Acute Leukemias
Acute leukemias are neoplastic disorders marked by uncontrolled proliferation of hematopoietic cells, with a predominance of immature lymphoid or myeloid cells, in the bone marrow and peripheral blood. Although leukemic cells do not divide more rapidly than normal marrow cells, they possess a growth advantage because the blasts fail to differentiate in response to normal hormonal signals and cellular interactions. The malignant cells eventually replace the marrow and invade other tissues and organs, leading to manifestations of the disease. Production of normal erythrocytes, granulocytes, and megakaryocytes is diminished, resulting in anemia, infection, and hemorrhage.
Approximately 18,000 cases of acute leukemia occur in the United States each year. The incidence increases with age. About 80% of leukemic children have acute lymphoblastic leukemia (ALL), the most common childhood malignant neoplasm, whereas 80% of adults with leukemia have acute myeloid leukemia (AML). The known etiologies include chromosomal damage from ionizing radiation or from chemicals (e.g., benzene and alkylating agents used in therapy), congenital disease (e.g., Down syndrome), chronic bone marrow diseases (e.g., myelodysplasia), congenital predisposition (e.g., identical twins with leukemia), and congenital immunodeficiency syndromes (e.g., ataxia-telangiectasia).
MORPHOLOGY AND BIOLOGY
Acute leukemias—defined as 20% or more blasts in blood or bone marrow under the World Health Organization (WHO) criteria—are classified according to the predominant neoplastic cell line and thus may be designated as lymphoblastic or myeloid. The WHO classification takes into account cytogenetic and molecular findings in addition to morphologic and histochemical criteria (see Tables 15.1 and 15.4 ). About 70% to 80% of cases can be classified on the basis of morphology alone, whereas an additional 10% to 15% of cases require histochemical determinations for specific diagnosis. In approximately 10% of cases, surface marker and cytogenetic studies are necessary for accurate classification, particularly in the “undifferentiated” acute leukemias. Use of monoclonal antibodies has revealed that about 20% of cases of ALL are of T-cell origin; most of the remainder are of B-cell or pre-B cell origin (see Table 15.3 ).
| Acute Myeloid Leukemia with Recurrent Genetic Abnormalities |
|
| Acute Myeloid Leukemia with Myelodysplasia-Related Changes |
| Therapy-Related Myeloid Neoplasms |
| Acute Myeloid Leukemia, not Otherwise Specified |
|
| Myeloid Sarcoma |
| Myeloid Proliferations Related to Down Syndrome |
|
| Acute Leukemias of Ambiguous Lineage |
|
| Precursor Lymphoid Neoplasms |
|
| B Lymphoblastic Leukemia/Lymphoma with Recurrent Genetic Abnormalities |
|
| T Lymphoblastic Leukemia/Lymphoma |
| Subtype | FAB type | Frequency (%) † | Morphology | MP | SE | NSE | Immune Markers | Cytogenetic Abnormalities ‡ | Genes Involved |
|---|---|---|---|---|---|---|---|---|---|
| Acute myeloblastic leukemia with minimal differentiation | MO | 3 | Rare granules; no Auer rods | – | – | – | CD11, CD13, CD33 HLA-DR | inv (3q26), t(3;3) | EVII |
