INTRODUCTION
SUMMARY
Blood cell production is an enormously complex process in which a small number of hematopoietic stem cells expand and differentiate into an excess of 1011 cells each day. Based on a number of strategies available to the experimental hematologist a hierarchy of hematopoietic stem, progenitor, and mature blood cells is emerging in which each successive developmental stage loses the potential to differentiate into a specific type or class of cells. The characteristics of the stem and progenitor cells that give rise to the cells of the blood are the subject of this chapter, including the roles played by transcription factors and external signals in lineage fate determination, the cytokines and cell adhesion molecules that support cell survival, self-renewal, expansion, and differentiation, and the cell surface properties that allow for their purification, and biochemical and genetic characterization. A thorough understanding of hematopoietic stem and progenitor cells and their supportive microenvironment can provide critical insights into developmental biology of multiple cell systems, favorably impact blood cell development for therapeutic benefit, impact genetic therapy for a number of blood and other human diseases, and potentially provide the tools necessary to allow the regeneration of multiple organs.
Acronyms and Abbreviations:
AGM, aorta-gonad-mesonephros; BFU-E, burst-forming unit–erythroid; BFU-MK, burst-forming unit–megakaryocyte; CAFC, cobblestone area-forming cell; CAR, CXCL12–abundant reticular; CFC, colony-formingcell; CFU-E, colony-forming unit–erythroid; CFU-GM, colony-forming unit–granulocyte-macrophage; CFU-MK, colony-forming unit–megakaryocyte; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; EBF, early B-cell factor; ECM, extracellular matrix; EGF, epidermal growth factor; EPO, erythropoietin; EPOR, erythropoietin receptor; FAK, focal adhesion kinase; FL, Flt-3 ligand; G-CSF, granulocyte colony-stimulating factor; G-CSF-R, granulocyte colony-stimulating factor receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GM-CSF-R, granulocyte-monocyte colony-stimulating factor receptor; GMP, granulocyte-macrophage progenitor; HSC, hematopoietic stem cell; Ig, immunoglobulin; IL, interleukin; IRF4, interferon regulatory factor 4; LEF, lymphoid-enhancer binding factor; LR, laminin receptor; LTC, long-term culture; LTC-IC, long-term culture-initiating cell; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony-stimulating factor; MEP, megakaryocyte-erythroid progenitor; MK, megakaryocyte; MSC, mesenchymal stem cell; PI3K, phosphoinositol 3′-kinase; R, receptor; RAG, recombination activating gene; ROS, reactive oxygen species; SCF, stem cell factor; SCL, stem cell leukemia; SDF-1, stromal-derived factor-1; SLAM, signaling lymphocyte activation molecule; TCF, T-cell factor; TGF, transforming growth factor; TPO, thrombopoietin; VCAM, vascular cell adhesion molecule; VLA, very-late antigen.
AN OVERVIEW OF HEMATOPOIESIS
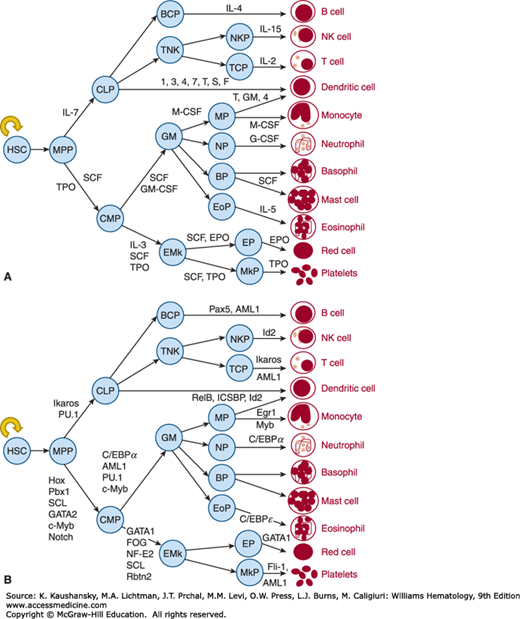
Blood cell production is an enormous and complex process. Based on the adult blood volume (5 L), the number of each of the blood cell types per microliter of blood, and their circulatory half-life, it can be calculated that each day an adult human produces 2 × 1011 erythrocytes, 1 × 1011 leukocytes, and 1 × 1011 platelets. These numbers can all increase approximately 10-fold in states of blood cell destruction or enhanced need. Over the past four decades experimental hematologists have developed a model of blood cell production in which a hierarchical developmental progression of primitive, multipotential hematopoietic stem cells (HSCs) gradually lose one or more developmental potentials and ultimately become committed to a single cell lineage, which matures into the corresponding blood cell type.1 Perhaps one of