- Type 2 diabetes mellitus (T2DM) is a heterogeneous disorder caused by a combination of genetic and environmental factors which adversely affect β-cell function and tissue insulin sensitivity.
- About 10% of patients with phenotypic T2DM actually have a late-onset form of autoimmune diabetes while up to another 5% have autosomal dominantly inherited forms such as maturity-onset diabetes of the young and mitochondrial diabetes. The remaining 85% of patients have “garden variety” T2DM which is a polygenic disorder.
- The preponderance of available information indicate that genetic factors primarily affect β-cell function whereas acquired factors (obesity, physical inactivity, glucose and lipid toxicity) are mainly responsible for the insulin resistance seen in this condition. Impaired β-cell function can be detected in genetically predisposed individuals (i.e. the monozygotic twin or first-degree relative of someone who has T2DM) at a time when they have normal glucose tolerance and normal tissue insulin sensitivity.
- T2DM develops because of a progressive deterioration in β-cell-cell function coupled with the addition of acquired insulin resistance for which the
- β-cell-cell cannot compensate. At time of diagnosis, β-cell-cell function is already reduced about 50% and most of the defects demonstrable in people with T2DM are already present in individuals with impaired glucose tolerance and those genetically predisposed to develop T2DM.
- Commonly found insulin secretion abnormalities include: absent first-phase and diminished second-phase release. Responses to ingestion of mixed meals and to non-glucose stimuli are reduced with a decrease in maximal secretory capacity. The secretory pulses, both rapid and ultradian, are smaller and less regular. The ratio of proinsulin to insulin release is also elevated.
- Normally there is a curvilinear hyperbolic relationship between β-cell function and tissue insulin sensitivity so that as tissue insulin sensitivity decreases (as in obesity and pregnancy), β-cell function increases in a compensatory fashion to maintain normal glucose tolerance. Prospective longitudinal studies have shown that people who have impaired glucose tolerance and progress to T2DM fall off this curve because of an inability of the β-cell to compensate for insulin resistance.
- Acquired factors also adversely affect β-cell function and contribute to the development of T2DM. These include adverse effects of hyperglycemia and increased plasma free fatty acids, respectively termed glucotoxicity and lipotoxicity, alterations in incretins (GLP-1, GIP) and malnutrition in utero and in early life which may affect programing of the β-cell with respect to glucose sensing, apoptosis, regeneration and the ability to compensate for insulin resistance. Endoplasmic reticulum stress-induced apoptosis and the effect interleukin-1β has on β-cell function and life cycle are recently receiving more attention for their role in the pathogenesis of T2DM.
- A pathologic feature present in 90% of patients with T2DM is the abnormal extracellular deposits of islet amyloid. It is composed of insoluble fibrils formed from the protein islet amyloid polypeptide (IAPP). Other histologic changes include a 30–50% decrease in islet mass and an alteration in the relative proportion of the islet cell population.
- The IAPP and its amyloid deposits have not been clearly found to be cytotoxic to β-cell and recent evidence from animal studies suggests that the formation of intracellular smaller IAPP oligomers is possibly the cytotoxic form associated with increased β-cell apoptosis.
Type 2 diabetes (T2DM) is a heterogeneous disorder, phenotypically, genotypically and pathogenetically. Approximately 10% of patients have a late-onset form of autoimmune diabetes which may represent a hybrid of type 1 and type 2 diabetes (see Chapter 9) [1]; up to another 5% of patients have one of the autosomally dominant inherited forms of maturity-onset diabetes of youth (MODY); another 1% may have rare genetic mutations involving insulin receptors and elements of the insulin signaling pathway (see Chapter 15). The remaining 85% of patients have “garden variety” T2DM which is the subject of this chapter.
Impaired β-cell function vs insulin resistance
The common variety of T2DM results from a combination of genetic and acquired factors which adversely affect β-cell function and tissue insulin sensitivity (see Chapter 11) [2,3]. For many years it was controversial whether impaired β-cell function or tissue insulin resistance was the underlying pathogenetic element. Until recently, it was generally thought that insulin resistance preceded β-cell dysfunction and was the primary genetic factor, while β-cell dysfunction was a late phenomenon brought about by exhaustion after years of compensatory hypersecretion [4–6]. During the past several years, however, the accumulation of evidence from sophisticated studies examining β-cell function and tissue insulin sensitivity, both cross-sectionally and longitudinally, have swung the pendulum over to the concept that impaired β-cell function is the primary underlying, probably genetic, defect [2,7–9].
