Leopoldo N. Segal, Michael D. Weiden, Harold W. Horowitz Keywords acute exacerbations of COPD (AECOPD); azithromycin; bronchitis; bronchodilators; chronic obstructive pulmonary disease (COPD); corticosteroid use in COPD; GOLD COPD summary; Haemophilus; lung inflammation; lung microbiome; microbiome; Moraxella; Streptococcus pneumoniae; therapy for COPD exacerbation; vaccinations Chronic obstructive pulmonary disease (COPD), including emphysema and chronic bronchitis, has been the third leading cause of death since 2008 in the United States.1 It will become the third leading cause of death in the world by 2020.2,3 COPD prevalence has increased owing to increased longevity and long-term exposure to common environmental risk factors such as inhaled particulate matter. By 2030, 10% of the general population and 50% of smokers will have COPD.4,5,6 Consequently, COPD is an important driver of increased health care utilization, producing significant social and economic costs.7–9 COPD is caused by multiple factors, including environmental exposures, infections, inflammation, and genetic predisposition. Tobacco smoking is the largest environmental risk factor in the United States. In other countries, occupational dust exposures, outdoor air pollution, and poor indoor air quality from burning biomass fuels are major COPD risk factors.10 Among immune-competent adults, increased airway bacteria produce airway inflammation and accelerate airway obstruction.11 Immunosuppression is an independent risk factor for COPD. Immunoglobulin (Ig)A-deficient individuals have repeated lower respiratory tract infections during childhood and poor adult lung function.12 Human immunodeficiency virus (HIV) infection promotes chronic pulmonary inflammation and increased pulmonary matrix metalloprotease expression, leading to smoking-related emphysema.13,14 Even after effective antiretroviral therapy, patients with the acquired immunodeficiency syndrome (AIDS) have an increased frequency of pulmonary infection and accelerated lung function decline.15,16 Genetic susceptibility is another risk factor for developing COPD. Mutation of the α1-antitrypsin gene leads to low serum antiprotease activity, causing a much higher risk for COPD in smokers and workers exposed to particulate matter.17 Homozygous α1-antitrypsin deficiency (PI*ZZ) occurs in 1% to 4.5% of COPD patients, and the heterozygous form (PI*MZ), with less severe deficiency, occurs in 17.8% of COPD patients.18 Ethnic origin is an important risk factor for COPD, raising the possibility of differential group-level genetic susceptibility to lung injury.19 For most individuals, predisposition to abnormal lung function is polygenic, with more than 20 risk genes currently identified.20,21 Poor socioeconomic status, chronic asthma, fetal growth retardation, poor nourishment, and history of pulmonary tuberculosis are other risk factors for COPD.19,22–24 Acute exacerbations of COPD (AECOPD) produce significant morbidity and mortality. Risk factors include viral and bacterial infections, change in environmental conditions such as smog, gastroesophageal reflux, lack of compliance with maintenance treatment, severity of baseline disease, and history of prior exacerbations.25 Seasonality has been demonstrated in AECOPD, which occurs in northern and southern latitudes approximately two times more frequently during winter than in summer. AECOPD are responsible for the greatest proportion of health care–related costs associated with COPD.7–9 The discussion in this chapter will focus on the impact of AECOPD on disease course, infectious causes of AECOPD, and treatment as well as prevention options. Patients with COPD usually present with progressive shortness of breath, cough, and sputum production. These symptoms are associated with accelerated decline in lung function, which may continue despite smoking cessation. The dyspnea usually starts during exercise but can occur with minimal exertion or at rest as disease progresses. Cough and sputum production are usually intermittent and more pronounced in the morning. Other chronic pulmonary diseases with similar clinical presentation and acute exacerbations should be differentiated from those of COPD because