the most compelling arguments supporting this model of hematopoiesis is derived from extensive purification schemes using cell surface markers that yield cells at each predicted developmental stage (Fig. 18–1).2 Although hematopoietic development is considered by most investigators as an irreversible stepwise and progressive loss of developmental potentials, studies now suggest that cells undergoing apparent differentiation steps might oscillate between different stages depending on their position in the cell cycle.3 But regardless of the precise relationships between different stages of hematopoietic development, the availability of this model and the data leading to its construction have provided important insights into the biology and clinical uses of hematopoietic stem and progenitor cells. This chapter focuses on our understanding of the molecular basis for blood cell development, beginning with the HSC and its offspring, the lineage-committed progenitor cells.
Figure 18–1.
The figure displays the hematopoietic progenitors that have been defined by in vitro assays or by more complex tissue-based assays. In (A) the growth factors responsible for cell survival and proliferation at each corresponding stage of hematopoietic development are shown, and in (B) the corresponding transcription factors are illustrated. See text for definitions, except that T,GM,4 represents tumor necrosis factor alpha (TNF-α), GM-CSF and IL-4, and 1,3,4,7,T,S,F represents IL-1, IL-3, IL-4, IL-7, TNF-α, SCF, and Flt3 ligand. Although a single type of macrophage is illustrated, the blood monocyte can differentiate into a plethora of tissue specific macrophage types, including the hepatic Kupffer cell, the brain microglia, and the bone osteoclast (details are provided in Chaps. 67 and 69). Similarly, a single dendritic cell is shown, but of two distinct origins, lymphoid or myeloid (Chap. 20).
DEVELOPMENTAL BIOLOGY OF HEMATOPOIESIS
Blood cell production begins in the yolk sac,4 where extraembryonic mesoderm develops into angioblasts and primitive erythroid precursors at day 7 postcoitum of the mouse; cells of the outer layer of the undifferentiated mesoderm at this time flatten and become endothelial cells, and the inner cells round up to become clusters of erythroid precursors,5 termed blood islands(Chap. 7). Like in the embryo proper, there is much evidence to suggest that these two cells are derived from a common precursor (the hemangioblast).6 Once adjacent blood islands begin to coalesce on day 8, the endothelial cells form vascular channels, which by day 8.5 connect with the embryonic vasculature, allowing yolk sac blood cells to exit the blood islands, complete their maturation, and enucleate in the embryonic bloodstream.7 In both mouse and man there is a stage of embryonic development where both primitive erythrocytes (as characterized by ζ globin phenotype) and definitive red cells are produced in the yolk sac, although the former appears only very transiently. Although not as well characterized, yolk sac myelopoiesis and thrombopoiesis also occur, perhaps as part of the development of multipotent progenitors that appear by day 8.5 postcoitum. Cells capable of differentiating into multiple cell lineages become recognizable early during yolk sac hematopoiesis.8 However, such cells reproducibly engraft only in the marrow of myeloablated embryonic animals and not in adults,9 making it unlikely that such cells are true HSCs, although this topic remains controversial. By day 11 postcoitum repopulating HSCs are clearly present in the yolk sac, but the relationship of these cells and the HSCs that are clearly demonstrable a day earlier in a region of the embryonic paraaortic splanchnopleure known as the aorta-gonad-mesonephros (AGM) is not certain. By day 12.5, postcoitum hematopoiesis in the murine yolk sac is eliminated.