The idea that insulin resistance could be the primary defect can be traced back to the classic studies of Himsworth & Kerr [10] more than 60 years ago, in which it was demonstrated that lean patients with early-onset diabetes were sensitive to exogenous injection of insulin, whereas obese patients with late-onset diabetes were resistant. Twenty-one years later, the first measurement of plasma insulin levels in patients with T2DM [11] found that they had greater than normal values in the fasting and postprandial state, providing further evidence for insulin resistance. Over the following 40 years, numerous studies (summarized in [4,5,12]) reported that patients with T2DM, people with impaired glucose tolerance (IGT), and first-degree relatives of individuals with T2DM who subsequently developed T2DM were hyperinsulinemic and insulin resistant [13,14].
The problem with interpretation of the results of these studies is that they failed to take the following into consideration:
- Importance of the dynamics of insulin secretion.
- Appropriateness of the plasma insulin level for the prevailing stimulus for insulin secretion (i.e. plasma glucose level).
- Relation of β-cell function to insulin resistance.
Importance of the dynamics of insulin secretion
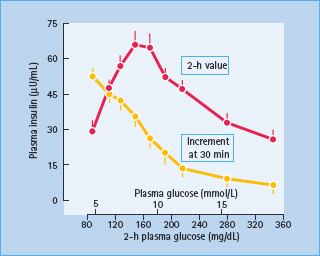
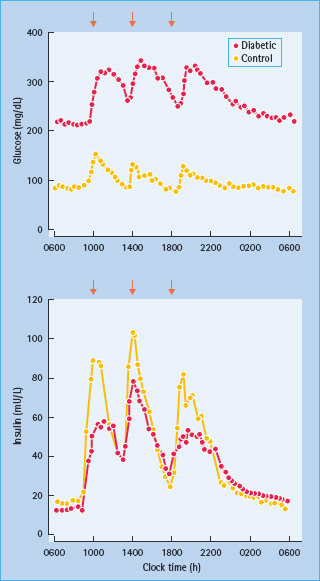
Although the finding was originally described by Yalow & Berson in 1960, it was not well appreciated until 1967 that, during oral glucose tolerance tests, the elevated 2-hour post-challenge plasma insulin levels in people with T2DM were accompanied by reduced early (30 minute) insulin responses [15]. These decreases in early insulin release are evident even in individuals with mild IGT (Figure 10.1). This reduction in early insulin response has been shown to diminish suppression of endogenous glucose production after glucose ingestion [16]; the resultant hyperglycemia provides a greater stimulus to the β-cell, explaining the late (2 hour) hyperinsulinemia. The latter had often been erroneously interpreted to be the result of insulin resistance [4–6]. Studies in normal volunteers, however, in whom early insulin responses were diminished by infusion of somatostatin so as to simulate the situation in people with IGT and T2DM produced late hyperin-sulinemia [17]. Moreover, restoration of early insulin responses either by insulin or an insulin secretogogue reduces late hyperin-sulinemia [18,19].
Figure 10.1 Relationship between early (30 minutes) and late (2 hour) plasma insulin levels and 2-hour plasma glucose levels during oral glucose tolerance tests (n = 294, mean ± SEM). Reproduced from Gerich [2], with permission from The Endocrine Society.
Appropriateness of the plasma insulin level for the prevailing glucose level
The major factor acutely regulating insulin is the plasma glucose concentration. In addition to glucose, increases in circulating amino acid, free fatty acid (FFA), gastrointestinal hormones (incretins: glucagon-like peptide [GLP-1], glucose-dependent insulinotropic peptide [GIP]) augment insulin secretion whereas increases in other factors such as catecholamines, cortisol, growth hormone, leptin, and tumor necrosis factor α(TNF-α)can reduce β-cell responses (see Chapter 6 ).
As glucose is the predominant stimulus for insulin secretion, the prevailing plasma glucose concentration must be taken into consideration in judging whether the plasma insulin concentration is appropriate. For example, the plasma insulin level in an individual with diabetes whose plasma glucose concentration is 180 mg/dL (10 mmol/L) may be twice as great as that of an individual without diabetes with a plasma glucose concentration of 90 mg/dL (5 mmol/L), but is clearly inappropriate because the non-diabetic individual would have a plasma insulin level four times as great at the same hyperglycemia [20]. Such evaluation is difficult with data from oral glucose tolerance tests (OGTT). Although equations exist that relate OGTT responses to those of hyperglycemic clamps [21], the latter are considered to be the gold standard for assessing β-cell function, because with this technique an identical stimulus for insulin release is used and both phases of insulin release can be evaluated.
