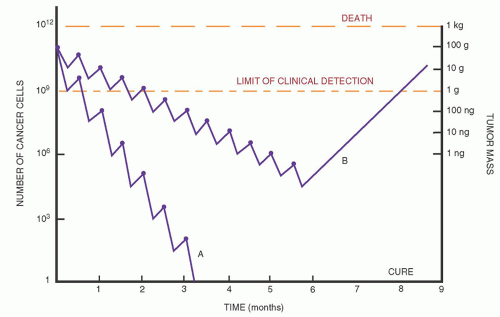
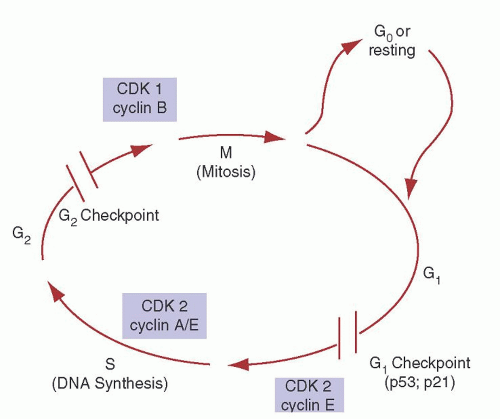
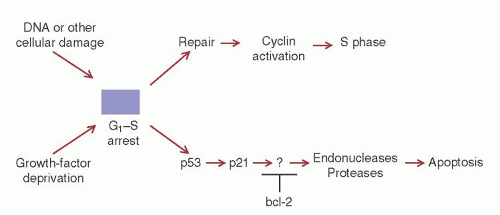
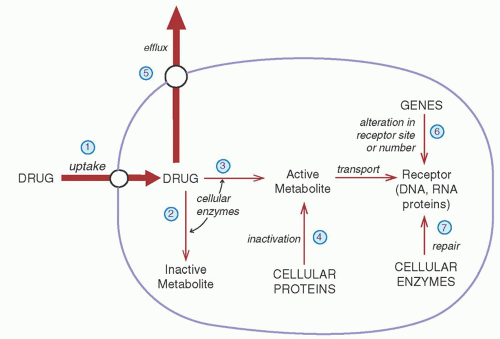
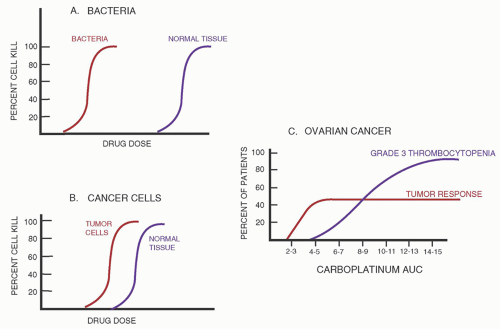
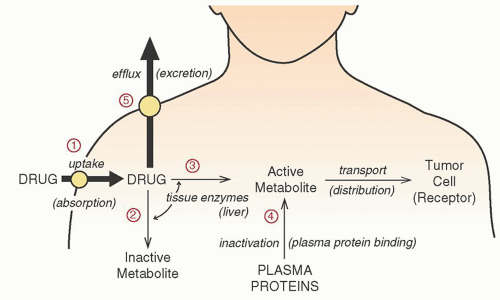
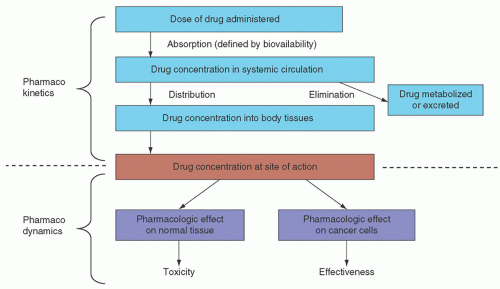
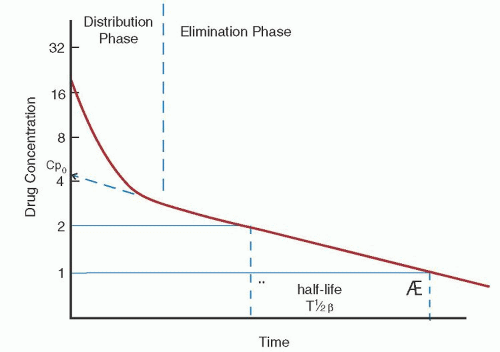
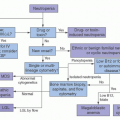
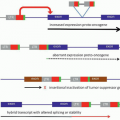
Name (Synonym) |
Drug Class |
Action |
Clearance Routea |
Major Toxicity |
Alemtuzumab (Campath®) |
Radioactive monoclonal antibody |
Binds to CD52 to target radioactivity |
Radioactive extinction |
Myelosuppression Hypersensitivity reaction Infection |
Altretamine (Hexalen®, hexamethylmelamine) |
Nonclassical alkylating agent |
Unknown, may alkylate DNA |
Hepatic metabolism |
Hypersensitivity reaction, deficient synthesis of key proteins (clotting factors, insulin), CNS depression, pancreatitis |
Anagrelide (Agrylin®) |
Phospholipase inhibitor |
Prevents megakaryocytes from maturing |
Metabolism via CYP/A2 |
Palpitations, headache, nausea, abdominal pain, dizziness |
Arsenic trioxide (Trisenox®, ATO) |
Targeted drug |
Degrades PML-RAR fusion protein |
Hepatic metabolism |
APL differentiation syndrome, Q-T prolongation, nausea, fatigue |
Asparaginase (Elspar®, Oncaspar®, pegasparaginase) |
Enzyme |
Breaks down the amino acid asparagine; sensitive lymphocytes lack ability to synthesize asparagine |
Hepatic metabolism |
N&V, neurotoxicity, myelosuppression, diarrhea, diabetes, anticoagulation |
Azacitidine (Vidaza®) |
Hypomethylating agent |
Inhibitor of DNA methylation |
Hepatic metabolism |
Myelosuppression |
Bexarotene (Targretin®) |
Retinoid |
Binds to the retinoid X receptor to induce cellular differentiation |
Oxidative hepatic metabolism |
Hepatotoxicity, hyperlipidemia, hypothyroidism, photosensitivity, teratogenicity |
Bendamustine (Trenda®) |
Alkylating agent |
Forms DNA cross-links |
Hydrolysis in plasma to inactive metabolites |
Nausea, fatigue, myelosuppression, fever |
Bevacizumab (Avastin®) |
Monoclonal antibody to VEGF |
Decreases angiogenesis |
Protein degradation |
Hypertension, headache, bleeding, thrombosis, proteinurea |
Bleomycin (Blenoxane®) |
Antibiotic |
Single-strand DNA breaks |
Renal |
Hypersensitivity reaction, pulmonary fibrosis, skin and mucocutaneous reactions, fevers |
Bortezomib (Velcade®) |
Proteosome inhibitor (targeted agent) |
Inhibits protein destruction blocking NFK-β |
Oxidative hepatic metabolism |
Nausea, fatigue, diarrhea, peripheral neuropathy, thrombocytopenia |
Brentuximab vedotin |
Monoclonal antibody |
Binds with CD30 antigen with toxin then internalized |
Hepatic |
Hypersensitivity reaction, neuropathy, fatigue, fever, diarrhea, neutropenia |