Although it was long believed that the developmental origin of the adult mammalian hematopoietic system was the yolk sac, subsequent research has shown that the first adult-type HSCs are derived from mesodermal cells within the ventral wall of the dorsal aorta of the AGM.10,11,12 The AGM remains a source of hematopoiesis between days 9.5 and 11.5 postcoitum in the mouse, and days 30 and 37 in the human.13,14 Of interest, the development of hematopoietic cells in this region (as well as in the yolk sac) occurs in a “reverse” direction, that is, single lineage-committed progenitors appear prior to multilineage progenitors, which appear prior to stem cells. This region also has cells that express a number of molecules in common with endothelial cells, including CD34, the transcription factors SCL and GATA-2, and the receptors c-KIT and FLK-1.15 Moreover, cell culture experiments have established that such cells display combined endothelial and hematopoietic potential, establishing them as “hemangioblasts,” the postulated combined endothelial cell–hematopoietic precursor.16
Approximately 2 days following the appearance of HSCs in the AGM region, hematopoiesis begins in the murine fetal liver. Careful dissection experiments of the 1970s indicate that fetal liver hematopoiesis is dependent on an exogenous source of hematopoietic cells,17 which populate the fetal liver in two waves, consisting of erythroid and multilineage progenitors around day 9 of murine gestation and committed progenitors and true HSCs at day 11.18 Although there is no direct proof, the temporal appearance of these cell types in the AGM approximately 1 to 2 days prior to their appearance in the fetal liver strongly suggests that the former is the source for populating the latter. In humans the fetal liver becomes the major source of blood cells around 5 weeks of gestation, and the marrow begins to populate with hematopoietic cells at 8 weeks of gestation. Unlike the random pattern of cells seen in the yolk sac, hematopoiesis in the fetal liver is well organized; erythroid cells are usually found in clusters surrounding a central macrophage and CD15+ myelopoietic cells localize mainly around portal triad vessels, although lymphoid precursors fail to demonstrate a specific localization pattern and are randomly found amongst hepatocytes.19 Up to 50 percent of the fetal liver is composed of hematopoietic cells at days 12 to 14 of murine embryonic life, a proportion that begins to decrease as hepatocytes replace hematopoietic cells and the latter shift to the marrow, prior to birth.
The final shift in the site of hematopoiesis occurs before birth; although the marrow begins to populate with liver derived hematopoietic cells at day 16 in the mouse and at 8 weeks gestation in the human, it is mostly myeloid in nature and contributes little to the circulating blood until just before birth.20 Hematopoietic stem and progenitor cells circulate in large numbers during fetal life, as clinically witnessed by the use of umbilical cord blood as a rich source of HSCs for transplantation. However, shortly after birth neonatal blood has very few primitive hematopoietic cells, as they begin to home to and lodge in the marrow. Genetic studies reveal that marrow localization of HSCs is dependent on the chemokine CXCL12 (previously known as stromal cell-derived factor [SDF]-1)21 as elimination of the chemokine or its receptor (CXCR4) leads to marrow hypoplasia.22 The shifts in localization of hematopoiesis during mammalian development are likely the result of changes both in the cell surface adhesion molecules on hematopoietic stem and progenitors that occur during ontogeny, and the characteristics of stromal cells of the yolk sac, AGM, fetal liver, and adult marrow that provide the microenvironmental support of HSC survival, homing and lodgment, self-renewal, proliferative expansion, and differentiation (Chap. 7).
