 88–2G).304,305,306,307 The eosinophils are abnormally large, and the eosinophilic myelocytes contain large basophilic granules. Macrophages with ingested Charcot-Leyden crystals may be present. This phenotypic variant of AML has been designated M4Eo in the FAB classification. Although this variant has an increased risk of CNS involvement, it carries a more favorable prognosis than the average case of AML. Fluorescence in situ hybridization (FISH) is a more accurate method for detection of cryptic 16q22 gene rearrangements and is useful in conjunction with conventional cytogenetics for patients with M4Eo AML. AML with t(6;9)(p23;q34) is an uncommon variant, occurring in approximately 1 percent of cases, and may express itself as acute myelomonocytic or acute myeloblastic leukemia. Anemia, thrombocytopenia, a variable white cell count, and increased myeloblasts are frequent. The myeloblasts often contain Auer rods. Marrow basophilia is present in about half the cases.208,280,448 The variant occurs at a younger age, has a poor prognosis, and has a tendency to trilineage dysmorphia and ringed sideroblasts.449
Prominence of erythroid cell proliferation in AML cases was noted by Copelli450 and DiGuglielmo451 in the early 20th century. Moeschlin452 used the term erythroleukemia. Dameshek453 suggested the name DiGuglielmo syndrome and dissected the disorder into three phases, depending on the decreasing prevalence of dysmorphic erythroblasts and the reciprocal increasing prevalence of myeloblasts. Erythroid leukemia makes up approximately 5 percent of AML cases and is referred to as M6 in the FAB classification.454 Familial erythroleukemia has been described.455,456 Erythroid leukemia is arbitrarily divided into three degrees of severity: (1) erythroleukemia in which more than 50 percent of the marrow cells are dysmorphic; (2) erythroblasts admixed with myeloblasts, the latter composing approximately 20 percent of non-erythroid cells or approximately 5 to 10 percent of total marrow cells; and (3) a form in which dysmorphic erythroblasts dominate the marrow, pure erythroid leukemia, in which more than 80 percent of marrow cells are dysmorphic erythroblasts with a trivial granulocytic proportion of cells and very few if any myeloblasts. This last form of the disease may start in as a milder variant, formerly called erythremic myelosis, in which granulopoiesis, and thrombopoiesis may be only mildly abnormal. This phase, dominated morphologically by bizarre dysmorphia of erythroblasts, can be protracted but eventually evolves into a dimorphic phase in which myeloblasts are more prominent, severe neutropenia and thrombocytopenia develop, and the patient progresses to erythroid leukemia. The disease may evolve further into polyblastic AML.457,458,459,460 In the erythremic myelosis variant, erythropoiesis is ineffective. However, some normal regulation may remain because hypertransfusion decreases both erythropoietin levels and the amount of abnormal erythropoiesis.461 Spontaneous growth of leukemic erythroid clonogenic cells is a feature of the disease.462 Periodic acid–Schiff (PAS)-positive erythroblasts are evident in almost all cases.457,460
The erythroid leukemias are characterized by a striking population of dysmorphic erythroblasts in marrow and red cells in blood (see Fig. 88–2I, J, and K). Anemia and thrombocytopenia are present in nearly all cases. Some patients may have elevated total leukocyte counts. The red cells show marked anisocytosis, poikilocytosis, anisochromia, and basophilic stippling. Nucleated red cells are present in the blood. The marrow erythroblasts are extremely abnormal, with giant multinucleate forms, nuclear budding, and nuclear fragmentation. Cytogenetic abnormalities are present in approximately 70 percent of patients and complex cytogenetic abnormalities are frequent. The frequency of erythroid leukemia is increased if methods for detecting erythroid differentiation more sensitive than light microscopy are used. These cell features include glycophorin A, spectrin, carbonic anhydrase I, ABH blood group antigens, and other antigens that occur on early erythroid progenitors, such as the transferrin receptor (CD71).463,464,465 Antihemoglobin antibody and antihuman erythroleukemic cell line antibody often are positive.458
Erythremic myelosis can have an indolent course and may be managed for a time without intensive chemotherapy. Treatment is warranted in patients with erythroleukemia and acute erythroid leukemia, and the results are approximately the same as with other phenotypes in patients of similar age.460 The more predominant the erythroid component and the lower the proportion of myeloblasts, the better the response to therapy.403
The association of an exaggerated hemorrhagic syndrome with certain leukemias was described by French hematologists in 1949.466 In 1957, Hillstad467 bestowed the appellation promyelocytic leukemia upon this morphologic-clinical subtype of AML. This variant, which is called M3 in the FAB classification and APL in the WHO classification, occurs at any age and constitutes approximately 7 percent of AML cases.290,291,468,469 APL occurs with greater frequency among Latinos from Europe and South and Central America.190,191 APL represents 19 percent of AML cases in the Chinese189 as compared to 8 percent among persons of European descent. APL is also increased among persons with an increased body mass index.470,471,472 Unlike all other major variants of AML, which increase in incidence logarithmically with age, the incidence of APL is constant over the human life span.188 Hemorrhagic manifestations are prominent including hemoptysis, hematuria, vaginal bleeding, melena, hematemesis, and pulmonary and intracranial bleeding, as well as the more typical skin and mucous membrane bleeding. In severely leukopenic patients, blasts may not be evident in the blood. Moderately severe thrombocytopenia (<50 × 109/L) is present in most cases. The marrow contains few agranular blast cells and some blast-like cells with scant granules. The dominant cells are promyelocytes, which comprise 30 to 90 percent of marrow cells (see Fig. 88–2D and E). Auer rods and cells with multiple Auer rods (1 to 10 percent) are present in nearly every case. Promyelocytes with multiple Auer rods have been referred to as faggot cells. Leukemic promyelocytes stain intensely with myeloperoxidase and Sudan black and express CD 9, CD13, and CD33, but not CD34 or HLA-DR.290,291,468,469
A variant type of promyelocytic leukemia is referred to as microgranular (M3v in the FAB nomenclature).473,474,475,476 Microgranular cases represent approximately 20 percent of patients with promyelocytic leukemia. The leukemic cells may mimic promonocytes with convoluted or lobulated nuclei. Auer rods may be present but are less evident. The majority of the leukemic cells contain azurophilic granules that are so small they are not visible by light microscopy, but the peroxidase stain usually is strongly positive. Typical hypergranulated promyelocytes usually are present on careful inspection. The total white cell count often is highly elevated, and severe coagulopathy is prominent in microgranular cases.474 Rarely, the cells contain eosinophilic or basophilic granules, but t(15;17) is present, and the response to all-trans retinoic acid (ATRA) persists,477,478,479 although the basophilic variant can be virulent.480
A translocation between chromosome 17(q21), which rearranges the RAR-α gene at band q21, and another chromosome is present in all cases of APL and in the acute promyelocytic transformation of CML; it is not found in other AML variants. The t(15;17)(q22;q21) is the most frequent cytogenetic abnormality (>95 percent), but variant translocations between chromosome 3, 5, or 11 and chromosome 17 or isochromosome 17, and other even less common variants have been described.290,468,481,482,483 In some cases, cytogenetic analysis is inadequate and Southern blot analysis is required to identify the rearrangement of the RAR-α gene. A functional distinction is that the t(15;17), PML–RAR-α fusion, the t(5:17), NPM–RAR-α fusion, and the t(3;17), TBLR1–RAR-α fusion confer retinoid therapy responsiveness, whereas t(11;17), PLZF–RAR-α fusion, usually is retinoid resistant. In cells with the t(11;17), Auer rods are absent and CD56 expression usually is present, offering some clinical variables to provoke special molecular investigations.484 The retinoid resistance may not always be present.485