Busulfan (Myleran®, Busulfex®) |
Alkylating agent |
Forms DNA cross-links. |
Metabolism |
Myelosuppression, hepatotoxicity (venoocclusive disease), pulmonary fibrosis |
Capecitibine (Xeloda®) |
Antimetabolite |
A 5-FU prodrug |
Hepatic metabolism |
Diarrhea, myelosuppession, palmar-plantar erythrodysethesia |
Carboplatin (CBDCA, Paraplatin®) |
Platinum complex |
Produces DNA cross-links |
Renal |
Thrombocytopenia, leukopenia, nephrotoxicity, ototoxicity, neuropathy, N&V |
Carmustine (BCNU) |
Nitrosourea |
Alkylates DNA at O6 position of guanine |
Hepatic metabolism |
Delayed (4-6 wk) myelosuppression, pulmonary toxicity, hepatotoxicity |
Cetuximab (Erbitux®) |
Monoclonal antibody |
Binds to the epidermal growth factor receptor |
Binding of antibody to receptor |
Anaphylactic reaction, skin rash, fevers |
Chlorambucil (Leukeran) |
Alkylating agent |
Cross-links DNA |
Metabolism |
Myelosuppression, pulmonary toxicity, hepatotoxicity |
Cisplatin (CDDP) (Platinol®) |
Platinum complex |
Produces DNA cross-links |
Protein binding |
Nephrotoxicity, N&V, ototoxicity, alopecia, neuropathy |
Cladribine (LeustatinTM) (2-chlorodeoxy adenosine) |
Antimetabolite (purine analog) |
Incorporation into DNA; NAD consumption |
Renal |
Myelosuppression, fever, renal toxicity (high-dose) |
Clofarabine (Clolar®) |
Antimetabolite |
Incorporates into DNA; inhibits DNA polymerase |
Renal |
Nausea, hepatotoxicity, palmar-plantar erythrodysesthesia |
Cyclophosphamide (Cytoxan®, Neosar®) |
Alkylating agent |
Cross-links DNA strands |
Hepatic metabolism (renal) |
Myelosuppression, N&V, cystitis, cardiac (high-dose) |
Cytarabine (Cytosar®, ara-C, cytosine arabinoside, DepoCytTM) |
Antimetabolite (pyrmidine analog) |
Incorporates into DNA; inhibits DNA polymerase |
Hepatic metabolism |
Myelosuppression, N&V, mucositis, ocular, hepatic |
Dacarbazine (DTIC) |
Nonclassical alkylating agent |
DNA methylation |
Renal (hepatic metabolism) |
Vesicant, myelosuppression, N&V, hepatic |
Dactinomycin (Cosmegen®) (actinomycin-D) |
Antibiotic |
DNA intercalation |
Biliary |
Myelosuppression, N&V, vesicant, mucositis |
Dasatinib (SprycelTM) |
Targeted agent, signal transduction inhibitor |
Inhibits the tyrosine kinase of several growth factor receptors including bcr-abl |
Hepatic metabolism (CYP 3A4) and biliary excretion |
Fluid retention, N&V, diarrhea, myelosuppression, hypothyroidism |
Daunorubicin (Cerubidine®, Dauno Xome®) |
Antibiotic (anthracycline) |
Topoisomerase inhibition, DNA intercalation, free-radical formation |
Biliary excretion, hepatic metabolism |
Myelosuppression, N&V, cardiomyopathy, vesicant, red urine, mucositis |
Decitabine (Dacogen®) |
Hypomethylating agent |