Relation of β-cell function to insulin resistance
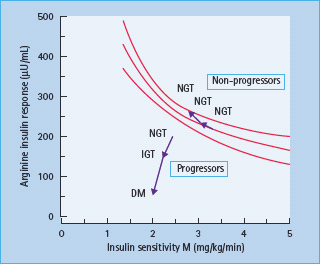
Obesity and other acquired factors that cause insulin resistance result in an adaptation of the β-cell so as to maintain normal glucose homeostasis [7,22]. This adaptation, first demonstrated in the case of obesity in 1974, results in an increase in the β-cell sensitivity to glucose [23]. The hyperbolic relationship between β-cell function and tissue insulin sensitivity, first demonstrated by Bergman et al. [24] in 1981 and subsequently confirmed in numerous other studies [7,22,25], exists in such a way that as tissue insulin sensitivity decreases, β-cell function increases to maintain normal glucose homeostasis (Figure 10.2). Thus, in addition to the prevailing stimulus for insulin secretion (e.g. plasma glucose concentration) the status of an individual’s insulin sensitivity must be taken into consideration in order to assess the appropriateness of β-cell function [7,8].
Figure 10.2 Curvilinear relationship between β-cell function (represented by the acute insulin response to intravenous arginine) and insulin sensitivity (represented by the glucose infusion rate necessary to maintain euglycemia [M] during a hyperinsulinemic clamp). People who maintained normal glucose tolerance (NGT) stayed on the curvilinear line. People whose glucose tolerance deteriorated to impaired glucose tolerance (IGT) and to type 2 diabetes (DM) fell below the line, demonstrating inadequate β-cell compensation for insulin resistance. Reproduced from Weyer et al. [22], with permission from the American Society for Clinical Investigation.
Role of β-cell defects in the natural history of type 2 diabetes
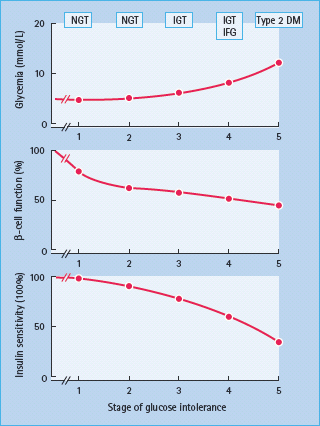
Longitudinal and cross-sectional studies indicate that most individuals destined to develop T2DM pass through five stages (Figure 10.3) [7,8,26].The first stage begins at birth, when glucose homeostasis is normal but individuals are at risk for T2DM because of genetic polymorphisms (diabetogenic genes) or an in utero environment predisposing the to become obese and limiting the ability of their pancreatic β-cells to compensate for insulin resistance. In genetically predisposed individuals with normal glucose tolerance, impaired β-cell function is demonstratable even when no insulin resistance is apparent [27–29]. During stage 2, decreases in insulin sensitivity emerge usually as a result of unhealthy lifestyles (environmental), and these, at least initially, are compensated for by an increase in β-cell secretion so that glucose tolerance remains normal. Nevertheless, despite this increase in insulin secretion, a reduction in β-cell function can be demonstrated even when individuals’ plasma glucose levels are in the normal range [27–31]. During stage 3, β-cell function deteriorates further to the point that when challenged, as during a glucose tolerance test or a standardized meal, postprandial glucose tolerance becomes abnormal. At this point, β-cell function is clearly abnormal but sufficient to maintain normal fasting plasma glucose concentrations. In stage 4, there is further deterioration in β-cell function noted at least in part from glucose toxicity as a result of postprandial hyperglycemia. This also can reduce insulin sensitivity. Fasting plasma glucose concentrations increase in this stage because of an increase in basal endogenous glucose production. Finally, in stage 5, as a result of further deterioration in β-cell function, both fasting and postprandial glucose levels reach levels diagnostic of diabetes.
Figure 10.3 The stages of glucose tolerance and associated β-cell function and insulin sensitivity, from normal glucose tolerance (NGT) through impaired glucose tolerance (IGT), with or without impaired fasting glucose (IFG), and finally type 2 diabetes mellitus (DM).