treatment differs. Examples of these include asthma, cystic fibrosis, bronchiectasis, diffuse panbronchiolitis, and obliterative bronchiolitis. Worsening symptoms, increased sputum volume, and transition of sputum color from clear to green or yellow suggests an acute exacerbation, which more commonly occurs during the winter.26 The main differential diagnosis of exacerbations in patients with COPD includes pneumonia and congestive heart failure, both of which are common comorbidities in these patients.4 COPD is characterized by progressive airflow obstruction, defined by reduction of forced expiratory volume after 1 second (FEV1) and a low ratio of FEV1/forced vital capacity (FVC) on pulmonary function tests. Chronic bronchitis and emphysema are the two major clinical subtypes. Because overlap between them is common and treatment is similar, there is little clinical importance to distinguish chronic bronchitis from emphysema. Chronic bronchitis is defined by a history of productive cough for 3 months per year in at least 2 successive years. Emphysema used to be a pathologic diagnosis with distal airspace enlargement accompanied by destruction of alveolar walls. In the modern era, high-resolution computed tomography (CT) is able to define the extent and distribution of emphysema. With increasing use of CT to screen for lung cancer, more asymptomatic smokers are diagnosed with emphysema.27 Severity of stable COPD is based on pulmonary function (Table 67-1).4 Initially, the results of standard pulmonary function tests are normal. Abnormalities in early disease may require more sensitive techniques that evaluate distal lung function, such as impulse oscillometry.28 With progression, patients suffer decreasing FEV1, decreasing FEV1/FVC ratio, and increased total lung capacity caused by air trapping during expiration. Alveolar destruction promotes airway collapse and reduces diffusing capacity of carbon monoxide (DLco) in the lung. Eventually severe airflow obstruction can lead to abnormal arterial blood gases with hypoventilation (Pco2 >40 mm Hg) and hypoxemia (Po2 <60 mm/Hg). Integration of physiologic parameters with symptoms are useful, validated predictors of mortality.29,30 One of the most commonly used composite criteria to accurately characterize COPD is called BODE. It includes body-mass index, airflow obstruction (as defined by abnormal FEV1), dyspnea, and exercise capacity (evaluated by a 6-minute walk test).31,32 BODE predicts response to rehabilitation, hospitalization, and mortality.31,33–35 Although there is no universal agreement on how to define or diagnose AECOPD, they are commonly defined as acute events with worsening respiratory symptoms beyond normal day-to-day variations. They usually require increased rescue β-agonist inhaler use to control symptoms.36 One widely used scale to diagnose the presence and severity of AECOPD requires patients to have at least one of the following clinical presentations: upper respiratory tract infection symptoms within the previous 7 days, increased wheezing, fever without another identified cause, or an increase in heart rate or respiratory rate greater than 20% from baseline. This scale then categorizes patients into three groups based on whether they have worsening dyspnea, increase in sputum purulence, and/or increase in sputum volume. A severe exacerbation has all three criteria, a moderate exacerbation has two, and a mild exacerbation has only one.37 Risk factors associated with COPD exacerbations include having three or more COPD exacerbations in the previous year, reduced FEV1, smoking, and nonadherence with oxygen therapy.38,39 The risk for severe exacerbation depends on each patient’s medical history, including baseline FEV1, number of prior exacerbations, prior need for mechanical ventilation, and comorbidities. Whereas the vast majority of AECOPD can be managed in the outpatient setting, the presence of high severity should prompt consideration of hospital admission.4 The clinical signs of risk for respiratory failure are the most important measure of the current exacerbation’s severity. Tachypnea (especially with a respiratory rate above 25 breaths/min), tachycardia, inability to speak full