Allows activation of tumor suppressor genes |
Hepatic deamination |
Myelosuppression, fatigue, nausea, teratogen |
Denileukin diftitox (Ontak®) |
Toxin-fusion protein |
Binds to the IL-2 receptor, where the diphtheria toxin is internalized |
Proteolytic degradation |
Infusion reactions (fever, hypotension, myalgias), skin rash, transaminitis, vascular leak syndrome, hypothyroidism |
Docetaxel (Taxotere®) |
Tubulin binder |
Mitotic spindle inhibitor |
Hepatic metabolism, biliary excretion |
Myelosuppression, hypersensitivity (steroids needed), fluid retention, neuropathy |
Doxorubicin (Adriamycin®, Rubex®, MyocetTM, Doxil®) |
Topoisomerase inhibitor (Anthracycline) |
Topoisomerase inhibition, free-radical formation |
Biliary excretion, hepatic metabolism |
Myelosuppression, N&V, cardiomyopathy, vesicant, red urine, mucositis |
Epirubicin (EllenceTM) |
Topoisomerase inhibitor (Anthracycline) |
Inhibits topoisomerase II |
Hepatic metabolism and excretion |
Nausea, vomiting, myelosuppression, cardiac toxicity |
Erlotinib (Tarceva®) |
Targeted agent |
Inhibits the tyrosine kinase of the epidermal growth factor receptor |
Hepatic oxidative metabolism |
Skin rash, diarrhea |
Etoposide (VePesid®, VP-16, Etopophos®, Toposar®, etoposide phosphate) |
Topoisomerase inhibitor |
Inhibits topoisomerase II |
Renal (hepatic metabolism) |
Myelosuppression, mucositis, hypersensitivity reaction |
Everolimus (Affinitor®) |
Targeted therapy |
Blocks oncogenic pathway through m-TOR inhibition |
Hepatic metabolism through CYP3A4 |
Edema, rash, stomatitis, diarrhea, myelosuppression, infection |
Fludarabine (fludarabine phosphate, Fludara®) |
Antimetabolite (purine analog) |
Inhibits DNA polymerase, incorporation into DNA and RNA, NAD depletion |
Renal |
Myelosuppression, mucositis, hypersensitivity reaction, neurologic |
Fluorouracil (5-FU, Adrucil®, FUDR®) |
Antimetabolite (pyrimidine analog) |
Inhibits thymidylate synthetase, incorporated into DNA and RNA |
Hepatic metabolism |
Myelosuppression (more with bolus), diarrhea & mucositis (more with continuous infusion), stomatitis, cardiac ischemia, CNS (cerebellar ataxia) |
Gefitinib (Iressa®) |
Targeted therapy |
Block the tyrosine kinase of EGFR |
Hepatic metabolism via CYP3A4 |
Rash, diarrhea |
Gemcitabine (Gemzar®) |
Antimetabolite |
Inhibits ribonucleotide reductase, incorporated into DNA as false nucleotide |
Metabolism |
Myelosuppression, nausea, diarrhea, hepatic, fever |
Hydroxyurea (Hydrea®, DroxiaTM, MylocelTM) |
Antimetabolite |
Inhibits ribonucleotide reductase |
Hepatic metabolism, renal |
Myelosuppression, mucositis |
Idarubicin (Idamycin®) |
Topoisomerase inhibitor (anthracycline) |
Similar to doxorubicin |