| Acute myeloblastic leukemia without maturation | M1 | 15–20 | A few azurophilic granules or Auer rods | + § | +/– | – | CD11, CD13, CD33 HLA-DR | t(9;22) +8t(v;11) −7e−5 or 5q | |
| Acute myeloid leukemia with maturation | M2 | 25–30 | Some maturation beyond promyelocytes; Auer rods | ++ | ++ | – | CD11, CD13, CD33 HLA-DR | t(8;21) ¶ t(9;22), t(6;9) +8 −7e−5 or −5q | AMLI-ETO DEK-CAN |
| Acute promyelocytic leukemia | M3 | 5–10 | Hypergranular promyelocytes; multiple Auer rods | +++ | +++ | – | CD11, CD13, CD33 | t(915;17)t(11;17) t(5;17) | PML-RAR PLZF-RAR NPM-RAR |
| Acute myelomonocytic leukemia | M4 | 20 | ≥20% monocytes; monocytoid cells in blood; Auer rods | ++ | ++ | ++ | CD11, CD13, CD14, CD33 HLA-DR | 11q23 inv (3q26), t(3;3) EVI t(6;9) +8−7−5 †† or 5q | MLL DEK-CAN |
| M4 ** | 5–10 | Eosinophilia; early eosinophils with large purple granules | ++ | ++ | ++ | CD2, CD13, CD14, CD33 HLA-DR | inv(16) ¶ , del(16) (q22)t(16;16)d | CBF-MYHII | |
| M5 | CD11, CD13, CD14, CD33 HLA-DR | ||||||||
| Acute monocytic leukemia | 2–9 | Monoblastic (M5A)Promonocytic (M5B), no Auer rods | – | – | +++ | 11q23 t(8;16) | |||
| Erythroleukemia | M6 | 3–5 | Predominance of erythroblasts; dyserythropoiesis; | + | – | – | +8 | MILL | |
| +8 | MOZ-CBP | ||||||||
| Auer rods in myeloblasts | −7 †† | ||||||||
| Acute megakaryoleukemia | M7 | 3–12 | “Dry” aspirate; biopsy specimen with blasts and dysplastic megakaryocytes; no Auer rods | – | – | – | CD33, glycophorin A | −5 or 5q t(1;22) | |
| CD33, CD41, CD61 HLA-DR | −7 †† | ||||||||
| −5 or 5q |
* Findings may be somewhat variable.
‡ Complex chromosome defects may be seen in M0, M1, M2, M4–M7.
¶ Associated with a more favorable prognosis.
** Eosinophilic variant of M4.
†† Associated with a less favorable prognosis.
| Subtype/Translocation | Molecular Alteration | Frequency (%) | FAB type | HLA-DR | CALLA | CD19/CD20 | c | sIg | T cell | TdT |
|---|---|---|---|---|---|---|---|---|---|---|
| B-precursor ALL † | L1, L2 | + | +/− | +−/+− | + | − | − | + | ||
| t(12;21)(p13;q22) | TEL-AML1 | 20–25 | ||||||||
| t(1;19)(q23;p13.3) | E2A-PBX1 | 5–6 | ||||||||
| t(17;19)(q22;p13.3) | E2A-HLF | <1 | ||||||||
| t(9;22)(q34;q11) | BCR-ABL | 4 | ||||||||
| t(4;11)(q21;q23) | MLL-AF4 | 4 | ||||||||
| Other 11q23 | Other MLL fusions | 1 | ||||||||
| t(5;14)(q31;q32) | IL-3 dysregulation | <1 | ||||||||
| B-cell ALL | 2 | L3 | + | + | +/+ | − | + | − | − | |
| t(8;14)(q24;q32) | MYC dysregulation | |||||||||
| t(2;8)(q12;q24) | ||||||||||
| t(8;22)(q24;q11) | ||||||||||
| T-cell ALL | 8 | L1, L2 | − | −/− | −/− | − | − | + | + | |
| t(1;14)(p32;q11) | TAL1 dysregulation | |||||||||
| t(1;7)(p32;q35) | TAL1 dysregulation | |||||||||
| t(7;9)(q34;q32) | TAL2 dysregulation | |||||||||
| t(7;19)(q34;p13) | LYL1 dysregulation | |||||||||
| t(10;14)(p24;q11) | HOX11 dysregulation | |||||||||
| t(7;10)(p35;q24) | HOX11 dysregulation | |||||||||
| t(11;14)(p15;q11) | LMO1 dysregulation | |||||||||
| t(7;11)(q35;p13) | LMO2 dysregulation | |||||||||
| t(11;14)(p13;q11) | LMO2 dysregulation | |||||||||
| t(1;7)(p34;q34) | LCK dysregulation | |||||||||
| t(7;9)(q34;q34) | TAN1 dysregulation |
* In children. Data from Pui et al. (1993).