sentences, and fatigue are indications for hospitalization. Oxygen saturation above 90% can be misleading because hypoxemia is frequently a late event in the progression to respiratory failure. In an acutely symptomatic patient, arterial blood gas analysis is more useful than oximetry because it can diagnose hypercapnia as well as hypoxemia. Use of accessory muscles with paradoxical breathing characterized by inward motion of the abdomen during inspiration indicates diaphragmatic fatigue and impending respiratory failure. Abdominal paradoxical breathing, progressive hypercapnia, or deteriorating mental status usually indicates the need for ventilatory support in an intensive care unit with noninvasive or invasive positive-pressure ventilation.4 Additional diagnostic tests include chest radiography to identify pulmonary infiltrates and electrocardiography to assess for cardiac ischemia and arrhythmias, particularly paroxysmal atrial tachycardia. The basic metabolic panel is helpful to assess severity for AECOPD. Elevated levels of sodium bicarbonate are a sign of chronic hypercapnia. Increased anion gap is a sign of anaerobic metabolism of the respiratory muscles or sepsis syndrome or both. Hyponatremia sometimes occurs as a result of the syndrome of inappropriate secretion of antidiuretic hormone. Hyperglycemia is a response to stress or systemic corticosteroids, and renal insufficiency is a manifestation of reduced cardiac output in end-stage pulmonary hypertension. The presence of these derangements may warrant hospitalization.4 Although chest radiography is an insensitive test to diagnose COPD, it is the usual first step in the evaluation of patients with progressive dyspnea and cough. It is useful to assess the symptomatic patient for advanced lung cancer, pulmonary fibrosis, or congestive heart failure. Sometimes COPD is suspected because the chest radiograph shows hyperinflation with flattened diaphragm or increased retrosternal space. If bullae are observed, then COPD is highly likely. CT is the imaging modality of choice for the evaluation of COPD and many other symptomatic pulmonary diseases. Data suggest that quantitative CT allows estimation of the risk for exacerbation frequency and determination of indices of disease impact such as BODE.40 Impaired lung function in COPD is caused by destruction and remodeling of large and small airways due to chronic inflammation caused by complex interactions between the ambient (toxic inhalants, changing microbiome) and the airway mucosa (host immune response). Both inflammatory changes and lung function deterioration become more prominent with each exacerbation (Fig. 67-1). Early pathology is produced by inflammation in bronchioles less than 2 mm in diameter followed by parenchymal remodeling.41,42 In early stages, the central airway walls are infiltrated with CD8+ lymphocytes, producing bronchial wall thickening evidenced on chest CT.43 Apoptosis and necrosis of epithelial and endothelial cells are also present, as are activated CD4+ T cells.44,45 As disease progresses, neutrophils become prominent and release neutrophil elastase, leading to parenchymal destruction.46,47 Persistent airway injury also produces squamous metaplasia with loss of cilia function in affected bronchial segments. Disease of both the large and small airways contributes to airflow obstruction and ventilation heterogeneity.48 Pulmonary hypertension due to loss of the pulmonary capillary bed can also develop in COPD. Chronic hypoxia also produces vasoconstriction, leading to fixed structural changes that worsen pulmonary hypertension.49 As disease progresses, frequent exacerbations of COPD further contribute to increased lung inflammation and persistent loss of lung function.50–54 COPD patients have a 20-fold increase in alveolar macrophages in alveoli, bronchioles, and small airways.55,56 Because alveolar macrophages are a major source of inflammatory cytokines and growth factors, recruitment of inflammatory cells into the lung accelerates the vicious inflammatory cycle produced by microaspiration. In spite of increased numbers, alveolar macrophages have impaired phagocytosis, reducing their ability to clear bacteria from the lower airway and