THE HEMATOPOIETIC STEM CELL
Although the concept of a common “mother cell” of all blood elements in the adult dates to Maximov in 1909, and its potential for participation in disease as proposed by Danchakoff in 1916,23 the basic concepts of a hierarchical organization of stem and progenitor cells leading to mature blood cell production were coalesced by Till and McCulloch using a spleen colony-forming assay, experimentally establishing the existence of multipotential hematopoietic cells.24 The capacity to transplant marrow cells and reconstitute all aspects of hematopoiesis in myeloablated recipients provided an in vivo assay for the HSC, but it was not until the development of clonal in vitro assays of lineage-committed progenitors that a coherent model of blood cell production began to emerge. The pioneering work of Pluznik and Sachs25 and of Bradley and Metcalf26 provided methods to enumerate and characterize marrow cells committed to the hematopoietic lineage. These investigators independently developed culture conditions that allowed colonies of leukocytes to develop from single progenitors. However, as a result of the more fastidious conditions required for erythropoiesis and megakaryopoiesis in vitro, the description of methods to culture these progenitors did not occur for another decade or more.27,28,29,30,31 Work using density fractionation, cell sorting, and fluorescent dye exclusion methods has yielded purified populations of stem cells,32,33,34,35,36 common myeloid37 and lymphoid38 progenitors, and lineage-restricted hematopoietic progenitors,39,40 methods that have greatly advanced our understanding of the cell and molecular biology of blood cell development. Figure 18–1 depicts a working model of this process.
Based on transplantation data indicating that there are a remarkably similar total-body number of HSCs in mice and cats, it has been estimated that all mammals, including humans, possess 2 × 104 stem cells,41 and because only a small fraction of these are cycling (and therefore contributing to blood cell production) at any given time, it is also clear that daily blood cell development from the few cycling stem cells to produce the approximately 4 × 1011 mature blood cells represents a massive amplification process. However, the capacity of HSCs to contribute to hematopoiesis changes with age (Chap. 9). The number of HSCs increases with age in some but not all strains of mice.42,43 Also, HSC differentiation in aged animals is skewed toward the myeloid rather than lymphoid lineage.44 The molecular basis for these changes are undergoing intense study.45,46,47,48
Another measure of stem cell kinetics is the time it takes for transplanted marrow cells to repopulate a lethally irradiated animal. Studies using retroviral markers suggest that HSCs can be divided into short-term and long-term repopulating cells, based on the timing of their appearance in the blood following intravenous transplantation (fewer than or more than 3 months following transplantation in mice).49 However, a rapidly repopulating stem cell has been identified using a direct marrow injection strategy, a cell capable of generating large numbers of erythroid and myeloid cells within 2 weeks of injection.50 Moreover, by transplanting luciferase-labeled single stem cells, a strategy that allows the serial tracking of the cells during life, initially detected foci were found to expand locally, seed other sites in the marrow or spleen, and then recede with different kinetics.51 From these experimental approaches it is clear that HSCs are heterogeneous.
Experimental transplantation in animals affords the clearest estimation of HSC properties as the capacity to durably regenerate all of hematopoiesis in an otherwise lethally irradiated animal remains the gold standard for the field; moreover, the technique can be made quantitative. Typically, either 2 × 105 genetically marked, whole murine marrow cells, or reduced numbers of variably purified cells are infused intravenously into recipient animals who had previously received 90 to 110 cGy of whole-body irradiation. Blood cells and marrow are monitored for hematopoietic recovery in the following weeks and months, and the success of the transplant is measured by survival, and long-range contribution to hematopoiesis in the recipient. The contribution of donor cells to recovery is established by analysis of the posttransplant blood or marrow cells; the most common method of distinguishing donor from residual recipient blood and marrow cells is the use of flow cytometry against isoforms of the cell membrane-bound phosphatase CD45, present on virtually all hematopoietic cells. In a more quantitative embodiment of the strategy, limiting numbers of the genetically distinct cells (e.g., CD45.1+) are mixed with a “just adequate” (for full recovery) number of alternately marked cells (e.g., CD45.2+) and the proportion of CD45.1 to total CD45.1+ plus CD45.2+ cells is assessed following transplantation, yielding a calculation of the number of stem cells in the initial inoculum, an approach termed competitive repopulation.52 Because there exist both “short-term” and “long-term” repopulating cells, the degree of donor cell chimerism is tested 3 or more months following transplantation, to be certain that only the latter are evaluated. For example, transplantation of megakaryocyte-erythroid progenitor (MEP) cells allows for survival in a lethally irradiated mouse, as these cells allow sufficient time for endogenous recovery of the small number of relatively radio-resistant HSCs in the recipient mouse.53 Consequently, survival alone following cell transplantation is not a sufficient measure of the presence of stem cells in a given population. Thus, with the appropriate caveats, this approach allows an assessment of the numbers or “quality” of HSCs in the test population (i.e., some genetically altered stem cell populations repopulate less robustly than wild-type cells as a consequence of defects in cytokine receptors or other genes that affect the self-renewal, survival, or proliferation of stem cells). Based on the use of these experimental tools, we know most about murine HSCs. Obviously, this approach is not available to assess human HSCs. Instead, a number of alternate experimental approaches have been developed.