The breakpoint on chromosome 17 is within the gene encoding the RAR-α, and the breakpoint on chromosome 15 is within the locus of a gene originally referred to as MYL and renamed PML (to indicate its relationship to promyelocytic leukemia).290,486 The gene encodes a unique transcription factor. The translocation results in two new chimeric or fusion genes: RAR-α–PML, which is actively transcribed in APL, and PML–RAR-α, which also is transcribed and may account for the aberrancy in hematopoiesis. The PML–RAR-α gene has two isoforms that produce a short- and a long-type fusion messenger RNA, respectively.487 Patients with the short isoform may have a worse outcome than those with the longer form. Polymerase chain reaction (PCR) for the mRNA of the fusion gene can be used to identify residual cells during remission and may predict relapse. The PML–RAR-α transgene can reproduce the disease in mice,488 although in some models a superimposed FLT3 mutation is required to express the disease. FLT3 mutations are frequently found in human disease, especially in the hypogranular variant.157
A propensity to hemorrhage is a striking feature of this subtype. The prothrombin and partial thromboplastin times are prolonged, and the plasma fibrinogen level is decreased in most cases. The disturbance in coagulation first was thought to principally result from intravascular coagulation initiated by procoagulant released from the granules of the leukemic promyelocytes. Elevated thrombin–antithrombin complexes, prothrombin fragment 1+2, and fibrinopeptide A plasma levels support that supposition. Increased levels of fibrinogen–fibrin degradation products, D-dimer, and evidence of plasminogen activation indicate fibrinolysis.489,490,491 Furthermore, decreased levels of plasminogen, increased expression of annexin II on the leukemic cells,492 and reports of responses to tranexamic acid support a role for fibrinolysis in the bleeding in APL.493 Release of nonspecific proteases may further contribute to fibrinogenolysis. Thus, the coagulopathy is now considered tripartite.494
Although APL responded to chemotherapy regimens for AML, especially those containing an anthracycline antibiotic,495 the cytologic pattern of response in the marrow often was paradoxical.496,497,498,499 Persistence of leukemic promyelocytes preceded remission in the absence of further therapy, whereas induction of marrow cell hypoplasia was classically considered a requirement for remission in patients with AML. Generally, if leukemic blast cells persist after therapy for AML, relapse ensues unless hypoplasia is induced by more cytotoxic therapy. The unusual pattern of response in APL was put into context by reports of successful treatment with isomers of retinoic acid, an agent that leads to maturation of leukemic promyelocytes in vitro.499 In 1988, the success of ATRA in remission induction was reported500,501 and confirmed.290,291 Relapse occurs invariably, however, so chemotherapy regimens or addition of arsenic trioxide also were required. Use of ATRA has decreased the risk of early hemorrhagic complications and death and has enhanced the long-term response to chemotherapy. Despite the improvement in therapy, approximately 5 to 10 percent of patients die during remission induction, most of hemorrhage, often into the brain. The prolonged remissions of patients with promyelocytic leukemia has been interrupted in approximately 3 percent of cases by the later appearance of oligoblastic myelogenous leukemia with deletions of all or part of chromosome 5 or 7 and no evidence of involvement of chromosome 17, compatible with a myelogenous leukemia secondary to cytotoxic chemotherapy.501,502,503 The responsiveness to arsenic trioxide has provided additional treatment approaches that are discussed in the “Therapy” section below.
Monocytic leukemia was first reported by Reschad and Schilling-Torgau504 in 1913. Approximately 8 percent of patients with AML present with monocytic leukemia, which is referred to as M5 in the FAB classification. Patients with monocytic leukemia have a higher prevalence (50 percent) of extramedullary tumors in the skin, gingiva, eyes, larynx, lung, rectum and anal canal, bladder, lymph nodes, meninges, CNS, and other sites than do other phenotypes (<5 percent). Hepatomegaly, splenomegaly, and lymphadenopathy are more frequent in monocytic leukemia.207,505,506,507
The proportion of monocytic cells is usually greater than 75 percent. The total leukocyte count is higher in a larger proportion of patients, and hyperleukocytosis occurs more frequently (approximately 35 percent) than in other variants.508,509,510 The marrow and blood cells may be largely monoblasts (acute monoblastic leukemia) or more mature-appearing promonocytes and monocytes (acute monocytic leukemia) (see Fig. 88–2H). When the blood contains more mature-appearing monocytic cells, the marrow contains a lower proportion of blast cells, approximately 15 to 50 percent. When the blood monocytes are largely blast cells, the marrow contains approximately 50 to 90 percent blasts. In nearly all cases, 10 to 90 percent of monocytic cells react for nonspecific esterase stains, α-naphthyl acetate esterase, and naphthol AS-D-chloroacetate esterase; in a cytochemical or chemoluminescence assay; or with monoclonal antibodies against monocyte surface antigens, especially CD14. Immunoreactivity of cells for lysozyme is characteristic. Serum and urine lysozyme levels are elevated in most patients. Serum lactic dehydrogenase and β2-microglobulin concentrations are increased in greater than 80 percent of patients.511 Plasminogen activator inhibitor-2 is present in the plasma and the cells of a high proportion of patients.512 Auer rods are absent when monoblasts dominate but may be present in cases where promonocytes and monocytes are prevalent in blood and marrow. Leukemic monocytes have Fc receptors and can ingest and kill microorganisms in some cases.513,514
There is an association between translocations involving chromosome 11, especially region 11q23, and monocytic leukemia.292,293,294 In particular, t(9;11) is found in leukemic monocytes.295,296,507,508 In t(9;11) the β1-interferon gene is translocated to chromosome 11, and the protooncogene ETS-1 is translocated to chromosome 9 adjacent to the α-interferon gene. The latter juxtaposition may be important in the pathogenesis of monocytic leukemia.515
The expression of FOS is closely correlated with monocytic maturation of cells in myelomonocytic and monocytic leukemia and in normal monocytopoiesis.516,517 Absence or markedly decreased expression of the retinoblastoma gene growth-suppressor product (p105) is present in approximately half of patients with monocytic leukemia. Patients express a more dramatic phenotype.518 A variant of acute monocytic leukemia in which the leukemic cells have monocytoid features and are positive for early and late monocytic lineage antigens and for TdT activity often occurs after prior radiotherapy or chemotherapy and is relatively resistant to treatment.519 A syndrome of acute monoblastic leukemia with t(8;16), resulting in MOZ-CBP fusion gene, is characterized by mildly granular promonocytes (simulating hypogranular promyelocytes), intense phagocytosis of red cells, erythroblasts, and sometimes neutrophils and platelets in blood and marrow, simulating macrophagic hemophagocytic syndrome, intravascular coagulation or primary fibrinolysis, and a high frequency of extramedullary disease.520
The management of monocytic leukemia is complicated by a greater incidence of CNS or meningeal disease either at the time of diagnosis or as a form of relapse during remission. Thus, examination of cerebrospinal fluid is often recommended, even in the absence of symptoms, when remission has been achieved.208,507,508 Some therapists recommend prophylactic intrathecal therapy with methotrexate or cytosine arabinoside for patients who enter remission after having presented with hyperleukocytic acute monocytic leukemia because of the risk of subclinical meningeal involvement. Others posit that high-dose cytarabine with CNS penetration potential used in consolidation chemotherapy suffices for this purpose. There are few data for guidance in this matter.