thereby causing further inflammation and oxidative stress.57–60 Neutrophils are also recruited and activated by nonresolving pulmonary inflammation, particularly during exacerbations. Neutrophil elastase and metalloproteinases lead to lung parenchyma destruction. Neutrophil elastase is also a potent mucous secretagogue leading to mucous gland hyperplasia.61 Because neutrophils have a short tissue life span, the airway neutrophilia observed in COPD requires continuous neutrophil recruitment.62 Smoking also impairs neutrophil phagocytic function.63 Collectins, pentraxins, and complement also are deficient,64 further impairing mucosal immunity and predisposing to lower respiratory tract infections. Pulmonary inflammation continues after smoking cessation, in part owing to permanent structural damage and in part owing to recovery of the proinflammatory capacity of epithelial cells.65 Bacterial colonization in the lower airways is also an important determinant of the degree of airway and systemic inflammation in stable COPD.66 Inflammatory mediators “spill over,” producing systemic inflammation with elevated levels of C-reactive protein (CRP), fibrinogen, or leukocyte count. An elevated CRP level is most strongly associated with ischemic heart disease in smokers. Systemic comorbidities such as vascular disease are major risk factors for mortality during COPD exacerbations.67,68 Systemic inflammation caused by COPD is also hypothesized to be associated with anemia, osteoporosis, depression, and the metabolic syndrome.69–71 Increased bacterial colonization of the lower airways in COPD occurs owing to chronic microaspiration, impaired clearance of bacteria, and frequent COPD exacerbations.72,73 COPD patients frequently have microaspiration owing to gastroesophageal reflux due to incoordination between breathing and swallowing.74–76 Impaired mucociliary clearance in smokers reduces the ability to clear oral microbes from the lower airways, exacerbating inflammation. This leads to chronic cough with progressive incoordination of breathing with swallowing. Inhaled medications may also carry oral bacteria into the lower airway. This vicious cycle could explain the association of poor oral health and increased airway bacterial load, COPD exacerbations, and reduced lung function.77–81 Viruses and bacteria in the lower airway perpetuate the inflammation in COPD, causing damage by a number of mechanisms.11,82,83 They may be ciliotoxic, invade epithelial cells causing apoptosis, increase mucin production, or degrade humoral immunity via secretion of IgA proteases.84,85 Bacterial molecules such as endotoxins, membrane lipoproteins, peptidoglycan fragments, and lipoteichoic acid activate the innate immune response, exacerbating inflammation.86 COPD patients whose airways are heavily colonized with bacteria have higher concentrations of inflammatory cytokines and neutrophils in respiratory secretions.83 High bacterial burden in the lower airway is associated with accelerated FEV1 decline, more comorbidity, more exacerbations, and worse symptoms during exacerbations.15,87,88–90 In stable COPD, the rates of positive routine bacterial cultures of sputum vary between 22% and 83%.91–93 However, interpreting sputum culture is difficult owing to upper airway contamination, which reduces specificity, and failure to grow fastidious bacteria from the complex lower airway microbiome, which reduces sensitivity. In stable COPD, nonpotential pathogenic microorganisms are isolated much more frequently than potential pathogenic microorganisms.91 Nonpotential pathogenic microorganisms are usually oropharyngeal microbes such as Corynebacterium spp., Neisseria spp., Enterococcus spp., coagulase-negative staphylococci, Streptococcus viridans, and fungi such as Candida spp.91 The most commonly isolated potential pathogenic microorganisms are Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis.52,91,94 Culture-independent technologies that assay microbial nucleic acids and antigens often have found potential pathogens in culture-negative respiratory specimens.95,96 For example, one third of stable COPD patients have Haemophilus influenzae within airway epithelial cells and alveolar macrophages.97 