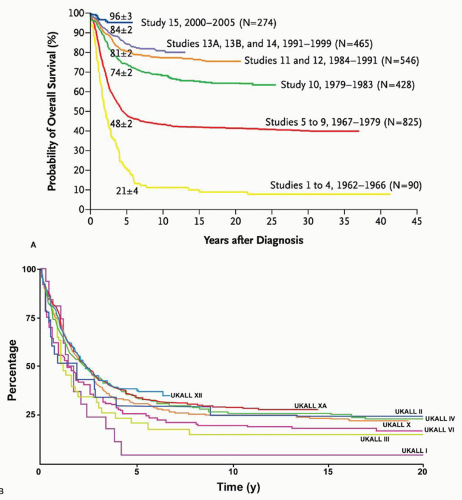
 FIGURE 74.1. Overall survival in successive acute lymphoblastic leukemia patient cohorts. A: Childhood acute lymphoblastic leukemia patients treated at the St. Jude Children’s Research Hospital. (With permission form Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354:166-178.) B: Adult acute lymphoblastic leukemia patients treated by the UKALL collaborative study group. (With permission from Durrant IJ, Richards SM, Prentice HG, et al. The Medical Research Council trials in adult acute lymphocytic leukemia. Hematol Oncol Clin North Am 2000;14:1327-1352.) |
which is the most common cytogenetic abnormality in adult ALL. ABL is a nonreceptor tyrosine protein kinase that enzymatically transfers phosphate molecules to substrate proteins, thereby activating downstream signal transduction pathways important in regulating cell growth and proliferation.25 Other gene rearrangements result in loss- or gain-of-function mutations involving transcription factors that are important for normal hematopoietic development.26 An example is the t(12;21) (p13;q22) chromosomal translocation, which juxtaposes the ETV6 (TEL) and RUNX1 (AML) genes.27 Excluding numerical aberrations, ETV6-RUNX1 is the most frequent cytogenetic abnormality in childhood ALL, although it is uncommon in adults. Another general mechanism of cancer formation involves loss or inactivation of tumor-suppressor genes, many of which have key regulatory functions in controlling cell cycle progression.28 Examples are p16(CDKN2A) and p15(CDKN2B). Stock et al. investigated the incidence of cell cycle regulatory gene abnormalities in adult patients with de novo ALL treated by the Cancer and Leukemia Group B (CALGB) study group.29 Deletions, microdeletions, and gene rearrangements involving p16(CDKN2A) and p16(CKDN2B) were common occurrences. Even more frequent was aberrant expression of RB1 and TP53, two other tumor-suppressor genes. Concurrent abnormalities involving two or more of these genes were found in one third of adult ALL patients.
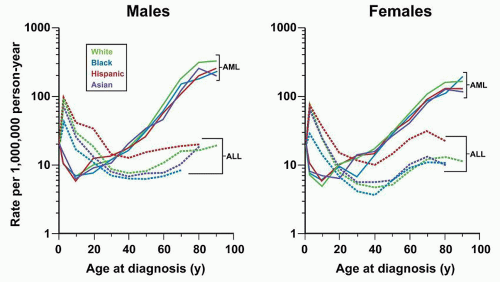 FIGURE 74.2. Age-specific incidence rates of acute lymphoblastic leukemia (ALL) (dotted lines) and AML (solid lines) according to sex and race/ethnicity, SEER-17, 2001 to 2007. (With permission from Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34-43.) |
leukemic blasts may produce bone pain and arthralgias, but marrow necrosis is much less frequently found in adults as compared with children who have ALL.35 Approximately one half of adult patients have hepatomegaly, splenomegaly, or lymphadenopathy at diagnosis that can be appreciated on physical examination. Mediastinal masses are detected by chest radiographs or computed tomography scans primarily in patients with T-lineage ALL, who also frequently have pleural involvement and may complain of chest pain.36 The fewer than 10% of ALL patients who have CNS involvement infrequently present with referable symptoms, such as headache, vomiting, neck stiffness, alteration in mental status, and focal neurologic abnormalities. Other sites of extramedullary involvement include testis, retina, and skin, although virtually any organ can be infiltrated by leukemic blast cells.
TABLE 74.1 CLINICAL FINDINGS AT DIAGNOSIS IN ADULTS WITH ACUTE LYMPHOBLASTIC LEUKEMIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 74.2 DIAGNOSTIC APPROACH FOR ADULT ACUTE LYMPHOBLASTIC LEUKEMIA | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
TABLE 74.3 LABORATORY FINDINGS AT DIAGNOSIS IN ADULTS WITH ACUTE LYMPHOBLASTIC LEUKEMIA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 74.4 CHROMOSOMAL ABNORMALITIES AT DIAGNOSIS IN ACUTE LYMPHOBLASTIC LEUKEMIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 74.5 PROGNOSTIC FACTORS FOR REMISSION DURATION IN ADULTS WITH ACUTE LYMPHOBLASTIC LEUKEMIA (ALL) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
patients with unfavorable abnormalities, as opposed to greater than 75% for those with favorable cytogenetic findings.60,65,90, 91, 92, 93 and 94 Additional reports from single institutions have provided similar outcome data.95,96 Cytogenetic findings commonly identified in adult ALL with an intermediate prognosis include normal karyotype, hyperdiploidy, and abnormalities at 6q or 9p. The t(8;14) (q24;q32)[MYC-IGH] and other MYC gene rearrangements are associated with the Burkitt leukemia subtype and are not prognostically unfavorable markers with optimal treatment.
to have persistent blast cells. These patients were less likely to achieve CR after 4 weeks of therapy. Even among only the patients who achieved CR within 4 weeks, an otherwise favorable feature, those who had persistent blasts at day 15 fared significantly worse. Among all complete remitters, the 5-year disease-free survival was 34% for those who cleared marrow blasts at day 15, compared with 19% for those who did not. Annino et al. and the Italian collaborative study group evaluated the early corticosteroid response by giving a 7-day course of prednisone immediately before induction.9 The pre-induction response was previously shown to have strong prognostic value in pediatric ALL.114,115 In the adult ALL study, prednisone response was defined by reduction of peripheral blood blasts to less than or equal to 1 × 109/L. Lack of a prednisone response was found to affect overall survival negatively, as it did in the pediatric ALL studies, and, among CR patients, it also adversely influenced remission duration.
(SCT) in first remission, given an available donor and eligibility status. Considerable clinical data suggest that this strategy has been effective, particularly with Ph+ patients. The ECOG/MRCXII trial also demonstrated the benefit of transplant in standard risk patients under age 50. Elderly ALL patients pose special treatment considerations that are discussed in this chapter. General issues relating to supportive care of the patient with leukemia are discussed in Chapter 69.
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