Insulin secretion normally decreases with age. The decline rate has been shown to be about 0.7–1% per year for basal and glucose-stimulated insulin secretion during adult human life in individuals with normal glucose tolerance [32–36]. The rate of decline is about twofold greater in people with IGT [33] and reaches approximately 6% per year in patients with T2DM [37,38]. Insulin sensitivity does not decrease per se with aging and decreases in insulin sensitivity when observed are most likely related to other factors such as changes in body composition and physical fitness.
Insulin secretion in type 2 diabetes, in IGT and in genetically predisposed individuals with normal glucose tolerance
Overview of insulin secretion in healthy individuals
Proinsulin, the insulin precursor, is converted to insulin and C peptide within the β-cell secretory granule. The conversion process requires the sequential action of three peptidase enzymes (prohormone convertases 2 and 3, and carboxypeptidase H) and produces four proinsulin conversion intermediates (32,33-split, 65,66-split, des-31,32-split, and des-64,65-split proinsulins) before ultimately yielding insulin and C peptide (see Figure 6.5). Normally, a small amount of intact proinsulin and its conversion intermediates, mostly the des-31,32-split proinsulin, are released into the circulation along with insulin and C peptide. They are estimated to constitute 10–20 % of total immunoreactive insulin measured in the circulation during the basal state [39,40].
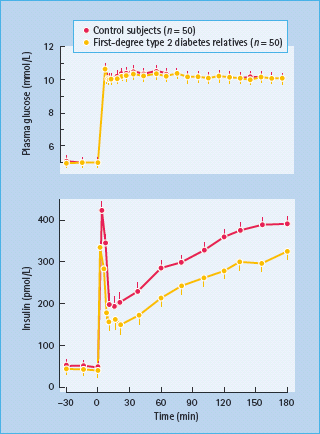
The events that lead to release of insulin from β-cells are complex (see Chapter 6). In response to an acute square wave of hyperglycemia such as that used in hyperglycemic clamp experiments in humans or in studies of perfused rat pancreas, insulin release is biphasic (Figure 10.4). It is characterized by an acute increase in insulin release lasting approximately 10 minutes, termed first-phase release, followed by a slowly increasing second phase of insulin release that is more sustained and typically persists as long as glucose is elevated. The first-phase release has been related to insulin-secretory granules located close to the β-cell plasma membrane (immediate releasable pool). In response to an increase in extracellular glucose, islet ATP and cyclic adenosine monophosphate (cAMP) levels increase, causing closure of ATP-sensitive potassium channels; this causes depolarization of the β-cell membrane and an influx of calcium through voltage-sensitive calcium channels; the resultant increase in intracellular calcium leads to movement of insulin-containing granules toward the β-cell membrane, where they merge/incorporate/melt into the membrane with release of the granules’ contents. The second-phase insulin release involves synthesis of new insulin molecules as well as ATP-dependent mobilization of granules from a storage pool into the rapidly releasable pool (see Chapter 6). Thus, the normal β-cell response to an increase in glucose concentration is dependent upon glucose entry in the β-cell and its metabolism, synthesis of insulin and insulin granules, cytochemical structures (microfilaments/microtubules) and other proteins necessary for moving granules toward the β-cell membrane and facilitating their melting into the membrane so that their contents can be released.
Figure 10.4 Biphasic plasma insulin responses during a square wave hyperglycemic clamp in normal volunteers with and without a first-degree relative with type 2 diabetes. Reproduced from Pimenta et al. [29], with permission from the American Medical Association.
The insulin secretion pattern throughout the day is more complicated than that seen during acute square wave of hyperglycemia in hyperglycemic clamp experiments. In vivo insulin secretion was found to be pulsatile, undergoing short (rapid) and long (ultradian) oscillations. The basis for these is still poorly understood, but there is evidence that the integrity of these responses is necessary for maintenance of normal glucose homeostasis [41]. The rapid oscillations correspond to serial secretory insulin bursts (Figure 10.5). These high frequency bursts occur every 5–15 minutes and account for the majority of insulin secreted in humans [42–44]. Additionally, enhanced insulin secretion following stimulation with GLP-1, sulfonylureas, or oral glucose can be accounted for by increase in the amplitude of these secretory pulses [45–48]. By contrast, inhibition of insulin secretion by somatostatin or insulin-like growth factor 1 (IGF-1) is accomplished by a decrease in the amplitude of these pulses [49,50]. These rapid oscillations are superimposed on slower and larger ultradian oscillations (Figure 10.6) which occur every 80–150 minutes. The ultradian oscillations are present during basal conditions and have clear amplification after meals. They also tend to closely follow similar oscillations of plasma glucose [51,52].