Hepatic |
Similar to doxorubicin |
Ifosfamide (Ifex®) |
Alkylating agent |
Cross-links DNA strands through alkyl groups |
Hepatic metabolism, renal excretion. |
Myelosuppression, N&V, neurologic, alopecia, cystitis (must be given with MESNA) |
Imatinib mesylate (GleevecTM, STI-575) |
Targeted agent |
Inhibits the tyrosine kinase of the bcr-abl and c-kit oncogenes |
Hepatic metabolism |
Nausea, diarrhea, fluid retention, abnormal LFTs, hypothyroidism |
Interferon-α (INF-α, Intron A®, Roferon®) |
Biologic |
Degradation of messenger RNAs, modulation of oncogene expression, increase in NK cells and other immunoregulatory elements |
Renal metabolism |
Fever, chills, myalgias, headache, fatigue, anorexia, myelosuppression, hepatic, CNS, depression |
Ibritumomab (ZevalinTM) |
Monoclonal antibody |
Antibody to CD20 coupled to Y90 |
Radioactive decay |
Myelosuppression, allergic reactions, hypothyroidism |
Irinotecan (Camptosar®, CPT-11) |
Topoisomerase I inhibitor |
Inhibits topoisomerase I |
Metabolism, biliary excretion |
Myelosuppression, diarrhea, pneumonitis, stomatitis |
Lenalidomide (Revlimid®) |
Immunomodulator |
Uncertain—possible TNF-α inhibitor; inhibits angiogenesis |
Renal |
Teratogenicity, myelosuppression, DVTs, diarrhea, fatigue |
Lomustine (CeeNU®, CCNU) |
Alkylating nitrosourea |
Same as carmustine |
Same as carmustine |
Same as carmustine |
Mechlorethamine (nitrogen mustard, Mustargen®) |
Alkylating agent |
Cross-links DNA via alkylation |
Tissue binding |
Vesicant, ototoxicity, myelosuppression, N&V |
Melphelan (Alkeran®, L-PAM, phenylalanine mustard) |
Alkylating agent |
Cross-links DNA strands via alkylation |
Spontaneous degradation, protein binding |
Myelosuppression, pulmonary fibrosis (rare), N&V (high-dose) |
Mercaptopurine (6-MP, Purinethol®) |
Antimetabolite (purine analog) |
Incorporation into DNA |
Hepatic metabolism |
Myelosuppression, hepatotoxicity |
Methotrexate (MTX) |
Antimetabolite (folic acid analog) |
Inhibits dihydrofolate reductase with decreased thymidylate and protein synthesis |
Renal excretion |
Myelosuppression, mucositis, hepatotoxicity (chronic low-dose), renal (high-dose), pulmonary |
Mitomycin (Mutamycin®) |
Antibiotic |
Cross-links DNA strands |
Hepatic metabolism |
Myelosuppression, N&V, vesicant, pulmonary, hepatic, renal |
Mitoxantrone (Novantrone®, DHAD) |
Anthraquinone |
Similar to doxorubicin |
Hepatic metabolism |
Similar to doxorubicin, blue-green urine |
Nelarabine (Arranon®) |
Antimetabolite (purine analog) |
Incorporated into DNA and blocks DNA replication |
Hepatic demethylation |
Neurotoxicity including somnolence, fatigue, dizziness, headache, myelosuppression |
Nilotinib (Tasigna®) |
Targeted therapy |