Severely immunocompromised mice can be engrafted by human HSCs, provided their survival can be supported in a strictly controlled animal care environment and that the experiments take place prior to the development of other untoward effects in such animals (e.g., tumor formation). The first assay employing this strategy relies on the combined immunodeficiency created by the severe combined immunodeficiency (SCID) and nonobese diabetic (NOD) genetic mutations.54 Subsequently, these mice were found to bear some ability to reject or alter the developmental characteristics of human cell repopulation, leading other investigators to add genetic defects to the NOD-SCID background that improve the engraftment of normal and pathologic human marrow cells, such as NOD-SCID/β2-microglobulin null,55 or the most commonly used NOD-SCID/γc null,56 or by crossing with mice that also express human hematopoietic cytokines.57 Such animal models have allowed the assessment of (1) stem cell numbers in human CD34+ cells from mobilized blood or umbilical cord blood,58 (2) assessment of the effects of gene therapy vectors,59,60 cell-cycle inhibitors,61 or cytokine cocktails designed to expand stem cell numbers58,59,60,61,62,63,64 on the retention of repopulating capacity, or (3) the study of fundamental biologic properties of human HSCs in vivo, such as the cell-cycle restriction of repopulating cells.65
Although in vivo assays remain the gold standard, NOD-SCID and more severely immunocompromised mice are difficult to maintain and remain expensive and quite cumbersome methods to assess human HSC quality and quantity. As a result, a number of culture-based methods were developed to more quickly and quantitatively evaluate human HSC function. Generally, each relies on long-term cell growth in culture and other special features to establish their validity as a model of the human HSC.
The ability to grow marrow cells in culture for extended periods of time provided an important tool to explore HSC biology.66 In long-term cultures human or murine marrow is incubated in serum-containing medium under defined conditions, and after several weeks the stromal layer that has developed is recharged with fresh marrow cells, which then produce mature blood cells and their progenitors for many months. Cell fractionation studies show that the HSC resides adherent to the stromal cell layer in such cultures,67 and that enzymatic disruption of the stromal layer will allow one to reseed a secondary stromal cell layer with the capacity to produce hematopoietic cells for a period of weeks to months, thereby defining an in vitro assayable cell termed the long-term culture-initiating cell (LTC-IC).68 A second assay that was developed based on similar principles is the cobblestone area-forming cell (CAFC), which, when evaluated by phase-contrast microscopy, gives rise to complex colonies of multiple hematopoietic cell types under the stromal cell layer of long-term cultures.69 Unfortunately, when careful comparisons are made between these assays and transplantation studies, the true HSCs comprise only a fraction of the repopulating cells found in marrow. Thus, conclusions about stem cell behavior from such in vitro assays cannot be considered rigorous.
Numerous investigators have used monoclonal antibodies to an increasing number of hematopoietic cell surface proteins to negatively and/or positively enrich for stem and primitive hematopoietic progenitor cells. Although the function of only a few of these stem cell markers is known, it has not impeded their use for research and/or therapeutic benefit. Others have taken advantage of the capacity of primitive hematopoietic cells to extrude fluorescent organic chemicals or on their buoyant density to obtain purified populations of these scarce marrow cells; most successful stem cell purification strategies employ several such techniques.