Figure 10.5 Insulin concentration profile in a canine portal vein illustrating the rapid secretory oscillations before and after glucose ingestion. There is a marked increase in pulse amplitude after glucose ingestion (arrow). Reproduced from Porksen et al. [47], with permission from the American Diabetes Association.
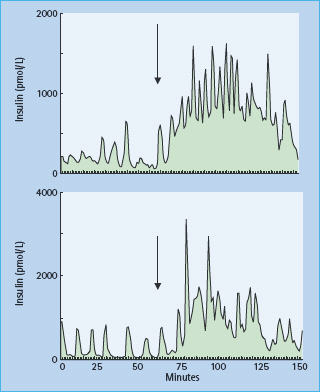
Figure 10.6 Twenty-four hour insulin secretory profi le showing ultradian oscillations in a normal weight subject. Meals were consumed at 0900, 1300 and 1800. Statistically signifi cant pulses of secretion are shown by the arrows. Reproduced from Polonsky et al. [51], with permission from the American Society for Clinical Investigation.
Abnormalities in type 2 diabetes
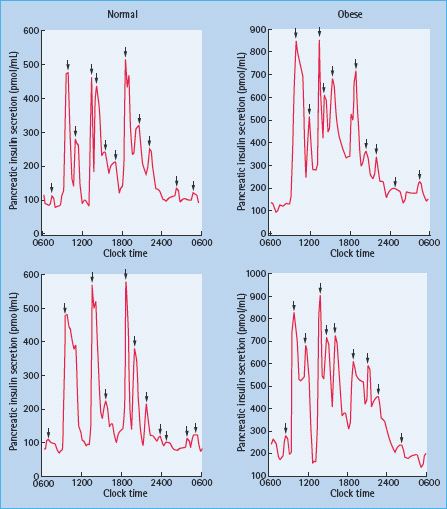
Defects in β-cell function are quite obvious by the time T2DM is diagnosed. For example, in the UK Prospective Diabetes Study, evaluation of β-cell function using the homeostasis model assessment (HOMA) indicated that β-cell function was at diagnosis already reduced by 50% and that there was subsequent further deterioration, regardless of therapy (Figure 10.7) [37]. Commonly found abnormalities include absent first-phase and diminished second-phase release in response to hyperglycemia in hyperglycemic clamp experiments [53–55]. Responses to ingestion of mixed meals (Figure 10.8) and to non-glucose stimuli are delayed or blunted with a decrease in maximal secretory capacity [55–58]. Abnormal oscillatory pattern is also observed. The pulses, both rapid and ultradian, are smaller and less regular. The ultradian pulses are less amplified after meals and less likely to follow plasma glucose oscillations [42,59].
Figure 10.7 β-Cell function as measured by the homeostasis model assessment (HOMA) method in patients with type 2 diabetes from the UK Prospective Diabetes Study (UKPDS). β-Cell function is already reduced to 50% at diagnosis and declines thereafter, despite therapy. Adapted from UK Prospective Diabetes Study Group [37].
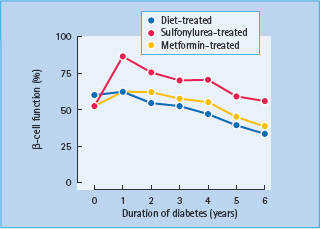
Figure 10.8 Plasma concentrations of glucose and insulin in subjects with type 2 diabetes and non-diabetic control subjects in response to mixed meals. Adapted from Polonsky et al. [165].
The ratio of proinsulin and its conversion intermediates to insulin in the circulation is at least twice as high in patients with T2DM than population norms. The increased ratio is noted in the basal and stimulated insulin secretion states and indicates less successful proinsulin to insulin conversion within β-cell secretory granules [39,40,60].