† Additional cytogenetic abnormalities include hyperdiploidy with >50 chromosomes (favorable prognostic factor), hyperdiploidy with 47–50 chromosomes, and hypodiploidy (unfavorable prognostic factor). The presence of t(9;22) or T(4;11) is associated with a poor prognosis as well.
Development of colony assays and new monoclonal antibodies has led to a clearer understanding of normal myeloid ontogeny. The patterns of expression of a variety of antigens during normal myeloid differentiation have been established. Immunophenotyping techniques have demonstrated that some cases of acute “undifferentiated” leukemia, or those considered on the basis of morphology to be ALL, show myeloid markers. Improvement of cytogenetic studies by the use of new banding techniques has revealed that almost all acute leukemias are characterized by chromosomal abnormalities, ranging from hypoploidy to polyploidy. Significant cytogenetic defects that have prognostic importance (e.g., are seen only with certain subgroups) have been identified, involving the ALLs as well as the AMLs. Genetic abnormalities in the acute leukemias can affect genes that encode proteins involved in signal transduction, transcription regulation, cellular differentiation, and apoptosis, as well as tumor suppression (antioncogenes).
In about 10% of adults with ALL, cytogenetic studies will show the presence of the Philadelphia chromosome (t(9;22) (q34;q11)), and the incidence increases with age. These Ph-positive patients tend to be older (median age 46 years vs 35 years) and to have a lower incidence of anemia and a higher incidence of leukocytosis than Ph-negative patients ( ). Ph-positive ALL patients are also likely to have FAB-L2 morphology (see Table 15.1 ), to be common ALL antigen-positive (CALLA-CD10-positive) and CD34-positive, and to have a worse prognosis than Ph-negative ALL cases. In a few patients with ALL and AML who are Ph-negative, molecular studies (e.g., polymerase chain reaction) will reveal the presence of BCR-ABL transcripts diagnostic of the Ph-chromosome abnormality not otherwise detected because of insufficient metaphases ( ).
CLINICAL MANIFESTATIONS
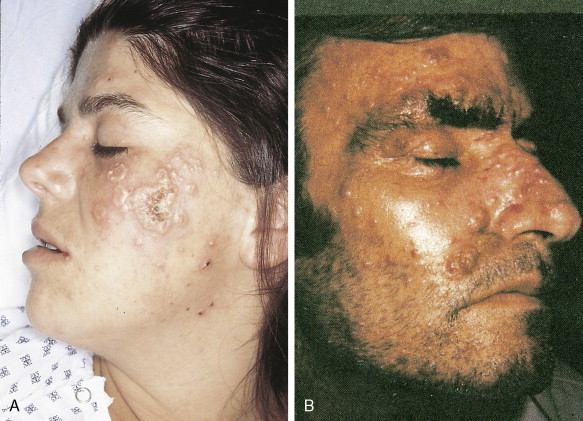
Patients can present with a variety of clinical manifestations, which correlate with the degree of marrow and other organ involvement. Among the more common symptoms are fatigue, bruisability, oral lesions, fever, and infection. Perirectal infections are particularly frequent in AML, and skin and gum infiltrates are seen mainly in AMLs. Joint swelling and bone pain with rheumatic symptoms occur commonly in ALL, which may also present as a meningitis-like syndrome. Diffuse lymphadenopathy and hepatosplenomegaly are more common in ALL (50% of cases) than in AML.