High-throughput sequencing of microbial 16S rRNA genes yields relatively unbiased estimates of the relative abundance of uncultivable and cultivable bacteria.98,99 The lower airways of normal individuals harbor low levels of oral bacteria such as Prevotella spp. and Veillonella spp.100,101 Culture-independent techniques have challenged the dogma that the lower airway is normally sterile and provide evidence that there are residential organisms, especially in individuals with already damaged lungs. Oral anaerobes likely modulate the pulmonary immune response in health and disease. This possibility is supported by a growing body of data linking periodontitis, COPD exacerbations, and reduced lung function.102 However, the technical challenges produced by contamination of lower airway samples with upper airway secretions needs to be resolved to better understand the role of airway microbiome in COPD pathogenesis. Respiratory viruses are also frequently found in patients with stable COPD. Using culture or polymerase chain reaction (PCR) techniques to assess sputum, the most common respiratory virus is respiratory syncytial virus (RSV), which is found in up to 23.5% of COPD patients, followed by non-RSV viruses, such as rhinovirus, coronavirus, and parainfluenza virus, in 16.2% of samples.103 Increased inflammation has also been reported with adenovirus.104 High-throughput complementary DNA sequencing can identify viral transcripts in an unbiased approach. A fuller understanding of the role of the virome in stable COPD and in AECOPD awaits resolution of the technical challenges of high-coverage RNA sequencing in lower respiratory tract samples. Two thirds of patients with a COPD exacerbation have bacteria or viruses or both cultured from lower airway secretions. Aerobic bacteria are isolated in half of patients, respiratory viruses are isolated in one third, and bacterial/viral coinfection is present in a fourth of patients with acute exacerbations.52,105 The increased incidence of AECOPD during the winter may reflect the significant role that viruses play in the pathogenesis of AECOPD. H. influenzae, S. pneumoniae, and M. catarrhalis are the bacterial pathogens most commonly isolated during COPD exacerbations. The same three also colonize stable COPD patients, and higher bacterial loads have been associated with AECOPD.52,94,95,106–109 In a series of studies using molecular biologic techniques, Sethi and associates have observed that acquisition of a new bacterial strain precedes AECOPD.83,96,106,109,110 The increase in total bacterial load during a COPD exacerbation is relatively small compared with the total bacterial load.109 In their model, the humoral and cellular immune responses to the new bacterial or viral strain likely drive the increased inflammation that causes the COPD exacerbation. Individuals with greater degrees of functional impairment, recent antibiotic use, or systemic corticosteroid therapy have higher rates of isolation of gram-negative bacteria, such as Pseudomonas aeruginosa, Stenotrophomonas maltophilia, and members of the Enterobacteriaceae, from sputum.111,112 Patients with an FEV1 greater than 35% of predicted value and no systemic corticosteroid or antibiotics within the preceding 3 months have a low probability of Enterobacteriaceae or P. aeruginosa in sputum culture.112 H. influenzae and P. aeruginosa are more common in patients with poorer lung function.108 Polymicrobial exacerbations occur with advanced pulmonary dysfunction and severe exacerbations.105,113 The role of “atypical” bacterial pathogens such as Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella pneumophila in exacerbation is poorly defined, but they are rarely found in AECOPD.50,103,114 Variations in reported rates of atypical pathogens could be related to the technical or geographic differences between reports.89,115 When serology or M. pneumoniae antigen detection has been used to indicate the presence of M. pneumoniae as a pathogen, PCR assay and culture frequently do not confirm its presence.103,116,117 Alternately, two reports from Italy and Greece demonstrate M. pneumoniae among between 7% and 9% of patients with AECOPD using serologic and PCR techniques.118,119 