The antigenic proteins and glycoproteins that exclusively or predominantly present on HSCs include (1) CD34, a 90- to 110-kDa type I glycoprotein that is postulated to mediate cell adhesion and/or cell-cycle arrest70,71,72; (2) CD90 (Thy1),73 a heavily glycosylated glycophosphoinositol-linked protein that participates in T-cell adhesion to stromal cells74; (3) CD117 (the c-Kit receptor),75 which supports primitive hematopoietic cell survival and proliferation76,77; (4) AA4,34 a murine molecule homologous to the human phagocyte C1q complement receptor78; (5) Sca1,79 a murine surface molecule shown by knockout studies to be necessary for normal stem cell development80; (6) CD133,81 a 115-kDa pentaspan cell-surface glycoprotein expressed on the apical surface of neuroepithelial and HSCs that has been proposed to function in establishing or maintaining plasma membrane protrusions82; (7) CD164,83 a cell-surface sialomucin that is present in several alternately spliced isoforms and that enhances blood cell homing and inhibits CD34+/CD38− cell proliferation,84 and CD150, a member of the signaling lymphocyte activation molecule (SLAM) family of lymphocyte proliferation receptors85; and (8) CD110 (the thrombopoietin [TPO] receptor c-Mpl)86 present on virtually all repopulating HSCs,87 and established to be vital for human HSC physiology as genetic elimination of the receptor leads to congenital amegakaryocytic thrombocytopenia at birth and aplastic anemia shortly thereafter (Chap. 117).
Many or most of the surface membrane proteins found on HSCs are also present on cells that have begun to differentiate toward specific lineages, precluding the exclusive use of positive selection alone for stem cell purification. Thus, a number of stem cell purification strategies include negative selection, based on cell surface markers absent on HSCs but present on mature blood cells and their corresponding unilineage-committed progenitors. Typically, cocktails of negatively selecting antibodies include CD38, HLA-DR, CD3, CD4, CD5, or CD8 for T lymphocytes; CD11b, CD14, or Gr-1 to exclude macrophages and granulocytes; CD10, CD19, CD20, or B220 to eliminate B lymphocytes; and glycophorin A or Ter119 to remove erythroid cells. The products that result from the use of such combinations of negative-selecting antibodies are termed Lin− cells.
A particularly difficult problem is presented by separating true HSCs from their progeny committed to the lymphoid or myeloid lineage, but not differentiated beyond that stage. Several studies clarify the cell surface profile of the common lymphoid progenitor (CLP) as Lin−/interleukin (IL)-7R(receptor)α+/Thy1−/Sca-110w/c-kitlow37 and the common myeloid progenitor (CMP) as Lin−/IL-7Rα−/c-Kit+/Sca-1−.36 The cell surface phenotype of human HSCs includes CD34+/CD38−/KDR(VEGFR2)+/Thy1+/CD133+/Lin−, although most of these markers require careful clinical assessment before their widespread use in patients can be considered. At present, at least for murine cells, that problem has been solved. The most effective cell sorting method for purifying murine HSC is the E-SLAM approach, negatively selecting for CD48 and positively selecting for CD45, CD201, and CD150. It is estimated that such a cell population is at least 50 percent pure HSC based on single-cell transplantation experiments in mice.
Integrins are a family of heterodimeric single-pass transmembrane proteins (18 α and 8 β subunits form at least 24 different cell surface adhesion receptors in humans) characterized by multiple immunoglobin (Ig)-like extracellular domains that allow two-way communication between a cell and its environment.89 A large number of cell types require contact for survival; in vitro, this is usually manifest as integrin-dependent cell adhesion, either to extracellular matrix protein(s) or to other cells. In such cultures, disruption of adherence causes programmed cell death; for example, endothelial cells undergo apoptosis upon forced detachment in vitro, as a result of disruption of multiple integrins.90 Integrins also influence the proliferation of cells by affecting the G1 to S phase transition of the cell cycle.91 These effects also operate in vivo; α1 integrin (a component of the α1β1 collagen receptor) null mice have a hypoplastic dermis, and the growth of α1 −/− fibroblasts on collagen is substantially reduced.92
Hematopoietic stem and progenitor cells express multiple integrins, including α4 β1 (also termed very-late antigen [VLA] 4), which binds to either vascular cell adhesion molecule (VCAM) 1 or fibronectin, and α5 β1 (VLA5), which binds to a region of fibronectin distinct from the β1β1 binding domain. Moreover, primitive hematopoietic cells are thought to express integrin αIIbβ3, the platelet fibrinogen receptor, based on the death of multiple hematopoietic lineages in mice expressing a suicide transgene under control of the integrin αIIb promoter.93 However, the physiologic significance of this finding is uncertain at present.