| Cytologic Features | L1 (Small Cell) | L2 (Large and Small Cell) | L3 (Burkitt Cell Type) |
|---|---|---|---|
| Cell size | Predominance of small cells | Large; heterogeneous in size | Medium; homogeneous |
| Nuclear chromatin | Homogeneous in any one case | Variable; heterogeneous in any one case | Finely stippled; homogeneous |
| Nuclear shape | Regular; occasional clefting or indentation | Irregular; clefting and indentation common | Regular; oval to round |
| Nucleoli | Not visible or small, inconspicuous | One or more present; often large | Prominent; one or more vesicular |
| Amount of cytoplasm | Scanty | Variable; often moderately abundant | Small to moderate |
| Basophilia of cytoplasm | Slight or moderate; rarely intense | Variable; deep in some cases | Very deep |
| Cytoplasmic vacuolation | Variable | Variable | Often prominent |
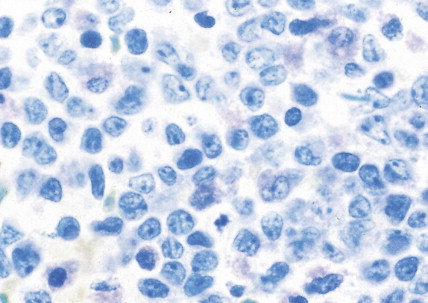
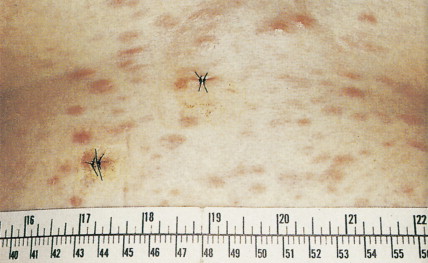
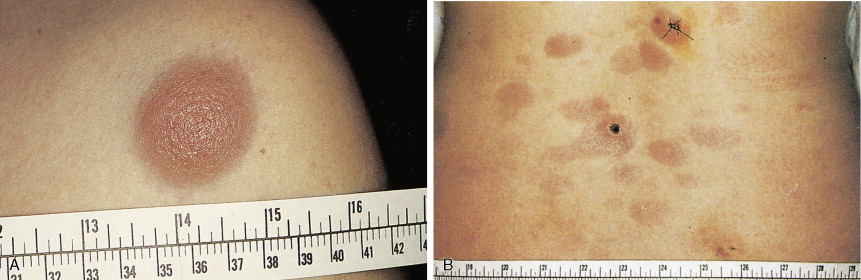
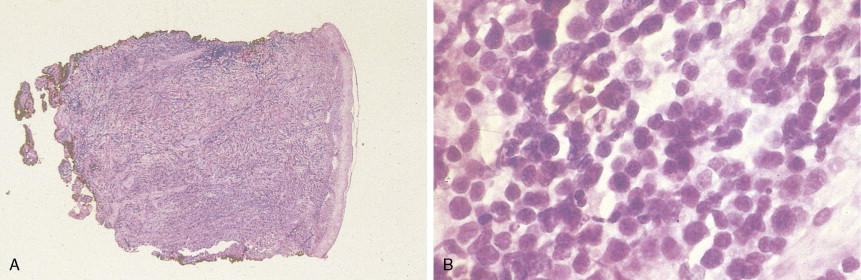
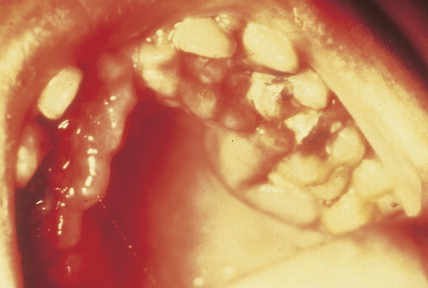
