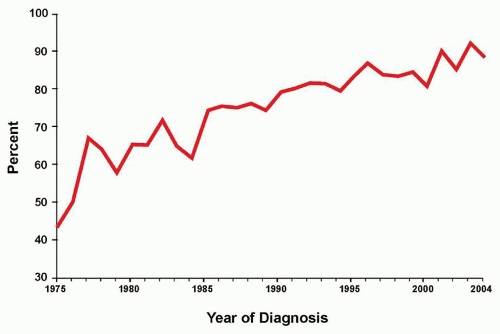
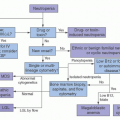
As therapy remains the single most important factor to influence outcome, the relative prognostic significance of characteristics at diagnosis (
Table 76.2) varies for the different treatment strategies delivered by national and international investigators. Consequently, evolving combinations of clinical, laboratory, and response variables have been used to guide therapeutic intensity over the years by those who conduct clinical trials for childhood ALL, overall with increasingly successful results. Indeed, the Children’s Oncology Group (COG) recently reported that sequential outcomes of over 21,000 patients (age 0 to 22 years) enrolled on COG trials between 1990 and 2005 culminated in survival rates of 90.4% for those children treated between 2000 and 2005 (
Fig. 76.3).
5 Concomitantly, studies conducted on long-term survivors of childhood cancer reveal that up to 62% of patients suffer from at least one chronic condition and 27% suffer from a grade 3 or 4 condition.
39 The goal of modern risk stratification, then, is to cure patients while minimizing acute and late toxicity, otherwise known as maximizing the therapeutic benefit.
Differences among the risk classification criteria used to deliver varied intensities of therapy by disparate groups have made accurate comparisons of outcomes difficult, if not impossible. To overcome this obstacle, an international workshop held in Rome in 1985
36 and a subsequent consensus workshop, sponsored by the United States National Cancer Institute in 1993, led to the development of common risk-based criteria.
40 In 1993, the NCI-Rome risk criteria were agreed upon by leaders among the major clinical consortia in an attempt to analyze different datasets using two of the most powerful predictors of outcome across all studies for B-lineage ALL: age and presenting WBC count at diagnosis. The standard-risk group (NCI-SR) includes those patients with B-precursor ALL ages 1 to 9 years with a WBC count less than 50 × 10
9/L. In 1995, NCI-SR patients were estimated to have EFS rates of approximately 80% and the remaining high-risk (NCI-HR) patients were estimated to have EFS of approximately 65%.
In the current era, approximately 85% of children with ALL will present with B-lineage disease and the remainder will present with T-lineage ALL. Two thirds of patients with B-lineage ALL present with NCI-SR features. Notably, the constellation of age and WBC count cannot reliably be used to predict outcome for patients with T-cell ALL, likely reflecting the different genomic and subsequent biochemical landscape of these malignancies. However, the unique biologic features of T-cell disease, the failure of prognostic factors effective in precursor B-cell ALL to predict outcome in T-cell ALL,
41 the differing patterns of MRD between B- and T-lineage disease,
42 and the advent of T-cell-specific therapies
43,44 all argue for the separate stratification of T-cell ALL in classification and treatment, an approach that has been adopted by many cooperative childhood leukemia groups.
In addition, genomic factors appear to be increasingly relevant for older adolescents and young adults with B-lineage ALL, and recent discoveries may affect not only prognosis but therapy. Finally, for patients less than 1 year of age with B-lineage ALL, the most important adverse prognostic factor remains the presence or absence of a rearrangement (not deletion) in the mixed lineage leukemia (MLL) gene, which is most commonly seen in infants less than 3 months of age.