Abnormalities in patients with IGT and in genetically predisposed individuals
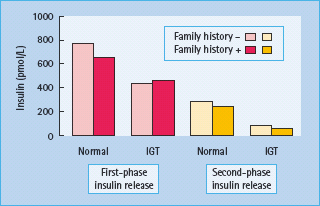
Most of the above abnormalities are also found in individuals with IGT, although at a lesser intensity. These include reduced first and second-phase responses to intravenous glucose (Figure 10.9) [61], reduced early insulin response to oral glucose [16], decreased responses to non-glucose stimuli [62], reduced ability of the β-cell to compensate for insulin resistance [7,22], alterations in rapid and ultradian oscillations of insulin secretion [63,64] as well as increased proinsulin : insulin ratio [65].
Figure 10.9 β-Cell function in individuals with normal glucose tolerance and impaired glucose tolerance (IGT), with or without a family history of diabetes. Data from Van Haeften et al. [61].
Perhaps the most convincing evidence that impaired β-cell function is the primary genetic defect comes from studies of first-degree relatives of individuals with T2DM who still have normal glucose tolerance but have reduced insulin responses in the absence of insulin resistance [2,61], especially studies of monozygotic twins [31,66–68].
Other than hyperinsulinemia to compensate for the increased insulin resistance, obese patients have insulin secretion pattern similar to non-obese subjects, and most of the above abnormalities are not present in obesity in the absence of comorbid diabetes or IGT conditions [51,69].
Possible mechanisms
Genetic causes
β-Cell dysfunction in T2DM results from a combination of genetic and acquired factors. Unlike MODY, “garden variety” T2DM is a polygenic disorder; this means that multiple genes (i.e. polymorphisms), each insufficient in themselves, must be present with or without acquired abnormalities in order to cause diabetes [70,71]. Such genes may affect β-cell apoptosis, regeneration, glucose sensing, glucose metabolism, ion channels, energy transduction, microtubules/microfilaments and other islet proteins necessary for the synthesis, packaging, movement and release of secretory granules [72,73]. To date, only a few polymorphisms have been identified as risk factors with confidence (see Chapter 12): one involves an amino acid polymorphism (Pro12Ala) in the peroxisome proliferator-activated receptor-γ (PPARγ), which is expressed in insulin target tissues and β-cells [74]; this apparently conveys susceptibility to adverse effects of FFA on insulin release. A second involves the gene encoding calpain-10, a cysteine protease that modulates insulin release as well as insulin effects on muscle and adipose tissue [75]. A third is the E23K variant of the KIR6.2 gene (potassium inwardly-rectifying channel J11 gene), which has been shown in a large association study to increase the risk of T2DM presumably through its effect on β-cells’ potassium channel and, in turn, on insulin secretion [76,77].
More recently, variants of the transcription factor 7-like 2 gene (TCF7L2) were found to be associated with increased risk of T2DM [78–80]. Insulin secretion is decreased in carriers of the at-risk alleles [78]. This is thought to be a result of impaired expression of GLP-1, a peptide encoded by the human glucagon gene (GCG) whose expression in gut endocrine cells is regulated by TCF7L2 [81].
Use of knockout techniques in mice has identified several elements of the insulin signaling cascade that might be potential sites where genetic polymorphisms may affect β-cell function, but to date none of these has been found to occur in people with T2DM [82].
Acquired factors
Several acquired and/or environment factors have been identified that may increase the risk for developing T2DM by impairing β-cell function.
Malnutrition in utero and early childhood
Animal experiments and retrospective studies in humans have provided evidence that malnutrition in utero and early childhood, as well as in utero exposure to hyperglycemia, is associated with an increased risk to develop T2DM later in life. Hales et al. [83,84] demonstrated an inverse relationship between weight at birth and at 1 year and the development of T2DM in adult life. It was proposed that malnutrition in utero and during the first few months of life may damage β-cell development; it is also possible that nutritional deficiency at this stage may program the β-cell so as to limit its subsequent ability to adapt to overnutrition. The latter possibility is supported by the fact that the strongest link was between those who were born thin and subsequently became obese. Recall that one of the hallmarks of individuals destined to develop T2DM is their failure to increase their insulin release appropriately to compensate for their acquired insulin resistance [85]. Nevertheless, because not all individuals with early life malnutrition subsequently develop T2DM if they become obese, this situation must be viewed as one risk factor among many that influence susceptibility.