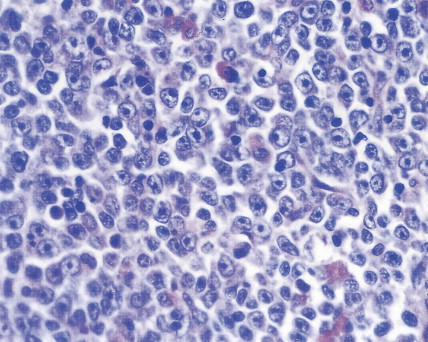
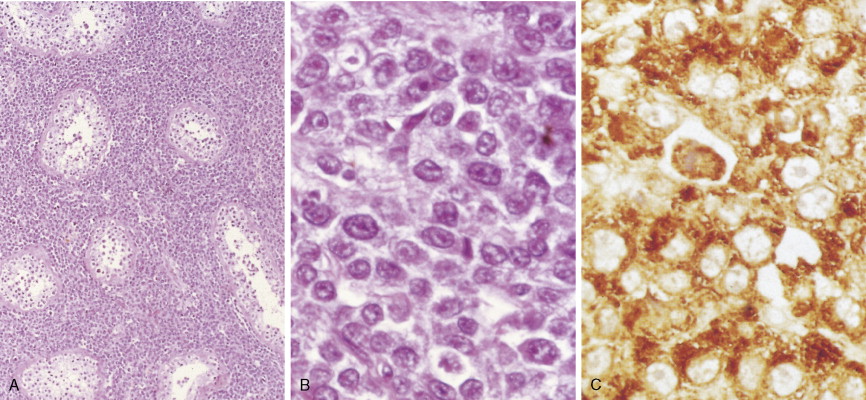
In patients with AML, unusual masses in soft tissues, nodal sites, or other areas may appear, representing collections of extramedullary immature myeloid cells. These lesions may precede overt marrow and peripheral blood invasion (and thus the diagnosis of AML) and may be mistaken for lymphoma or metastatic carcinoma. When they are localized these extramedullary leukemic infiltrates have been called myeloblastomas; the older term, granulocytic sarcoma, is inappropriate. Another term, chloroma, has been used when the tumor appears green, because of the presence in the leukemic cells of enzymes capable of metabolizing heme products; the green color rapidly disappears after oxidation on exposure to air. Myeloblastomas can occur anywhere, but the most common sites are soft tissues, skin, periosteum, bone, and lymph nodes. Diagnosis can be rapidly established on a touch prep stained with Wright-Giemsa and examined for Auer rods or azurophilic granules. The latter are peroxidase- and specific esterase–positive.