The avidity of progenitor cell–integrin interactions can be altered by external effectors; numerous cytokines and chemokines, including cytokines critical for stem cell function (stem cell factor [SCF], TPO, and CXCL12), enhance integrin-mediated binding.94–96 Counterreceptors for both integrins, such as VCAM1 and fibronectin (FN), are highly expressed in the marrow matrix and on marrow stromal cells (see “Matrix Proteins” in “The Hematopoietic Microenvironment” later). Integrin-based interactions with the stroma are responsible for homing and retention of stem and primitive progenitor cells in the marrow, as antibodies that interfere with the interaction can mobilize stem and progenitor cells into the blood.97 However, it is uncertain whether integrins can influence the survival or growth of HSCs, or affect their ultimate developmental fate.
One of the hallmarks of HSCs is their resistance to chemotherapy-induced cytotoxicity. A primary reason for this property is high-level expression of drug efflux pumps of the multidrug resistance class of proteins.98 The presence of these verapamil-sensitive efflux pumps has enabled the separation of HSCs based on their low-level retention of various fluorescent markers such as rhodamine 123 and Hoechst 33342, the “Rhlo/Holo” population of murine cells and the side population (SP) of cells in human marrow.99 However, before such maneuvers can be used for clinical stem cell enrichment procedures, the lack of toxicity of the fluorescent dyes must be confirmed. Nevertheless, such experimental strategies continue to shed important insights into HSC biology.
As a consequence of residing physically removed from the marrow vasculature, the stem cell microenvironment is hypoxic (see “Anatomy” in “Hematopoietic Microenvironment”). The metabolic consequence is that HSCs display a low metabolic state, with most of its ATP generation derived from glycolysis. As the HSC rarely undergoes cell division, it can “afford” the low metabolic state of cells dependent on glycolysis. One of the transcription factors critical for HSC physiology, MEIS1,100 appears responsible for this metabolic profile, by driving expression of hypoxia-inducible factor (HIF)-1α, that upregulates expression of the glycolytic enzyme machinery.101 In addition, HIF-1α also induces pyruvate dehydrogenase kinases (PDK1-4) that prevent mitochondrial pyruvate oxidation by suppressing pyruvate dehydrogenase complex, the first step that fuels the Krebs cycle. Stem cell down-modulation of mitochondrial oxidative phosphorylation acts to reduce the generation of reactive oxygen species (ROS), which is highly toxic to HSCs. It has been suggested that this property, the avoidance of ROS generation, contributes to the longevity of the stem cell pool in any individual.
As HSCs mature into committed progenitors, they migrate toward the marrow vasculature, and into a higher oxygen tension atmosphere. This would appear to be required to gain the additional metabolic rate afforded by oxidative phosphorylation. As one component of this “metabolic switch,” marrow hematopoietic progenitor cells express lower levels of PDKs, reducing PDK-mediated suppression of oxidative phosphorylation seen in HSCs.102
Adult hematopoietic cells display altered engraftment capacity dependent on their phase in the cell cycle. Using primitive hematopoietic cell populations several investigators have demonstrated that only quiescent G0/G1 phase cells engraft into lethally irradiated recipient animals; cells in the S and early G2 phase display minimal engraftment capacity,102,103 a situation that can be experimentally manipulated; elimination of p21, a key cell-cycle progression gene, enhances stem cell expansion.104 This finding correlates well with findings that the profile of expressed genes in a highly selected population of primitive hematopoietic cells shifts when they are induced from G0/1 phase into the cell cycle.105 However, even though this cell-cycle dependence of engraftment of stem cells is true for adult cells, the corresponding cell populations derived from umbilical cord blood or fetal liver is not cell-cycle dependent.106 A better understanding of these findings is very likely to shed important new insights into the genes that regulate engraftment.