Glucotoxicity
There is abundant evidence that both prolonged [86] and acute hyperglycemia [87] can adversely affect β-cell function. Moreover, improving glycemic control with the use of secretogogues, insulin sensitizers and even insulin will improve β-cell function in patients with T2DM [88–91]. The mechanisms by which hyperglycemia exerts its adverse effects on β-cells are complex and multifactorial. There is evidence that these mechanisms involve increased production of reactive oxygen species (ROS) within β-cells, oxidative stress-induced alteration of genes transcription and proteins expression, and increased β-cell apoptosis [92]. Nevertheless, because impaired β-cell dysfunction is clearly evident in genetically predisposed individuals with normal glucose tolerance [2] and because optimization of glycemic control does not completely reverse impaired β-cell function in T2DM, this so-called glucotoxicity is clearly a secondary phenomenon but one that could accelerate deterioration over time.
Lipotoxicity
In addition to elevated levels of plasma glucose, individuals with T2DM have increased circulating levels of FFA as do people who are obese [93]. Obese individuals have greater plasma FFA levels primarily because of their greater fat mass [94]. Obese people with T2DM have somewhat greater circulating FFA levels than obese people without T2DM, not because of greater release of FFA into the circulation, but because of decreased FFA clearance [95]. A number of in vitro and animal studies have demonstrated that prolonged elevation of plasma FFA impairs β-cell function [96,97]. Evidence suggests that fatty acids inhibit glucose stimulated insulin secretion, impair insulin gene expression and, more importantly, promote β-cell apoptosis [92]. One proposed mechanism involves the uncoupling protein-2 (UCP-2), a mitochondrial carrier protein that uncouples substrate oxidation from ATP synthesis [98]. FFA increase UCP-2 activity in β-cells. This impairs ATP generation from glucose metabolism and consequently decreases glucose stimulated insulin secretion [99,100]. FFA impair insulin gene expression by increasing ceramide generation which reduces the binding activity of pancreas-duodenum homeobox-1 (Pdx-1) and MafA, two transcription factors essential for regulation of multiple islet-associated genes including insulin gene [101]. In healthy human volunteers, however, it has not been possible to demonstrate consistently a deleterious effect of acute elevation of plasma FFA on β-cell function. Moreover, the effect of chronic elevation of plasma FFA on β-cell function has not been studied in humans [102,103]. Some animal and in vitro studies suggest that FFA exert their adverse effects on β-cell function only in the presence of hyperglycemia, hence the suggestion to use the term glucolipotoxicity instead of lipotoxicity [92]. It is clear that increased plasma FFA is not a primary cause of T2DM, but it is possible that a certain genetic background may prevent some individuals from responding to the adverse effects of FFA on β-cell function and thus increase their risk for developing T2DM.
Obesity
Obesity is associated with insulin resistance and is the most predictive acquired risk factor for development of T2DM (see Chapter 14) [104]. This may be mediated by a variety of factors released from adipose tissue that can adversely affect β-cell function, including elevated levels of FFA, TNF-α [105], resistin [106], leptin [107], adipsin [108] and amylin [109], as well as tissue accumulation of lipid [110,111]. Most obese individuals, however, compensate for their insulin resistance with an appropriate increase in insulin release and do not develop diabetes.
Inadequate stimulation by incretins
Incretins are hormones released by the gut in response to food ingestion, which augment insulin release in what is known as the incretin effect (i.e. more insulin responses after oral than intravenous glucose despite comparable glycemia). Two major incretins are GLP-1 and GIP (see Chapters 6 and 30). In addition to promoting the biosynthesis and secretion of insulin [112], they increase β-cell replication and inhibit its apoptosis in vitro and were found, in rodent model, to increase β-cell mass [113–116].
Patients with T2DM have higher plasma GIP levels after meal ingestion when compared with healthy subjects [114,117]. Results are conflicting in regard to GLP-1 and, if found, the differences in levels between patients with T2DM and healthy subjects have been generally modest [118–120]. The incretin effect, nevertheless, is impaired in T2DM [121,122]. Insulin responses to both GLP-1 [123] and GIP [124] are reduced. It is not clear whether the defective incretin effect is merely a manifestation of a generalized reduction in β-cell function as is the reduced insulin response to arginine [56,57,121,125]. Antidiabetic drugs, however, that enhance incretins activity or level (GLP-1 analogues and dipeptidyl peptidase-IV inhibitors) have proven to be effective in lowering glucose in T2DM [126].