Inhibits the tyrosine kinase of BCR/ABL |
Hepatic metabolism via CYP3A4 |
Myelosuppression, QT prolongation, N & V, hepatic toxicity, edema |
Ofatumumab (Arzerra®) |
Monoclonal antibody |
Binds to CD20 |
Proteolytic degradation |
Infusion reaction, infection, myelosuppression, HBV reactivation |
Oxaliplatin (Eloxatin®) |
Platinum complex |
Produces DNA cross-links |
Renal and tissue binding |
Hypersensitivity reaction, neuropathy, hepatitis, pulmonary fibrosis |
Paclitaxel (Taxol®, Abraxane®) |
Plant alkaloid |
Mitotic spindle inhibitor, stabilizes microtubulin. |
Hepatic metabolism, biliary excretion |
Myelosuppression, hypersensitivity syndrome (use with steroids and antihistamines), mucositis, neuropathy, myalgia |
Panitumumab (VectibixTM) |
Monoclonal antibody |
Binds to EGFR |
Proteolytic degradation |
Rash, infusion reaction, diarrhea |
Pazopanib (Votrient®) |
Targeted agent |
Inhibits the TKI of VEGF receptors |
Hepatic metabolism via CYP3A4 |
Hypertension, hair color change, diarrhea, myelosuppression, QT prolongation, hepatotoxicity |
Pemetrexed (Alimta®) |
Antimetabolite |
An antifolate that inhibits dihydrofolate reductase, and thymidylate synthetase |
Renal |
Myelosuppression, fatigue, N&V |
Pentostatin (Nipent®, 2-deoxycoformycin) |
Antimetabolite (purine analog) |
Adenosine deaminase inhibitor. |
Renal |
Myelosuppression, fever, rash, hepatotoxicity, pulmonary, CNS |
Prelatrexate (Folotyn®) |
Antimetabolite |
Inhibits DHFR (see methotrexate) |
Renal |
Myelosuppression, mucositis |
Procarbazine (Matulane) |
Nonclassical alkylating agent |
Alkylates DNA; DNA strand breaks |
Hepatic metabolism |
Myelosuppression, N&V, CNS (confusion, depression), MAO inhibition, hepatic, pulmonary |
Rituximab (Rituxan®) |
Monoclonal antibody |
Binds to CD20 on lymphocytes and initiates complementmediated cytotoxicity |
Proteolytic degradation |
Fevers, hypersensitivity reaction, Hepatitis B reactivation, infection |
Sorafenib (Nexavar®) |
Targeted therapy |
Inhibits the tyrosine kinase of VEGER, PDGFR, c-kit, and FLT-3 |
Heptic metabolism: Oxidation and glucuronidation |
Fatigue, palmar-plantar erythrodysethesia, hypertension, hyperphosphatemia, rash proteinuria |
Streptozocin (Zanosar®) |
Alkylating nitrosourea |
Methylation of O6-guanine of DNA |
Renal |
Myelosuppression, N&V, renal, diabetes, vesicant |
Sunitinib (Sutent®) |
Targeted therapy |
Inhibits the tyrosine kinases of VEGFR, PDGFR, c-kit; inhibits angiogenesis |
Hepatic oxidative metabolism |
Hypertension, bleeding, diarrhea, mucositis, fatigue |
Temozolomide (Temodar®) |
Atypical alkylating agent |
Methylates DNA guanine resulting in strand breaks |
Metabolism by hydrolysis |
Myelosuppression, N & V, fatigue |