Rare cases of dendritic cell or Langerhans cell phenotype have been described (Chap. 71).521,522 Uncommon cases of histiocytic sarcoma are the tissue or extramedullary variant of monocytic leukemia (Chap. 71).523,524 The outcome of treatment, once thought to be less favorable than with other forms of AML, is comparable to the outcome of other subtypes.525
In 1963, Szur and Lewis526 reported patients with pancytopenia, low percentages of blast cells, and intense myelofibrosis but an absence of teardrop red cells, splenomegaly, leukocytosis, and thrombocytosis, the usual features of primary myelofibrosis. They designated the syndrome malignant myelosclerosis.526 Reports of similar cases ensued, with some investigators referring to the syndrome as acute myelofibrosis.527 The development of methods to phenotype megakaryoblasts indicated the cases were variants of AML rather than of primary myelofibrosis and have been designated acute megakaryocytic or acute megakaryoblastic leukemia.391,528,529 This leukemia is referred to as M7 in the FAB classification. The prevalence of this phenotype is approximately 5 percent of all AML cases if appropriate cell markers are used in the diagnosis, and is at least twice that frequency in childhood AML.530,531 The syndrome is an especially prevalent variant of AML that develops in patients with Down syndrome398,532 or in patients with mediastinal germ cell tumors and coincident AML.425,426,427,428,429
Leukemic megakaryoblasts and promegakaryocytes can be difficult to identify by light microscopy using polychrome staining. However, with experience, heightened suspicion can be engendered by blasts in the blood with abundant budding cytoplasm or blasts having a lymphoid appearance, especially if the marrow cannot be aspirated because of intense myelofibrosis, the latter evident on the marrow biopsy. Initially high-resolution histochemistry for platelet peroxidase and identification of the demarcation membrane system using transmission electron microscopy were required for diagnosis. Now antibodies to von Willebrand factor or to platelet glycoprotein Ib (CD42), IIb/IIIa (CD41), or IIIa (CD61) can be used to identify very primitive megakaryocytic cells.528,529 A small proportion of megakaryoblasts may be present in other cases of AML, but in megakaryocytic leukemia they are the prominent or the dominant leukemic cells (see Fig. 88–2L through O). Moreover, the other key features of the syndrome usually are present, especially severe myelofibrosis.530
Patients usually present with pallor, weakness, excessive bleeding and anemia, and leukopenia. Lymphadenopathy or hepatosplenomegaly is uncommon at the time of diagnosis. High leukocyte and blood blast cell counts may be present initially or may develop later. The platelet count may be normal or elevated in many patients at the time of presentation. Abnormal platelets or megakaryocytic cytoplasmic fragments may be found in the blood. Marrow aspiration often is unsuccessful (“dry tap”) because of extensive marrow fibrosis in most cases, although not all. The marrow biopsy contains small blast cells, large blast cells, or a combination of both. The former have a high nuclear-to-cytoplasmic ratio, have dense chromatin with distinct nucleoli, and resemble lymphoblasts. Cases have been mistaken for ALL. The larger blasts may have some features of maturing megakaryocytes with agranular cytoplasm with cytoplasmic protrusions, clusters of platelet-like structures, or shedding of cytoplasmic blebs. The blast cells are peroxidase negative and tend to aggregate. Confirmation of their megakaryoblastic maturation requires immunocytologic studies for the presence of von Willebrand factor and the immunoreactivity to CD41, CD42, or CD61. The more mature megakaryocytes, which often coexist in the marrow, stain with PAS reagent, contain sodium fluoride-inhibitable nonspecific esterase, and fail to react for α-naphthylbutyrate esterase or myeloperoxidase. The thrombopoietin receptor gene (MPL) is expressed in megakaryocytes (CD116) and exhibits the gain-of-function point mutation W515K/L in approximately 25 percent of cases of acute megakaryoblastic leukemia.533
The serum lactic acid dehydrogenase level frequently is strikingly increased and has an isomorphic pattern unlike that seen with other acute leukemias. Complex chromosome aberrations are common.534 An association of megakaryoblastic leukemia in infants with t(1;22)(p13;q13) has been reported.534,535,536,537 Abnormalities of chromosome 3 have been linked to clonal hemopathies expressing a prominent megakaryocytic phenotype.538,539 Progression of primary myelofibrosis or essential thrombocythemia to AML may have the phenotype of acute megakaryocytic leukemia. Paradoxically, in children with Down syndrome the disease can be treated with modified doses of chemotherapy, with a very high remission rate and long-term event-free survival.540–542 The result is thought to be related to the exquisite sensitivity of the leukemic cells to drug-induced apoptosis,475 whereas the long-term remission rate as a result of chemotherapy in children without Down syndrome or in adults are not as good.543,544
Acute eosinophilic leukemia is rare. Increased eosinophils in the marrow but not in the blood is a variant of acute myelomonocytic leukemia and inversion 16 or other abnormalities of chromosome 16 but is not considered an acute eosinophilic leukemia.303,304,305,306 First described in 1912,545 acute eosinophilic leukemia is a distinct entity that can arise de novo as AML, with 50 to 80 percent of eosinophilic cells in the blood and marrow.546,547,548,549 Anemia, thrombocytopenia, and blast cells in blood and marrow are present. There is apparent eosinophilic differentiation in striking proportions. The eosinophilic cells are dysmorphic and the cytoplasm hypogranulated with smaller than normal eosinophilic granules. The granules stain less intensely and are less refractile with polychrome stains. These findings are the result of the loss of the central crystalloid in the eosinophilic granules that can be identified with electron microscopic analysis. Biopsy of skin, marrow, or other sites of eosinophil accumulation often shows Charcot-Leyden crystals. A specific histochemical reaction, cyanide-resistant peroxidase, permits identification of leukemic cells with eosinophilic differentiation and diagnosis of acute eosinoblastic leukemia in some cases of AML with fewer identifiable eosinophils in blood or marrow.550 Eosinophilia, not part of the malignant clone, may be a feature of occasional patients with AML, an uncommon reactive phenomenon. In many cases, idiopathic eosinophilia (hypereosinophilic syndrome) is a monoclonal disorder representing a spectrum of more indolent chronic or subacute eosinophilic leukemia to more progressive acute leukemia (Chaps. 62 and 89).551 Acute eosinophilic leukemia may develop in patients having the chronic form of a hypereosinophilic syndrome. Overexpression of WT gene expression has been proposed as a means of distinguishing acute eosinophilic leukemia from a polyclonal, reactive eosinophilia.552
Patients with acute eosinophilic leukemia do not usually develop bronchospastic signs, neurologic signs, and heart failure from endomyocardial fibrosis as is seen in chronic eosinophilic leukemia, probably because those tissue changes are the result of release of toxins in the granule crystalloid, absent in most eosinophils in acute eosinophilic leukemia and because of the shorter duration of survival in acute eosinophilic leukemia. Hepatomegaly, splenomegaly, and lymphadenopathy are more common than in other variants of AML. The treatment approach is similar to other types of AML. A combination of cytarabine and an anthracycline antibiotic is an appropriate choice for treatment. Response to treatment is approximately the same as in other types of AML.550
First described in 1906,553 basophilic differentiation as a feature of AML is an uncommon event, occurring in approximately one in 100 cases of AML.549 Most cases of acute basophilic leukemia evolve from the chronic phase of CML,554 but de novo acute basophilic leukemia, in which the cells do not contain the Ph chromosome, does occur.549,535–560 The cells stain with toluidine blue, and the basophilic granules can be most striking in myelocytes. In some cases of acute myelomonocytic leukemia associated with t(6;9)(p23;q34), basophils may be increased in the marrow but not in the blood. Because CML with t(9;22)(q34;q11) has the same breakpoint (q34) on chromosome 9 as AML with t(6;9) and both diseases are strongly associated with marrow basophilia, a gene(s) at the breakpoint on chromosome 9 may influence basophilopoiesis.448
Anemia, thrombocytopenia, and blast cells in the blood are present at the time of diagnosis. The blood leukocyte count usually is elevated, and proportions of the cells are basophils. The marrow is cellular with a high proportion of blasts and early and late basophilic myelocytes. Special staining with toluidine blue or Astra blue often is necessary to distinguish basophilic from neutrophilic promyelocytes and myelocytes. Immunophenotyping may show myeloid markers (CD33, CD13) that are not specific. Presence of CD9, CD25, or both is characteristic of basophilic differentiation. Cells may have granules with ultrastructural features of basophils and mast cells.558 Electron microscopy can be useful in identifying basophilic granules in cases where no granules are evident by light microscopy and the phenotype simulates M0.558 Basophilic leukemia can be confused with promyelocytic leukemia if the basophilic early myelocytes are mistaken for promyelocytes.561 On the contrary, promyelocytic leukemia may have basophilic maturation and can be mistaken for basophilic leukemia. However, if the cells contain t(15;17), the disease should respond to ATRA and an anthracycline antibiotic.474,477,478 Prolonged clotting time, intravascular coagulation, and hemorrhage are uncommon presenting features in patients with basophilic leukemia, but are common in patients with promyelocytic leukemia. Coagulopathy can occur after chemotherapy. Cluster headaches, skin rashes, often with an urticarial component, and gastrointestinal symptoms may be present. Elevated blood and urine histamine and urinary methylhistamine levels are characteristic features. Rare cases of a chronic course in BCR-ABL–negative basophilic leukemia preceding the onset of rapid progression have occurred.562 Treatment for acute (Ph-negative) basophilic leukemia is similar to that for other variants of AML.