Controversy also exists regarding the role of C. pneumoniae in COPD exacerbations. Several well-conducted studies detected no C. pneumoniae or L. pneumophila,103,117 whereas others detected C. pneumoniae in 6% to 9% of those with AECOPD.89,118 Chronic colonization with C. pneumoniae occurred in one third of patients and was associated with worse pulmonary function in one study.89 Unlike stable COPD, rhinovirus is the virus most frequently associated with AECOPD.103,105,120 Coronavirus, parainfluenza, adenovirus, influenza virus, and human metapneumovirus also occur but are less prevalent.113,120 Coinfection with viruses and bacteria produce higher bacterial burden, more sputum eosinophils, greater lung function impairment, and longer hospitalization.105,113 It is likely that viruses and bacteria induce independent inflammatory pathways, accounting for more severe presentations and poorer outcome of patients with coinfection. Treatment of stable COPD should improve the patient’s symptoms and functional status, reduce the risk for exacerbations, and slow the decline of lung function.4 Smoking cessation and avoidance of environmental exposure are the most important interventions to prevent disease progression and should be encouraged at every medical visit. Multiple different behavioral and pharmacologic treatments should be explored, including varenicline, nicotine replacement, and bupropion.121–124 Pneumococcal, influenza, and combined tetanus/diphtheria/acellular pertussis (Tdap) vaccination is also recommended for every COPD patient.4,125 β2-Agonist and anticholinergic bronchodilators (usually long-acting formulations) are the mainstays of symptom control (Table 67-2).4 These drugs relax airway smooth muscle, producing bronchodilation, and also have anti-inflammatory activity, although the clinical significance of this is not clear.126 Anticholinergic medications also reduce sputum production in patients with chronic bronchitis. Corticosteroids in steady-state COPD are restricted to adjunctive therapy complementing long-acting bronchodilators in more severe cases of COPD. Inhaled corticosteroids do not reduce mortality but do reduce COPD exacerbations,127 decrease inflammation, and stabilize lung function in moderate and severe COPD.128 Unfortunately, inhaled corticosteroids increase the risk for pneumonia, which must be balanced with the benefit of less frequent exacerbations.127,129 Other drugs, such as nonselective and selective phosphodiesterase inhibitors (e.g., theophylline and roflumilast, respectively), may also be considered in patients with severe COPD with frequent exacerbations that are not adequately controlled by long-acting bronchodilators.130,131 TABLE 67-2 Modified Global Initiative for Chronic Lung Disease (GOLD) Treatment Recommendations According to Stable-State Patient-Group Category
Acute Exacerbations of Chronic Obstructive Pulmonary Disease
Epidemiology
Clinical Manifestations
Diagnosis
Acute Exacerbations of COPD
Radiology
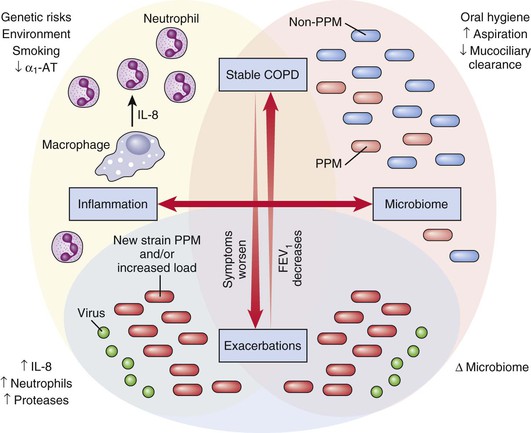
Pathophysiology
Mucosal Inflammation
Microbes in Stable COPD
Microbes in Acute Exacerbations of COPD
Nonantimicrobial Therapy for Steady-State COPD
PHARMACOLOGIC OPTIONS
Patient Group
First Choice
Second Choice
Alternative Choice
A
FEV1 >50%
Mild symptoms
≤1/yr Exacerbations
SABA prn
or
SAMA prn
LABA or LAMA
or
SABA and SAMA
Theophylline
B
FEV1 >50%
Moderate symptoms
≤1/yr Exacerbations
LABA
or
LAMA
LAMA and LABA
SABA and/or SAMA
Theophylline
C
FEV1 ≤50%
Mild symptoms
≤1/yr Exacerbations
ICS + LABA
or LAMA
LAMA and LABA
PDE-4 inhibitor
SABA and/or SAMA
Theophylline
D
FEV1 ≤50%
Moderate symptoms
≥1/yr Exacerbations
ICS + LABA
or LAMA
ICS and LAMA
or ICS + LABA and LAMA
or ICS + LABA and PDE-4 inhibitor
or LAMA and LABA
or LAMA and PDE-4 inhibitor
Carbocysteine
SABA and/or SAMA
Theophylline
Azithromycin
NONPHARMACOLOGIC TREATMENT OPTIONS
Patient Group
Essential
Recommended
A
Smoking cessation
Physical activity
Influenza and pneumococcal vaccination; Tdap
B-D
Smoking cessation
Pulmonary rehabilitation
Physical activity
Influenza and pneumococcal vaccination; Tdap![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Acute Exacerbations of Chronic Obstructive Pulmonary Disease
67