The successful intensification of therapy for high-risk ALL has weakened the power of many historically used adverse predictive factors (
Fig. 76.4). Although the NCI-Rome criteria represented a major advance in risk classification of childhood ALL, these criteria did not adequately take into account more current biologic features, such as molecular genetic alterations, that likely contribute to the age and presenting WBC count. In addition other powerful predictors of outcome include early response to therapy, as measured by either flow cytometric or polymerase chain reaction methodologies to detect minimal residual disease (MRD). In particular, the strongest predictor of outcome for B-lineage disease across all studies in multivariable analyses is the presence or absence of MRD at the end of the first or second phase of therapy. Indeed, many of the large international consortia have devised complex algorithms to direct therapy, the separate components of which will be presented and then reintegrated into current risk stratification schemes used by the major consortia groups.
Molecular Genetic Alterations Contributing to Modern Risk Stratification
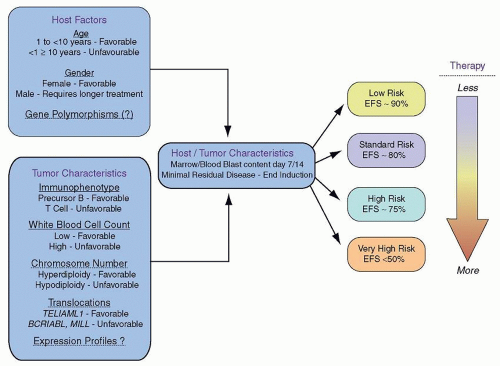
The prognostic significance of various genetic features of leukemic blasts has long been known (
Fig. 76.5). In particular, very high risk features for relapse include the presence of the
BCR/ABL, or Philadelphia chromosome (3% of B-lineage patients), extreme hypodiploidy (less than 44 chromosomes or a DNA index of less than 0.81; 3% of B-lineage patients), and intrachromosomal amplification of 21 (iAMP21; 1% to 3% of B-lineage patients).
45,46,47,48,49,50 Recent analyses of 610 patients with Ph-positive ALL who did not receive tyrosine kinase inhibitor (TKI) therapy and who were treated between 1995 and 2005 revealed improved 7-year overall survival in contrast to those who were treated between 1986 and 1996 (OS 44.9 ± 2.2% vs. 36.0 ± 2.0%,
P = 0.017).
46 Therapeutic benefit was seen with the use of maximally intensive, myeloablative HSCT. However, the addition of continuous imatinib, a TKI to the backbone of dose-intensive chemotherapy in COG AALL0031 demonstrated for the first time that adding a targeted agent could provide a survival benefit for a disease that was otherwise only optimally treated using HSCT.
45,51 (See Philadelphia chromosome-positive ALL in “Unique Patient Subgroups.”)
Similar improvements in outcome have not been established for patients diagnosed with hypodiploid ALL. Identified through either characteristic karyotype findings or DNA indices, rare cases of near-haploid ALL may escape detection due to the presence of a double hypodiploid clone, leading to “masked” near haploidy or hypodiploidy. However, in these cases, one clue may be the retention of two or four copies of chromosome 21, as this chromosome is never lost in a true hypodiploid genome. Experienced cytogenetic review can aid in the identification of these cases. Recent discoveries in the molecular genetics of hypodiploid ALL reveal a high frequency of genes regulating the RAS pathway (
NF1, NRAS, KRAS, PTPN11, FLT3, and
PAG1),
IKZF3 (encoding the lymphoid transcription factor), and a histone gene cluster at 6p22 in near haploid ALL (<32 chromosomes), and a similarly high proportion of
TP53,
RB, and
IKZF2 mutations in low hypodiploid ALL (32 to 39 chromosomes).
52 Currently, although no specific targeted agents are known directly to inhibit the various perturbed pathways listed above, additional work is being performed in preclinical models in order to further identify optimal therapeutic strategies.
Intrachromosomal amplification of a region on chromosome 21 has also been associated with poor prognosis by several groups, with EFS rates below 60%.
48,49,50 The detection of iAMP21 is relatively easy to distinguish using fluorescence-in-situ hybridization assays which are frequently performed to detect the
ETV6/RUNX1 fusion gene. In these cases, not only is the favorable
ETV6/RUNX1 fusion absent, but ≥5
RUNX1 (4 or more on a derivative chromosome) signals are present, signifying this amplification.