Mast cell leukemia is a rare manifestation of systemic mast cell disease (Chap. 63).549,563 It can be related to a mutation of the KIT gene.507 The leukemic mast cells are KIT (CD117) positive, naphthol AS-d-chloracetate esterase positive, tryptase positive, myeloperoxidase negative, and CD25-negative.564,565 Plasma tryptase is elevated. In some cases, electron microscopy of the granule-containing cells, which demonstrates the characteristic scroll-like granules of mast cells, may aid in distinguishing basophils from mast cells (Chap. 63). Extensive, apparently reactive, mast cell tissue infiltrations may be provoked by cytokines during the course of AML.566,567
The key laboratory distinctions between acute basophilic leukemia and acute mast cell leukemia are that the cells in the former are naphthol AS-d-chloracetate esterase negative, CD11b positive, CD117 negative or weakly positive, CD123 positive, have no increase in cell or plasma tryptase, and have basophilic-like granules on electron microscopy; whereas, the cells in mast cell leukemia are naphthol AS-d-chloracetate esterase positive, CD11b-negative, CD117-positive, CD123-negative, have an increase in cell and plasma tryptase, and have mast cell-like granules on electron microscopy.549
Chapter 71 discusses histiocytic and myeloid dendritic cell leukemia.
DIFFERENTIAL DIAGNOSIS
Acute leukemia in infants with Down syndrome should be differentiated from TMD (see “Neonatal Myeloproliferation and Leukemia” above). In adults, the term pseudoleukemia has been applied to circumstances that mimic the marrow appearance of promyelocytic leukemia. Recovery from drug-induced or Pseudomonas aeruginosa–induced agranulocytosis is characterized by a striking cohort of promyelocytes in the marrow, which upon inspection of the marrow aspirate or biopsy mimics promyelocytic leukemia.568,569,570
In pseudoleukemia, the platelet count may be normal; the degree of leukopenia often is more profound (<1.0 × 109/L) than usually seen in AML511,512; promyelocytes contain a prominent paranuclear clear (Golgi) zone not covered with granules; and promyelocytes do not have Auer rods.570,571,572 Similar reactions have been reported after granulocyte colony-stimulating factor (G-CSF) administration.573 In patients suspected of having pseudoleukemia, observation for a few days usually clarifies the significance of the marrow appearance, because progressive maturation to segmented neutrophils normalizes the marrow and leads to an increased blood neutrophil count.
In patients with hypoplastic marrows, careful examination of specimens is required to distinguish among aplastic anemia, hypoplastic acute leukemia,350,351,352 and hypoplastic oligoblastic myelogenous leukemia (MDS).574 Leukemic blast cells are evident in the marrow in hypoplastic leukemia, and islands of dysmorphic cells, especially megakaryocytes, are present in hypoplastic oligoblastic leukemia.
Leukemoid reactions and nonleukemic pancytopenias can be distinguished from AML by the absence of leukemic blast cells in the blood or marrow.575 In older children and adults, myeloblasts usually do not constitute more than 2 percent of marrow cells except in patients with a myeloid neoplasm, and the proportion of blast cells usually decreases in the marrow as a result of exaggerated expansion of the myelocyte compartment with neutrophilic leukemoid reactions.
THERAPY
The usual treatment of AML includes an initial program termed the induction phase. Induction may involve the simultaneous use of multiple agents or a planned sequence of therapy called timed sequential treatment. Once a remission is obtained, further treatment is indicated to preserve the remission state. Remission is defined as elimination of the leukemic cell population in marrow as judged by microscopy and flow cytometry and the restitution of marrow hematopoiesis resulting in a normal or virtually normal white cell, hemoglobin, and platelet concentrations in the blood. The postinduction treatment can consist of cytotoxic chemotherapy, HSC transplantation, or low-dose maintenance chemotherapy, depending upon patient performance status and risk factors. If relapse occurs, treatment options may include different chemotherapy regimens, allogeneic HSC transplantation, or other investigational regimens, often as part of a clinical trial.
Most patients with AML should be advised to undergo treatment promptly after diagnosis. Patients younger than 60 years of age have a poorer outcome as the time from diagnosis to treatment lengthens.576 Although remission rates are lower in older patients, a significant proportion enter remission. Occasionally, very elderly patients refuse treatment or are so ill from unrelated illnesses that treatment may be unreasonable. Age per se is not a contraindication to treatment, and septuagenarians and octogenarians who are fit can enter remissions. Treatment can be tailored to the decreased tolerance of older patients, some of whom have a smoldering course (see “Treatment of Older Patients” below). Associated problems, such as hemorrhagic manifestations, severe anemia, or infections, should be treated in parallel.
Orientation of the patient and the family should provide them with an understanding of the disease, the treatment planned, and the adverse effects of treatment, as well as information about long-term prognosis to the extent this can be provided while awaiting cytogenetic and molecular markers. Socioeconomic status and distance from the treatment center have minimal effects on survival in AML,577 but impaired Karnofsky performance status and instrumental activities of daily living score do impact outcomes.578
Pretreatment laboratory examination should include blood cell counts, cytochemistry analysis and immunophenotyping of leukemic cells from blood or marrow, marrow examination including cytogenetic and molecular analyses to include FLT3 ITD, NPM-1, CEBPα, and KIT mutation status in CBF leukemias, if available. If these are not available, they can performed later as required based on AML subtype from a cryopreserved specimen. Blood chemistry studies, chest radiography, electrocardiogram, and determination of partial thromboplastin time, prothrombin time, and fibrinogen level should be obtained. More extensive evaluation of coagulation factors should be made if (1) clotting times are abnormal, (2) bleeding is exaggerated for the level of the platelet count, or (3) APL or acute monocytic leukemia is the phenotype. Early HLA typing is useful so that compatible platelet products can be provided if alloimmunization (Chap. 139) occurs and for patients who will become marrow transplantation candidates (Chap. 23). Herpes simplex virus and cytomegalovirus serotyping may be helpful, especially if transplantation is a consideration. HIV and hepatitis serology is indicated in patients with appropriate risk factors, and patients should have a baseline cardiac scan to determine ejection fraction prior to administration of an anthracycline antibiotic.