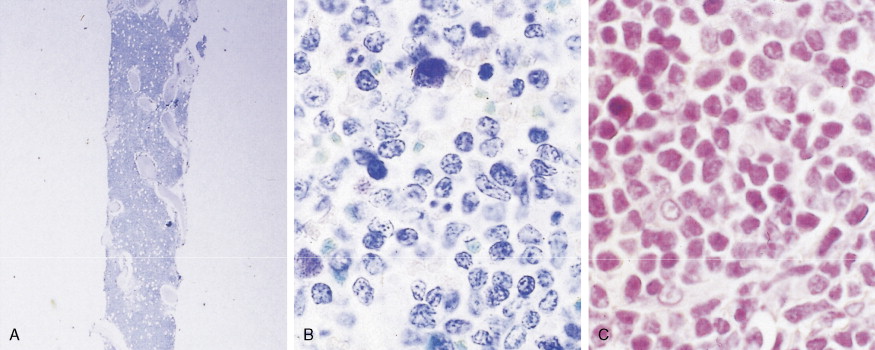
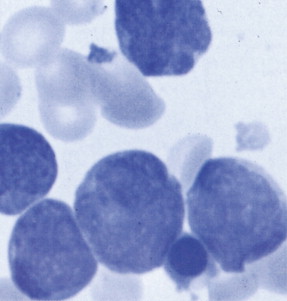
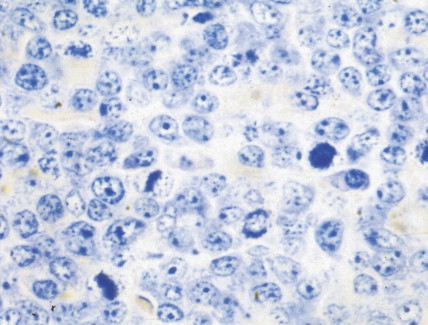
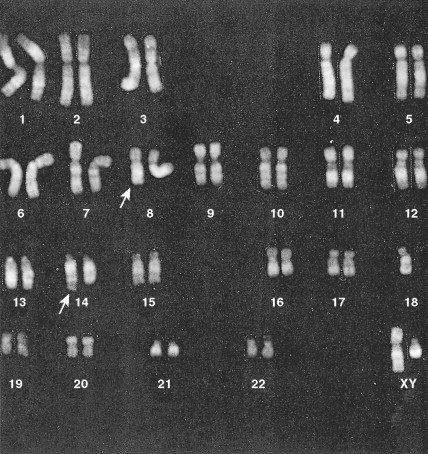
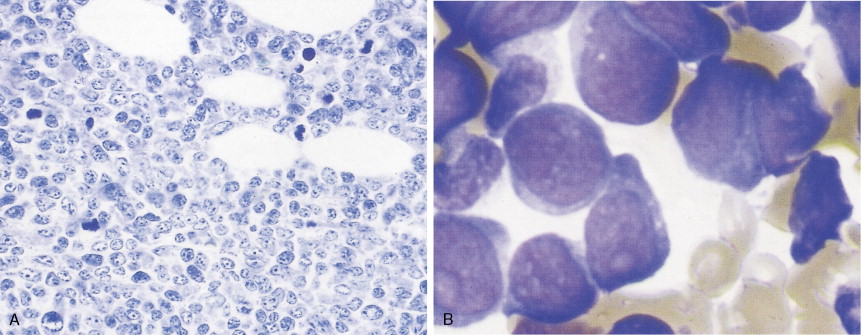
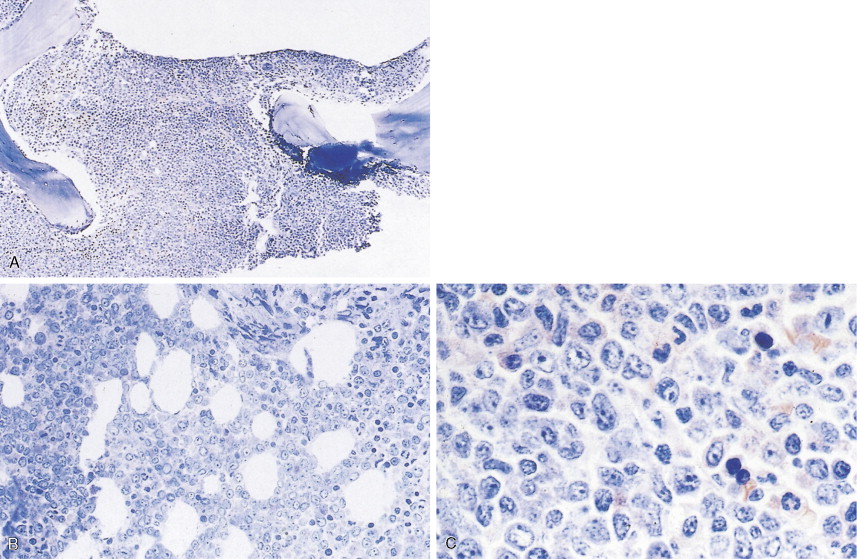
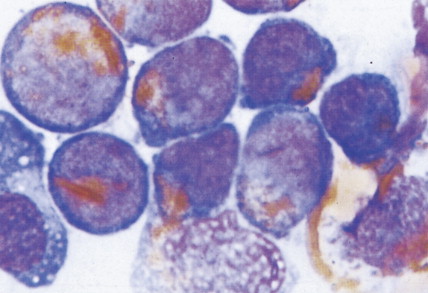
Hematologic Disorders with a Tendency to Leukemic Transformation
MYELODYSPLASTIC SYNDROMES
The myelodysplastic syndromes (MDSs) are a group of disorders that primarily occur in the elderly, who present with an anemia that proves refractory to treatment, with progressive neutropenia and thrombocytopenia, or with various combinations of these. In the past these syndromes, particularly those with normal numbers of blasts (<5%) in the marrow, have been referred to as “preleukemia.” To varying degrees there is a tendency to progression to acute leukemia, ranging from progression in approximately 10% of cases of refractory anemia and refractory anemia with ringed sideroblasts to progression in more than 40% of cases of refractory anemia with excess blasts.