Islet amyloid
A characteristic feature present in over 90% of patients with T2DM is the deposition of islet amyloid (see below). Amyloid is composed of insoluble fibrils formed from a protein called amylin or islet amyloid polypeptide (IAPP) [127,128]. Normally, IAPP is co-secreted with insulin from β-cells at the molar ratio of 1:10–50 [129]. Although the mature form of human IAPP has been reported to impair insulin release and to be β-cell cytotoxic [130], it is unlikely to have a primary role in the pathogenesis of T2DM because it is not present in all patients with T2DM and is actually found in up to 20% of islets in elderly individuals with normal glucose tolerance [131]. Moreover, most obese individuals who hypersecrete insulin do not develop T2DM even though IAPP is co-secreted with insulin.
Recent evidence from in vitro and animal studies suggests that small membrane permeant oligomers, but not the mature IAPP fibrils, are the cytotoxic form of IAPP. These oligomers form and act inside the β-cell, possibly causing endoplasmic reticulum stress-induced apoptosis [132,133]. In human, there is still no evidence that patients with T2DM have these toxic oligomers present in their pancreatic islets. However, the S20G mutation of the IAPP gene that increases the susceptibility of IAPP to form oligomers is linked to a rare familial form of T2DM [134–136].
Cytokines
β-Cell function and life cycle are known to be influenced by cytokines especially the proinflammatory cytokine interleukin-1β(IL-1β). IL-1β is known for its cytokine-meditated β-cell destruction in type 1 diabetes [137,138] but, more recently, it has received attention for its role in the pathogenesis of T2DM. In vitro studies have shown that β-cells are capable of producing IL-1β in response to high glucose and leptin levels [139,140]. IL-1β expressing β-cells were found in pancreatic sections of patients with T2DM but not in healthy subjects [140]. At low concentrations, IL-1β enhances human β-cell proliferation and decreases its apoptosis, whereas at higher concentration it impairs β-cell insulin release and increases its apoptosis [140–142]. Apoptosis here is thought to be mediated by Fas, a member of the tumor necrosis factor receptor family that triggers apoptotic cell death independent of TNF-α. IL-1β upregulates Fas expression within β-cells, ultimately enabling Fas-induced apoptosis [142,143]. An IL-1 receptor antagonist (IL-1Ra), which protected human β-cell from IL-1β adverse effects in vitro [140], improved glycemic control in patients with T2DM through enhanced β-cell secretory function in a preliminary study [144].
Other inflammatory changes and immunologic abnormalities have been reported in patients with IGT and T2DM. In addition to increased systemic cytokines, these abnormalities involve acute-phase proteins and mediators associated with endothelial cell activation [145]. Most of these changes are small and their contribution to β-cell failure is unclear.
Islet and islet cell changes in type 2 diabetes
Pancreatic islets represent 3–5% of the adult normal pancreas and β-cells comprise 60–80% of the islet cell population (see Chapter 6).
Histology of islets in type 2 diabetes
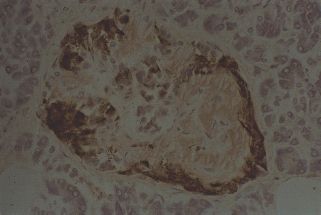
A pathologic feature present in 90% of patients with T2DM is the abnormal extracellular deposits of islet amyloid (Figures 10.10 and 10.11). These deposits are localized in the pancreas and are not part of a systemic amyloidosis disorder. The islet amyloid is formed from IAPP or amylin, a 37 amino acid peptide [127,128]. IAPP is normally produced by the β-cell, stored along with insulin in its secretory granules, and then co-secreted with insulin following β-cell stimulation [129,146]. IAPP has no known physiologic function in human. In patients with T2DM, the soluble IAPP peptides assume a β-sheet structure and oligomerize to form insoluble amyloid deposits [147,148]. It is still not clear what triggers this process and whether this simply reflects islet cell deterioration and destruction. Similar deposits, nevertheless, can be found in a minority of people without diabetes, especially with aging [131]. The IAPP and its amyloid deposits have not been clearly found to be cytotoxic to β-cells and recent evidence suggests that the formation of intracellular smaller IAPP oligomers is possibly the cytotoxic form associated with increased β-cell apoptosis in animal studies [132,133].
Figure 10.10 Amyloid deposition in islets of a patient with type 2 diabetes. Amyloid formed from islet amyloid polypeptide (IAPP; stained pink with Congo red) occupies more than 50% of the islet mass and is largely in the centre of the islet. Insulin-containing β-cells (labeled brown with immunoperoxidase) are clustered towards the periphery of the islet and are very reduced in number. Original magnifi cation +400.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