A peripherally inserted central catheter or a tunneled central venous catheter should be placed. This access to the circulation facilitates administration of chemotherapy, blood components, antibiotics, and other intravenous fluids and medications. It also permits sampling blood for analysis without patient discomfort or concern about venous access. Meticulous skin care at the catheter exit site is required to minimize tunnel infections. Central venous catheters have become a major source of infection during neutropenia, especially with Gram-positive organisms.579 In some patients with severe coagulopathy such as those with APL, a tunneled catheter may be best deferred to avoid significant bleeding or vessel activation during insertion. In those with neurologic symptoms, a head computed tomographic study or MRI followed by a lumbar puncture should be obtained. Before procedures, adequate platelet counts and control of coagulopathy should be achieved, if possible.
Therapy for hyperuricemia is required if (1) the pretreatment uric acid level is greater than 7 mg/dL (0.4 mmol/L), (2) the marrow is packed with blast cells, or (3) the blood blast cell count is moderately or markedly elevated. Allopurinol 300 mg/day orally should be given. Allopurinol can cause allergic dermatitis and should not be used if the uric acid level is less than 7 mg/dL and the total white cell count is less than approximately 20 × 109/L, as long as hydration is adequate and urine flow is high (>150 mL/h). The dermatitis may appear when antibiotics are instituted. This concurrence may confound the decision to continue antibiotics. Thus, allopurinol should be discontinued after the risk of acute hyperuricosuria or tumor lysis has passed (usually 4 to 7 days). Recombinant urate oxidase (rasburicase) can be used to prevent urate-induced nephropathy. This preparation, although costly, can reduce plasma urate levels by approximately 80 percent within 4 hours of the first drug dose. It is well tolerated, and the recommended dose of rasburicase is 0.2 mg/kg daily for 5 to 7 days intravenously, although shorter courses are usually effective.580
Attention to decreasing pathogen exposure by assiduous hand washing and meticulous care of catheter and intravenous sites is important, especially when the total neutrophil count is less than 0.5 × 109/L. Care of the patient in a single room is advisable to provide privacy during periods of intensive care and to help decrease the risk of exogenously acquired infection until the neutrophil count recovers.
The cytotoxic therapy of AML rests on two tenets: (1) two competing populations of cells are present in marrow—a normal polyclonal and a leukemic monoclonal population; and (2) profound suppression of the leukemic cells to the point they are inapparent in the marrow aspirate and biopsy is required to permit restoration of polyclonal hematopoiesis.581,582 Although these two principles hold in most cases, two deviations from these guidelines are (1) the predisposition of patients with APL to enter remission despite cellular posttherapy marrow583 and (2) the rare presence of monoclonal hematopoiesis in some cases of AML during remission (see “Results of Treatment” below). AML is a heterogeneous disease, and subgroups with different prognosis can be identified. In the future, incorporation of knowledge about the biology of the particular AML subtype may be utilized for adapted therapies, but at present, all subtypes of AML classified by cytogenetics or molecular changes with the exception of APL are approached similarly during induction, and often induction therapy must be started before knowledge of cytogenetic and molecular factors is available.584
The goal of induction therapy in AML is achievement of complete remission (<2 percent blasts in the marrow), a neutrophil count greater than 1000/μL, and a platelet count greater than 100,000/μL. An International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards has redefined outcomes in an effort to standardize reporting and comparison of data (see “Course and Prognosis: Results of Treatment: Definition of Remission” below).585 Other treatment guidelines have been published.586,587 The majority of adults enter remission with standard induction therapy, but for patients with high-risk disease, consideration can be given to an experimental approach, and complete remission rates do not reach 100 percent, so clinical trial participation can be considered during induction chemotherapy. How durable a complete remission will be attained in an individual patient often is difficult to predict at diagnosis. Gene-expression profiling can separate some patients into prognostic groups that may indicate patients with a high risk of not responding to standard approaches.105
Anthracycline Antibiotic or Anthraquinone and Cytarabine Current standard induction treatment for non-APL AML involves drug regimens with two or more agents that include an anthracycline antibiotic or an anthraquinone and cytarabine (see “Special Therapeutic Considerations: Acute Promyelocytic Leukemia” below for therapy of APL).588–617 Remission rates in the studies cited range from approximately 55 to 90 percent in adult subjects, depending on the composition of the population treated (Table 88–6). The two most important variables are the age of the patients and the proportion of patients with therapy-induced leukemia or an antecedent clonal myeloid disease. In the studies listed in Table 88–6, the median age of the patient populations was much younger (approximately 50 years) than the median age of the population of AML patients at large (approximately 70 years); thus the results cannot be generalized (see “Treatment of Older Patients” below). A combination of anthracycline and cytarabine has been the standard induction therapy since 1973.11,12 A now classic standard induction regimen is cytarabine 100 mg/m2 daily by continuous infusion on days 1 through 7 and daunorubicin at 45 to 90 mg/m2 on days 1 through 3, the “7 plus 3” regimen. Dose or schedule modulation of the anthracycline or cytarabine, addition of other agents such as etoposide, in various schedules of administration, represent attempts to improve upon results obtained with “7 plus 3” therapy.