Although the poor outcomes of infants harboring
MLL translocations have long been established,
53 the outcomes for older patients have been controversial. Recent data from the COG on the outcomes of 155 patients older than 1 year (2.2% of the entire population studied) reveal that the majority of these patients present with NCI-HR features (4.3% of NCI-HR vs. 1.1% of NCI-SR patients). More than half of the
MLL rearrangements were t(4;11), and the majority of the remainder were t(11;17) (16%), t:3;11) or t(11;19)(13% each). Although
MLL-rearranged patients did poorly overall compared with non-
MLL-rearranged patients, the presence of an
MLL rearrangement was not statistically significant in a multivariable model. Instead, among
MLL-rearranged patients who had a rapid response to induction therapy, EFS rates approached 81.1%, considerably better than anticipated. It should be noted, however, that in these clinical trials (AALL0331 and AALL0232), patients with
MLL rearrangements were treated with maximally intensive augmented BFM therapy, thus illustrating the concept of “treating” away adverse prognostic factors.
Likewise, several leukemic blast genetic factors have been shown to be strong predictors of favorable outcomes among many consortia, including the
ETV6/RUNX1 (formerly
TEL/AML1) translocation and the presence of trisomy 4 and 10. Fortunately, these are some of the most common genetic features that occur in childhood ALL, with
ETV6/RUNX1 occurring in approximately
25% and trisomy 4 and 10 occurring in approximately 18% of any risk childhood ALL. Some groups do risk stratify based on the presence of either or both of these factors; however, a multivariate analysis performed on >2,000 COG 9900 patients revealed that double trisomy of 4 and 10 retained independent significance whereas
ETV6/RUNX1 did not. However, among patients with these genetic features and a rapid response to induction therapy within the first week, more than 98% were long-term survivors, leading COG investigators to incorporate response with leukemia-specific genetic factors in their current approaches to risk stratification.
Additional genetic factors that may affect risk stratification and therapy in the future, and are thus beyond the scope of this chapter include those patients who have been recently identified to display a Ph-like gene expression profile (GEP) without harboring
BCR/ABL (15% of NCI HR patients). These patients frequently harbor genomic rearrangements in
CRLF2 with subsequent overexpression of the TSLP receptor, JAK family mutations, or novel kinase fusion genes. It is important to note that the Ph-like GEP is an independently poor prognostic factor in multivariable analyses when analyzed among NCI HR patients treated on AALL0232, and thus supports identifying alternative approaches, similar to the TKI+ chemotherapy approach used for Ph+ ALL, to maximize outcomes for these patients.
53a
Response to Induction and Minimal Residual Disease
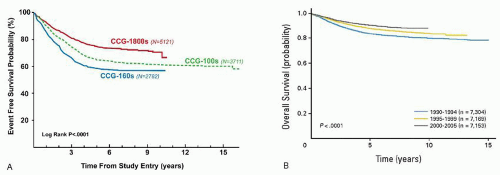
Response to induction has long been valued as a predictor of outcome. In 1983, investigators in the Berlin-Frankfurt-Münster (BFM) group evaluated the use of a 7-day prophase of prednisone and a single dose of intrathecal methotrexate to determine which patients should receive more intensive therapy. Prednisone poor responders (PPR), or those who had more than 1,000 circulating blasts day 8 of therapy, comprised 10% of their population and had an outcome that was 50% as good as the prednisone good responders (PGR). In subsequent protocols beginning in 1986, therapeutic questions were asked of the PPR versus PGR group, and this continues currently. In parallel efforts, the legacy
Children’s Cancer Group (CCG) also determined that delivering augmented therapy to those NCI HR patients with a poor bone marrow morphology response (day 7 M2 (5% to 25% blasts by morphology) or M3 (>25% blasts by morphology) improved their outcome.