Classification and Cytogenetics
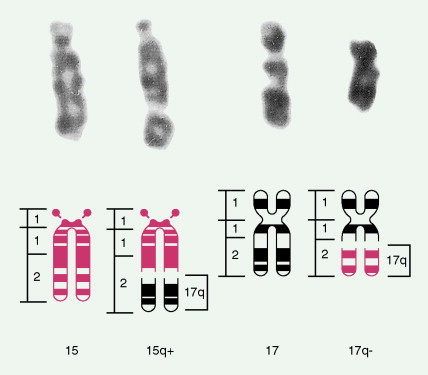
Given the variable presentation and prognosis of these disorders, various classification schemes have been devised to better divide and define these disorders. According to the French-American-British (FAB) classification scheme, the MDSs have been classified into five subgroups, which have been modified by the WHO into seven subgroups (see Fig. 15.54 ). The blood film abnormalities in each subgroup are highly variable, although general features include macrocytic red cells, qualitative granulocytic and monocytic changes, and giant platelets. Whereas patients with refractory anemia may show no gross changes in blood morphology, patients with refractory anemia with ringed sideroblasts frequently show a dimorphic red cell population. Leukoerythroblastic changes are common in patients with refractory anemia with excess blasts. “Chronic myelomonocytic leukemia,” a diagnostic entity included with the MDSs by the FAB classification scheme, is marked by abnormal myelomonocytic cells and monocytosis of more than 1.0 × 10 9 cells/L, with or without splenomegaly, and is considered separately by the WHO classification. A former category, RAEB-T, is no longer an entity under the WHO classification and is considered a progression to AML.

The bone marrow in the MDSs is typically hypercellular and shows morphologic abnormalities, often in all three series of hematopoietic cells. Cytogenetic abnormalities are common, particularly in secondary MDS (related to prior radiation therapy or therapy with alkylating agents) ( Table 15.5 ). They include –5 or –5q, –7 or –7q, –20 or –20q, +8 and –9q. Patients with complex cytogenetic abnormalities (the most common chromosomal abnormality observed) or –7 or –7q typically have an aggressive clinical course. There is usually evidence of dyserythropoiesis with nuclear atypia, some megaloblastosis, and ringed sideroblasts. In some cases reticulin is increased, whereas occasional cases are hypocellular. Granulocytic abnormalities include hypogranular or agranular myelocytes, metamyelocytes and neutrophils, pseudo-Pelger-Huët cells, and hypersegmented or polypoid neutrophils. Megakaryocyte abnormalities include small mononuclear or binuclear forms or large megakaryocytes with multiple, round nuclei and large granules in the cytoplasm. In the more advanced MDSs the blast cell population is also increased, but by definition these cells constitute less than 20% of the marrow cell total. When the level of blast cells exceeds this figure, it is assumed that a transformation to AML has occurred.
| Abnormality | Incidence (%) |
|---|---|
| Loss of all or part of chromosome 5 | 13 |
| Loss of all or part of chromosome 7 | 5 |
| Trisomy 8 | 5 |
| del 17p | <1 |
| del 20q | 2 |
| Loss of X or Y chromosome | 2 |
Clinical Manifestations and Prognosis
Clinically, patients present with symptoms related to bone marrow failure, with frequent episodes of infection and with bleeding abnormalities. These complications of severe neutropenia or thrombocytopenia result in death in many patients, but in others the disease progresses to frank AML. Typically, the liver, spleen, and lymph nodes are not enlarged. Gum hypertrophy and skin deposits do not usually occur. The prognosis is variable, and prognostic schemes have been devised. The most widely accepted and adopted has identified cytogenetics, the number of cell lines affected, and the number of blasts in the bone marrow to be important predictors of prognosis (see Table 15.6 ).
| Overall Score * | Median Survival (Years) |
|---|---|
| Low (0) | 5.7 |
| Intermediate | |
| 1 (0.5 or 1.0) | 3.5 |
| 2 (1.5 or 2.0) | 1.2 |
| High (≥2.5) | 0.4 |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