| Cytarabine | Anthracycline Antibiotic ± Another Agent | No. of Patients | Age Range in Years (Median) | Complete Remissions (%) | Year of Report | Reference |
|---|---|---|---|---|---|---|
| 100 mg/m2, days 1–7 | DNR 50 mg/m2 days 1–5 | 407 | 15–64 (47) | 77.5 | 2011 | 596 |
| 100 mg/m2, days 1–7 | IDA 12 mg/m2 days 1–3 | 525 | 15–64 (47) | 78.2 | 2011 | 596 |
| 100 mg/m2, days 1–7 | DNR 45 mg/m2, days 1–3 | 330 | 17–60 (47) | 57 | 2009 | 593 |
| 100 mg/m2, days 1–7 | DNR 90 mg/m2, days 1–3 | 327 | 18–60 (48) | 71 | 2009 | 593 |
| 200 mg/m2, days 1–7 | DNR 60 mg/m2, days 1–3 | 200 | 16–60 (45) | 72 | 2004 | 611 |
| 200 mg/m2, days 1–7 | DNR 60 mg/m2, days 1–3 Cladribine 5 mg/m2, days 1–5 | 200 | 16–60 (45) | 69 | 2004 | 611 |
| 200 mg/m2 twice per day for 10 days (some in this report received FLAG-IDA vs. H-DAT) | DNR 50 mg/m2, days 1, 3, 5 Thioguanine 100 mg/m2 twice per day, days 10–20 Gemtuzumab ozogamicin 3 mg/m2, day 1 | 64 | 18–59 (46.5) | 91 | 2003 | 609 |
| 3 g/m2 every 12 h for 8 doses | 60 mg/m2 DNR daily for 2 days | 122 | Adults | 80 | 2000 | 603 |
| 100 mg/m2 daily for 7 days (2 courses always given) | IDA 12 mg/m2 daily for 3 days | 153 | NR | 63 | 2000 | 589 |
| 500 mg/m2 by continuous infusion, days 1–3, 8–10 | Mitoxantrone 12 mg/m2 for 3 days Etoposide 200 mg/m2 days 8–10 | 133 | 15–70 (43) | 60 | 1996 | 606 |
| 100 mg/m2 daily for 7 days | DNR 45 mg/m2 for 3 days | 113 | NR (55) | 59 | 1992 | 588 |
| 100 mg/m2 daily for 7 days | IDA 13 mg/m2 for 3 days | 101 | NR (56) | 70 | 1992 | 588 |
Choice of Anthracycline Development of drug resistance is reduced with idarubicin relative to other anthracyclines. Idarubicin does not induce P-glycoprotein expression, but daunorubicin, doxorubicin, and epirubicin do.590 Idarubicin 12 mg/m2 gives better complete remission rates in younger adults than does daunorubicin 45 mg/m2, each given for 3 days. Amsacrine, aclarubicin, and mitoxantrone give improved results over standard-dose daunorubicin. In older adults, mitoxantrone may reduce cardiotoxicity, but this is controversial.591 In two randomized studies, high-dose daunorubicin (90 mg/m2) for 3 days resulted in superior complete remission rates as compared to 45 mg/m2 for 3 days when combined with cytarabine.592,593 When idarubicin 12 mg/m2 was compared to daunorubicin 80 mg/m2 for 3 days in patients 50 to 70 years of age, the remission rate with idarubicin was 83 percent compared to 40 percent with daunorubicin.594 Another analysis of idarubicin compared with high-dose daunorubicin in patients with AML showed idarubicin to result in a higher remission rate but not overall survival.595 In contrast, a randomized study showed no difference in remission and long-term efficacy between idarubicin 12 mg/m2 daily for 3 days as compared to daunorubicin, 50 mg/m2 daily for 5 days.596 In light of these studies, many therapists, when using daunorubicin, use the 90 mg/m2 dose for 3 days in younger patients, and this is in keeping with the current National Comprehensive Cancer Network (NCCN) guidelines.597 This benefit of higher dose applies only to younger and favorable or intermediate-risk patients.593 Dexrazoxane may be given during induction to reduce the risk of cardiotoxicity in patients at higher than usual risk because of a history of coronary artery disease or congestive heart failure, but this is rarely used in adults.598 Other regimens that incorporate fludarabine with cytarabine can be used in those patients for which an anthracycline would not be ideal.
High-Dose versus Standard-Dose Cytarabine High-dose cytarabine does not increase complete remission rates and increases toxicity compared to conventional doses, especially in older patients (for doses of these regimens, see “Intensive Consolidation Therapy” below). Patients receiving high-dose cytarabine have more leukopenia, thrombocytopenia, gastrointestinal problems, and eye toxicity. Disease-free survival and overall survival may be better than that achieved with standard therapy, leading some investigators to suggest use of high-dose therapy for induction in patients younger than age 50 years, but this approach is not a standard one, and these studies do not take into account the role of high-dose cytarabine in postremission therapy.599 Some studies show that marrow blast clearance is higher after an induction with high-dose cytarabine and that there is an improvement in disease-free survival for patients 50 years of age or younger.600 When high-dose cytarabine was compared to intermediate doses in induction therapy, no improvement in outcome was noted, and higher incidences of grades 3 and 4 toxic effects were noted.600 A trial in younger patients with multiple arms; fludarabine, high-dose cytarabine, and G-CSF (FLAG regimen) with idarubicin resulted in a higher remission rate than did standard daunorubicin plus cytarabine with or without etoposide. Relapse rates were also less with the high-dose cytarabine induction (38 vs. 55 percent).601 A superior remission rate and survival was achieved in younger patients (<46 years) induced with a regimen containing high-dose cytarabine, 82 versus 76 percent rate of remission and a 52 versus 43 percent rate of overall survival. These differences were also seen in secondary AML cases and in those with FLT3-ITD mutations.602 Also, complete remission rates of greater than 60 percent have been noted with high-dose cytarabine in patients with poor-risk cytogenetics.603,604
Timed Sequential Therapy and Other Drugs Timed sequential therapy, which uses agents in a scheduled sequence rather than concurrently, may prolong remission duration.605,606,607 Timed sequential chemotherapy combining mitoxantrone intravenously (IV) on days 1 to 3, etoposide IV on days 8 to 10, and cytarabine IV on days 1 to 3 and 8 to 10 resulted in a complete remission in 60 percent of patients, but treatment-related death in 9 percent of patients. Median disease-free survival was 9 months.605
Adding ATRA,608 gemtuzumab ozogamicin,609 fludarabine,610 cladribine or topotecan611,612 to induction regimens has not improved results significantly. A recent randomized study showed that the addition of the purine analogue cladribine, but not fludarabine, to daunorubicin and cytarabine improved the remission rate and prolonged survival in patients younger than 60 years of age.613 The addition of bortezomib to daunorubicin and cytarabine in those 60 to 75 years of age resulted in a remission rate of 65 percent. This was a single-arm trial with dose escalation of bortezomib.614 There are preliminary reports suggesting that the addition of gemtuzumab ozogamicin to standard induction chemotherapy may increase disease-free survival in patients with low- and standard-risk cytogenetic abnormalities,615 and inhibitors of FLT3 ITD are now being examined, but no data are available regarding utility of this approach.616 A recent prospective comparison of five different treatment strategies, adjusted for differences in prognostic characteristics, did not show clinically relevant differences in outcome when compared to a standard cytarabine and anthracycline containing arm.617 Thus, the standard practice guideline for AML, other than promyelocytic leukemia, recommends standard-dose cytarabine plus an anthracycline antibiotic as treatment.587
Hematopoietic Cytokines to Enhance Chemotherapy G-CSF and granulocyte-monocyte colony-stimulating factor (GM-CSF), when used in untreated leukemia, can increase the percentage of leukemic cells in the DNA synthetic phase, resulting in blast population expansion during short-term administration. This process could render the cells more sensitive to simultaneous chemotherapy, but clinical benefit from growth-factor priming has not been observed618,619 despite an increased ratio of intracellular cytosine arabinoside triphosphate to deoxycytidine-5′-triphosphate and enhanced cytarabine incorporation into the DNA of AML blasts.619 Remission rates or overall survival did not differ among adult patients who received cytarabine plus idarubicin or cytarabine plus amsacrine with or without G-CSF given concurrently, but relapse rates decreased in patients who received G-CSF.620 GM-CSF priming in a younger patient group treated with timed-sequential therapy increased complete remission rates but did not impact overall survival.621 Thus, these growth factors are not generally considered useful as enhancers of chemotherapy. A study did, however, suggest that an improved event-free survival and overall survival was noted in patients treated with high-dose cytarabine during remission induction,622 and complete remissions have occurred in hypoplastic AML after G-CSF treatment without chemotherapy.623