54The technological ability of measuring response to induction using residual blasts grew out of these initial observations. The ability to detect small numbers of leukemic cells in peripheral blood or bone marrow samples from patients in clinical remission by highly sensitive methods such as the polymerase chain reaction
55,56,57,58,59 or multiparametric flow cytometry
60,61,62 has enabled the monitoring of MRD, with the possibility of intensifying therapy for patients at higher risk of relapse. A number of studies have demonstrated the ability to identify impending relapse in subsets of patients, based on correlation of detection of MRD during or following completion of therapy.
55, 56, 57 and 58,63 Although variations in the sensitivity and specificity of the methods used to detect and to quantify MRD, as well as questions raised by the unexpected finding of persistent genetic abnormalities related to the leukemic clone in patients in apparent sustained remissions
58,64,65 initially delayed the implementation of MRD assessment in patient management, recent studies using improved methodologies have shown that detection of MRD by either multiparametric flow cytometry analysis of leukemia-associated antigens
66,67,68 or polymerase chain reaction detection of leukemia-specific immunoglobulin and T-cell receptor rearrangements
61,69, 70 and 71,72 definitively correlates with outcome. Indeed, the presence of MRD early in the treatment of ALL patients is highly prognostic of outcome and currently is a key variable to determine the intensity of postinduction therapy for many groups. In the COG 9900 series of clinical trials, end induction bone marrow MRD and day 8 peripheral blood MRD were the most powerful predictors of outcome in a multivariable analysis.
68 It should be noted, however, that MRD is not the sole predictor of outcome in this model; NCI risk group and genetic factors also remain prognostic, thus justifying the current approach that the COG is using in determining risk stratification. Other groups, including the St. Jude Children’s Research Group (SJRCH) and the Dana-Farber Cancer Institute (DFCI) incorporate measurements of MRD at the end of induction to intensify patients for additional therapy. Despite MRD being the most powerful predictor of outcome, up to 50% of children who relapse do not have evidence of disease using these sensitive methodologies, implying that additional features, such as critical genetic lesions, affect outcome.
The clinical use of MRD at the end of the first phase of therapy (traditionally deemed “induction”) or the second phase of therapy (“consolidation”) remains somewhat consortium dependent. In T-cell ALL, the prognostic significance of MRD appears in at least one report to be more powerful at the end of the second phase of therapy than at the end of the first phase.
73Finally, outcomes for the small number (<3%) of pediatric patients who fail to attain remission following induction therapy, although predictably poorer than other patients, are not as dismal as might be expected.
74 An intergroup study of 1,041 patients who failed induction reported a 10-year survival of 32±1%. Age ≥10 years, T-cell leukemia, the presence of 11q23 rearrangement, and ≥25% blasts at end of induction had a poor outcome. However, children <6 years of age with precursor B-ALL and without adverse genetics had a 10-year survival at 72±5% when treated with chemotherapy only. Patients with T-cell leukemia had improved survival with allogeneic HSCT.
Current Approaches to Staging/Stratification
Based on the collective information above, the COG has developed a risk-stratification system that incorporates key clinical features including National Cancer Institute (NCI) risk group
75 [Standard Risk (SR): age 1 to 9.99 years and initial WBC count <50 × 10
9/L; High Risk (HR): age 10+ years and/or initial WBC ≥50 × 10
9/L], immunophenotype, presence/absence of central nervous system or testicular leukemia, presence/absence of specific sentinel genetic lesions [good risk:
ETV6–
RUNX1 fusion or hyperdiploidy with trisomies of chromosomes 4 and 10; poor risk:
MLL-rearrangements (
MLL-R), hypodiploidy, intrachromosomal amplification of chromosome 21 (iAMP21), Philadelphia chromosome positive (Ph+) ALL], and early minimal residual disease response for risk stratification and treatment allocation.
51,68,76 The current risk stratification system (AALL08B1) builds on that developed for the first generation COG ALL trials (AALL03B1) with key differences being changing the MRD threshold used to define poor response from ≥0.1% (AALL03B1) to ≥0.01% (AALL08B1), and incorporating day 8 peripheral blood [and day 29 bone marrow (BM)] MRD measurements while eliminating day 8/15 BM morphology
77 (
Table 76.3).