Teniposide (VM-26, Vumon®) |
Microtubulin inhibitor |
Binds to topoisomerase II, causing DNA strand breaks |
Hepatic metabolism |
Myelosuppression, hypersensitivity reactions |
Temsirolimus (Torisel®) |
Targeted agent |
Inhibits m-TOR |
Hepatic metabolism and biliary excretion |
Edema, hyperlipidemia, myelosuppression, hepatic toxicity, hyperglycemia |
Thalidomide (Thalomid®) |
Immunomodulatory agent |
Suppresses TNF, blocks angiogenesis, increases IL2 and interferon |
Nonenzymatic hydrolysis |
Birth defects, thrombosis, fatigue, somnolence, neuropathy |
Thioguanine (6-thioguanine, 6-TG, Tabloid®) |
Antimetabolite (purine analog) |
Incorporates into DNA as fraudulent nucleotide |
Hepatic metabolism |
Myelosuppression, hepatic venoocclusive disease |
Thiotepa (Thioplex®) |
Alkylating agent |
Trifunctional alkylating agent, cross-links DNA |
Metabolism |
Myelosuppression, stomatitis |
Topotecan (Hycamtin®) |
Topoisomerase I inhibitor |
Inhibits the enzyme topoisomerase I, causing DNA stand breaks |
Renal |
Myelosuppression, N&V |
Tositumomab (Bexxar®) |
Monclonal antibody |
Radioactive drug that binds to CD20 |
Proteolytic degradation |
Myelosuppression, fever, nausea, hypersensitivity reaction |
Trastuzumab (Herceptin®) |
Monoclonal antibody |
Binds to the Her2neu oncogene, resulting in apoptosis |
Protein binding and proleotytic degradation |
Hypersensitivity reaction, cardiomyopathy, fever, diarrhea |
Tretinoin (ATRA, Vesanoid®) |
Targeted retinoid therapy |
Induces maturation of promyelocytes |
Hepatic oxidative metabolism and glucuronidation |
Fever, dyspnea, pulmonary infiltrates (retinoic acid symdrome), headache, fever, neurologic, hepatic |
Venmurafanib (Zelboraf®) |
Targeted agent |
Inhibits the tyrosine kinase of the BRAF oncogene |
Hepatic metabolism |
Rash, erythema, squamous cell skin cancers, photosensitivity, abnormal LFTs, fatigue |
Vinblastine (Velban®, VLB) |
Microtubulin inhibitor |
Binds to tubulin, prevents formation of mitotic spindle |
Hepatic |
Myelosuppression, vesicant, neurotoxin |
Vincristine (Oncovin®, Vincasar®, VCR) |
Microtubulin inhibitor |
Binds to tubulin, prevents formation of mitotic spindle |
Hepatic |
Neurotoxin, vesicant, CNS |
Vinorelbine (Navelbine®) |
Microtubulin inhibitor |
Binds to tubulin, prevents formation of mitotic spindle |
Hepatic |
Myelosuppression, vesicant, neuropathy |
Vismodegib (Erivedge®) |
Targeted agent |
Inhibits the Hedgehog signaling pathway |
Hepatic metabolism and excretion |
Teratogenic, fatigue, muscle spasms, diarrhea, alopecia |
Vorinostat (Zolinza®) |
Histone deacytylase inhibitor |
Inhibits histone deacytelase to unmask DNA methylation |
Hepatic |
Fatigue, diarrhea, N&V pulmonary emboli |