Reinduction Therapy Patients who have persistent leukemia after the first course of induction chemotherapy generally are given the same regimen a second time. The effect is usually assessed by marrow aspirate and biopsy 7 to 10 days after completion of chemotherapy (the “14-day marrow” examination). For those with hypocellular marrow and no evidence of residual leukemic blasts, recovery of normal counts is awaited, and for those with a hypocellular marrow and a small number of residual blasts, additional therapy may be delayed until count recovery or until another marrow assessment. For those with significant amounts of leukemic cells remaining, repeating the original induction therapy or use of a high-dose cytarabine regimen can be considered. The patient’s long-term outcome is worse if two courses of treatment are required, even if a complete remission is achieved. Approximately 40 percent of patients with persistent AML after one course of induction therapy have a complete remission after a second course,624 and disease-free survival at 5 years is approximately 10 percent. In some European centers, two courses of induction chemotherapy are given routinely, but the impact on remission rates or overall survival is uncertain.625 The longer the time to remission after the first induction therapy, the shorter the duration of disease-free survival.626 High-risk cytogenetic abnormalities, antecedent hematologic disorders, and other poor prognostic factors can be used to assign nonresponders to an experimental chemotherapy regimen designed to treat refractory disease, rather than repeating induction therapy. In one study, overall response to reinduction was 53 percent. Those patients with poor risk cytogenetics and those with a marrow blast percentage of 60 percent or greater following the 7-plus-3 regimen induction treatment were found to have a low probability of achieving a complete remission with reinduction.627 Mortality during induction therapy correlates with age628 and, perhaps, leukocyte count.629
Hyperleukocytosis Patients with blast counts greater than 100 × 109/L require prompt treatment to prevent the most serious complications of hyperleukocytosis: intracranial hemorrhage or pulmonary insufficiency. Hydration should be administered promptly to maintain urine flow greater than 100 mL/h/m2. Cytoreduction therapy can be initiated with hydroxyurea 1.5 to 2.5 g orally every 6 hours (total dose 6 to 10 g/day) for approximately 36 hours. Appropriate remission-induction therapy should be initiated as soon as possible after the leukocyte count has been decreased significantly. Simultaneous leukapheresis can decrease blast cell concentration by approximately 30 percent within several hours331,630,631 without contributing to uric acid or cellular phosphate release. Leukapheresis may improve acute disturbances resulting from the vascular effects of blast cells, but the procedure may not alter the long-term outcome with current therapeutic programs.339,340,630 Inhaled nitric oxide may improve the hypoxemia related to hyperleukocytosis.631
Antibiotic Therapy Pancytopenia is worsened or induced shortly after treatment is instituted. Absolute neutrophil counts less than 100/μL (0.1 × 109/L) are expected and are a sign of effective drug action. The patient usually becomes febrile (>38°C), often with associated rigors. Cultures of urine, blood, nasopharynx, and, if available, sputum should be obtained. Because the inflammatory response is blunted by severe neutropenia and monocytopenia, evidence of exudates on physical examination or imaging studies may be minimal or absent. Antibiotics should be started immediately after cultures are obtained.632 Chapter 24 describes antibiotic usage in the setting of intensive chemotherapy. Infections remain a major cause of therapy-associated morbidity and mortality.633,634 Gram-positive bacterial isolates now outnumber Gram-negative organisms.634 Cultures are often negative, but if fever and other signs are present, antibiotic therapy should be continued until neutrophil recovery.
Some centers use prophylactic antibacterial, antifungal, and/or antiviral antibiotics, whereas other centers do not. Antifungal prophylaxis can consist of low-dose amphotericin or azoles such as fluconazole, itraconazole, posaconazole, or voriconazole.635,636 In a randomized study in patients undergoing induction therapy, posaconazole was more effective in preventing invasive fungal infections than fluconazole or itraconazole.637 Voriconazole was not included in the comparison. Acyclovir, valacyclovir, or famciclovir prophylaxis during remission-induction therapy of patients with AML does not affect the duration of fever or the need for antibiotics. The incidence of bacteremia is not reduced, but acute oral infections are less severe.638 Liposomal amphotericin, the caspofungins and azoles are available for treatment of established fungal infections.639 Some centers use outpatient supportive therapy, including oral antimicrobials, immediately after induction therapy administration in adult AML.640
Hematopoietic Growth Factors to Treat Cytopenias Cytokine therapy as an adjunctive treatment for AML remains controversial.641 GM-CSF and G-CSF accelerate neutrophil recovery; neither GM-CSF nor G-CSF reproducibly decreases major morbidity or mortality. However, one study has shown decreased mortality from fungal infections in older patients.642 Use of cytokines during periods of cytopenia following induction therapy is safe, and nearly all trials have shown a modestly reduced duration of severe neutropenia with a variable effect on the incidence of severe infections, antibiotic usage, and duration of hospital stays. Although no increase in relapse has been noted when growth factors are started after completion of chemotherapy, no consistent enhancement of remission, event-free survival, or overall survival has been noted.643 Therefore, the cost-effectiveness and clinical effectiveness of growth factor usage is doubtful. Also, growth factor usage can cloud marrow interpretation when used during induction.
Component Transfusion Therapy Red cell transfusions should be used to keep the hemoglobin level greater than 7.0 g/dL, or higher in special cases (e.g., symptomatic coronary artery disease; Chap. 138). Platelet transfusions should be used for hemorrhagic manifestations related to thrombocytopenia and prophylactically if necessary to maintain the platelet count between 5 × 109/L and 10 × 109/L.644 Patients without coagulation abnormalities, anticoagulant use, sepsis, or other complications usually can maintain hemostasis with platelet counts of 5 to 10 × 109/L. Initially, random donor platelets can be used, although single-donor platelets or HLA-matched platelets may be preferable products and should be tried if random-donor platelets do not raise the platelet count significantly A no-prophylaxis platelet-transfusion strategy for blood cancers has been examined, but data support the need for prophylactic platelet transfusions.645 Family members may be effective donors, if allogeneic HSC transplantation is not being considered (Chap. 139). There are data that fever should result in increasing the platelet count used as a transfusion threshold, and there is some suggestion that higher hemoglobin values protect against bleeding related to thrombocytopenia.646
All red cell and platelet products should be depleted of leukocytes, and all products, including granulocytes for transfusions, should be irradiated to prevent transfusion-associated graft-versus-host disease (GVHD) in this immunosuppressed population (Chaps. 138 and 139).
Granulocyte transfusion should not be used prophylactically for neutropenia but may be used in patients with high fever, rigors, and bacteremia unresponsive to antibiotics, with blood fungal infections, or with septic shock. G-CSF administration to a volunteer donor increases neutrophil yield fourfold and results in posttransfusion blood neutrophil increments for more than 24 hours after transfusion.647 There is still ambiguity about the usefulness of this approach. GM-CSF administration may be warranted for treatment of major fungal infections (Chap. 24).
Jehovah’s Witnesses and others who refuse blood product support can survive tailored chemotherapy.648 In general, phlebotomy is minimized, and antifibrinolytics, hematinics, and growth factors are used to support such patients during severe cytopenias.
Therapy for Hypofibrinogenemic Hemorrhage Patients with evidence of intravascular coagulation (Chap. 129) or exaggerated primary fibrinolysis (Chap. 135) should be considered for platelet and fresh-frozen plasma administration before antileukemic therapy is started. Infusion of cryoprecipitate can be used for fibrinogen levels under approximately 125 mg/dL. If the findings are equivocal, patients should be monitored closely with measurements of fibrinogen levels, fibrin(ogen) degradation products, D-dimer assay, and coagulation times. Intravascular coagulation or primary fibrinolysis may occur in patients with APL and acute monocytic leukemia, but also may occur in occasional patients with other AML subtypes.
Management of Central Nervous System Disease CNS disease occurs in approximately one in 50 cases at presentation.649 Prophylactic therapy usually is not indicated, but examination of the spinal fluid after remission should be considered in (1) monocytic subtypes,508 (2) cases with extramedullary disease, (3) cases with inversion 16254 and t(8;21)263,266 cytogenetics, (4) CD7- and CD56-positive (neural-cell adhesion molecule) immunophenotypes,650 and (5) patients who present with very high blood blast cell counts. In these situations, the risk of meningeal leukemia or a brain myeloid sarcoma is heightened, but prophylactic intrathecal chemotherapy is not recommended if high-dose cytarabine is used for consolidation. Patients who present with neurologic symptoms should have a head computed tomogram or MRI to rule out hemorrhage or mass effect. If negative, a lumbar puncture should be performed. Treatment of meningeal leukemia can include high-dose intravenous cytarabine (which penetrates the blood–brain barrier), intrathecal methotrexate, intrathecal cytarabine, cranial radiation, or chemotherapy and radiation in combination.649 If CNS leukemia is present, intrathecal therapy is often given twice per week until blasts are cleared, and then once per week for 4 to 6 weeks. This therapy can be accomplished via the lumbar puncture route or through placement of an Ommaya reservoir. If there is a mass present, radiation or high-dose cytarabine with glucocorticoids can be considered.651 Systemic relapse commonly follows relapse in the meninges, and concurrent systemic treatment usually is indicated. Long-term success is unusual unless allogeneic HSC transplantation is possible. Unless the patient has neurologic symptoms, lumbar puncture generally is deferred until blood blast cells have cleared. No consensus exists on a trigger for platelet transfusion in adults with AML undergoing lumbar puncture, but a platelet count less than 20 × 109/L has been proposed as such a trigger,652 but many therapists use a higher platelet count (e.g., 50 × 109/L) as a safety threshold for lumbar puncture.
Management of Nonleukemic Myeloid Sarcoma Some patients present with myeloid (granulocytic) sarcomas without evidence of leukemia in the blood or marrow (see “Myeloid [Granulocytic] Sarcoma” earlier). Myeloid sarcoma may be the presenting finding in approximately 1 percent of patients with AML. Such patients should receive intensive AML induction therapy.262 Intensive therapy results in a longer nonleukemic period than patients who have undergone surgical resection or resection followed by local irradiation.250 Whether such patients should undergo allogeneic HSC transplantation in first remission irrespective of other factors has not been determined.653,654 Median relapse-free survival is approximately 12 months after AML-type chemotherapy.262 Patients with trisomy 8 have poorer survival rates.260
General Considerations Postremission therapy is intended to prolong remission duration and overall survival, but no consensus exists regarding the best approach. Postremission chemotherapy that does not produce profound prolonged cytopenias, closely simulating intensive induction therapy, has produced on average only slight prolongation of remission or life. Regimens that fall between these intensities have been used, with equivocal results. Intensive consolidation therapy after remission results in a somewhat longer remission duration and, more significantly, a subset of patients who have a remission of more than 3 years. The issue of postremission therapy and its impact is complicated by the large proportion of patients with AML who are older than 60 years of age and have limited tolerance for intensive therapy. In addition, a very small pool of leukemic stem cells may sustain the process, and elimination of these cells may require approaches other than intensive chemotherapy, especially in adults.
Several randomized trials have studied whether AML patients in first remission should receive consolidation chemotherapy alone, autologous transplantation, or allogeneic HSC transplantation, without reaching a consensus. Allogeneic transplantation was compared to autologous transplantation using unpurged marrow and two courses of intensive chemotherapy in 623 patients who had a complete remission after induction chemotherapy.655 Disease-free survival was 53 percent at 4 years for those receiving allogeneic transplantation, 48 percent for those receiving autologous transplantation, and 30 percent for patients receiving intensive chemotherapy. Overall survival after complete remission was similar in all three groups because patients who relapsed after chemotherapy could be rescued with allogeneic HSC transplantation. No significant difference in the 4-year disease-free survival between allogeneic HSC transplantation (42 percent) and other types of intensive postremission therapy (40 percent) has been found.656 In another study, only patients younger than 35 years of age with poor-risk cytogenetics had improved disease-free survival if they had a sibling donor and underwent allogeneic transplantation (43.5 percent vs. 18.5 percent at 4 years).657 Thus, in several studies, the early mortality after allogeneic HSC transplantation and the chemotherapy-induced remissions in patients who relapse following autologous transplantation or chemotherapy have led to comparable overall survival rates. However, leukemia-free survival was greater after allogeneic transplantation.658 In the last decade, treatment-related mortality from transplantation has declined and matched unrelated donor transplantations are as effective as those from a matched sibling donor, so currently, transplantation is recommended for all but good-prognosis patients (CBF leukemias or those with NPM1 mutation without a FLT3 mutation).659 A Markov decision analysis has shown that patients treated with allogeneic HSC transplantation have a longer life expectancy compared with those treated with chemotherapy among patients with an intermediate- or unfavorable-risk prognosis.660 A prospective matched-pairs analysis has also concluded that allogeneic HSC transplantation is the most effective postremission therapy for AML, especially for those 45 to 59 years of age and/or with high-risk cytogenetics.661 When quality of life was measured for patients in complete remission for 1 to 7 years, those treated with chemotherapy had the highest quality of life, whereas those who underwent allogeneic HSC transplantation had the lowest.662
The decision to utilize autologous or allogeneic HSC transplantation or high-dose cytarabine alone for consolidation should be individualized, based on the patient’s age and other prognostic factors, such as high-risk cytogenetic findings and antecedent hematologic disease. Patients with good-risk cytogenetics should receive up to four cycles of high-dose cytarabine. Patients with poor-risk cytogenetics should be considered for allogeneic HSC transplantation as soon as feasible. A meta-analysis has also shown that compared with nonallogeneic therapies, allogeneic HSC transplantation has superior relapse-free survival and overall survival for cases of AML classified intermediate and poor-risk, but not for cases considered good-risk AML in first remission.663
Intensive Consolidation Therapy For patients who do not receive high-dose chemotherapy with autologous or allogeneic transplantation in first remission, consolidation chemotherapy regimens containing high-dose cytarabine provide better results than intermediate-dose cytarabine,664,665 but these regimens are not universally accepted.666 Patients who are to have allogeneic HSC transplantation do not require four cycles of high-dose cytarabine, and may not benefit from even one, if a donor is readily available.667 RAS mutations are associated with benefit from high-dose cytarabine therapy.668 Patients with CBF leukemias such as t(8;21) also have particularly favorable responses to repetitive cycles of high-dose cytarabine. In patients who received three or more cycles, a relapse rate of 19 percent was reported.669
Other regimens, such as those containing gemtuzumab ozogamicin and fludarabine, have been used in postremission therapy, but whether they provide benefit over use of high-dose cytarabine has not been studied.670 Long-term disease-free survival at 5 years generally is approximately 30 percent when two to four cytarabine-containing regimens are administered.671,672 Adding mitoxantrone or amsacrine to high-doses cytarabine has not improved treatment outcomes in consolidation,673 and timed sequential chemotherapy used in consolidation did not improve outcome as compared with high-dose cytarabine.674 Most centers use four cycles of therapy. A cycle is 3 g/m2 twice daily on days 1, 3, and 5, providing six doses per cycle, with cycle durations dependent on normal blood count recovery. The optimal number of cycles for this therapy is not known.675 High-dose cytarabine can be administered at a dose of 3 g/m2 in a 1- to 3-hour intravenous infusion every 12 hours for up to 6 days (12 doses), but this schedule is almost never used because of its toxicity. There is some evidence that two cycles of intermediate-dose cytarabine (1 g/m2 every 12 hours for 6 days) may be a viable alternative to the 3 g/m2 for six doses schedules.676 When 36 g/m2 total dosing was compared with 12 g/m2
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