It can be argued that the most critical feature of the HSC is its ability to quantitatively balance its three fates—apoptosis, self-renewal, and differentiation—into the mature elements of the blood. Moreover, the undifferentiated cell must express (at the least) the initiating genes responsible for all possible developmental lineages. A useful conceptual framework for this process can be constructed by considering the gene expression profiles of stem and committed hematopoietic progenitors that develop into the multiple hematopoietic differentiation pathways. At each developmental step genes associated with the adopted pathway should remain expressed or be upregulated, while the genes that specify the alternate lineage(s) are likely silenced. A thorough understanding of these gene expression profiles should help to explain the circuitry of specific aspects of hematopoiesis, and of developmental biology in general.
Initial studies using immortalized multipotent hematopoietic cell lines reinforced this conceptual framework; pluripotency is characterized by the expression of multiple genes associated with multiple cell fates.107 Studies of purified HSCs and lineage-committed progenitors have also strengthened this hypothesis, revealing coexpression of several different lineage-affiliated gene sets in single primitive hematopoietic cells.108 In contrast, the downstream progenitors of HSCs were found to express only lineage-appropriate transcripts, such as for the granulocyte colony-stimulating factor receptor (G-CSF-R) in granulocyte-macrophage progenitors (GMPs), or β-globin and the erythropoietin receptor (EPOR) in committed erythroid progenitors.36 Similar findings were reported for lymphoid committed cells, although some promiscuity was detected in B-cell progenitors.109
With these principles established, more ambitious efforts to catalogue all the genes expressed by each stage of hematopoietic development have been made possible by advances in microarray and whole transcriptome sequencing approaches to gene expression.110 On an even broader scale, and as might be expected, comparisons of different types of stem cells (e.g., liver, skin, neural) reveals an overlap in the expressed genes, supporting the hypothesis that the mechanisms responsible for critical stem cell properties, such as self-renewal, are shared among the cells derived from multiple organs.111 This observation also provides a powerful tool to identify such proteins. Such studies have also begun to identify novel genes expressed in HSCs, potentially allowing our better understanding of their role in hematopoiesis.
An important goal of modern cell biology is to provide a molecular explanation for the gene or sets of genes required to orchestrate specific developmental events. Fundamental to this process is an understanding of the proteins present in cells that regulate gene transcription in a lineage-, ontogenic stage-, and developmental level-specific manner. Unlike what is claimed for many organ-specific programs, no single lineage-unique family of master regulators exerts executive control over hematopoiesis. Rather, an assemblage of specific and nonunique factors and signals converge to determine lineage and differentiation patterns. Several transcription factors have been identified in stem cell populations or have been shown to affect stem cell differentiation into the lymphoid and myeloid lineages. In addition to transcription factors that regulate HSC expansion, a number of epigenetic and microRNA changes have been identified that affect gene expression in these cells. The BMI1 gene encodes a protein that forms part of a polychrome group repressor complex, which represses a number of important target genes including the cell-cycle regulator p16/INK4a, a pathway that regulates HSC function in normal and malignant hematopoiesis.112 In addition, methylation can affect HSC gene expression, as the DNA methyltransferases DNMT3A and DNMT3B affect HSC self-renewal.113 And microRNA (miRNA) species are regularly being identified that regulate transcription and translation of critical HSC genes. For example, 9 miRNA were overexpressed and 22 downregulated in CD34+/CD38− HSCs compared with CD34+/CD38+ cells. Among the most upregulated miRNAs in the more primitive cells was miR-520h, predicted to target ATP-binding cassette, subfamily G (ABCG2) gene, known to be involved in stem cell maintenance. Transduction of miR-520h into CD34+ cells increased the numbers of several progenitor cell types (colony-forming unit–erythroid [CFU-E], burst-forming unit–erythroid [BFU-E], and colony-forming unit–granulocyte-macrophage [CFU-GM]) as well as the total number of CD34+ cells (reviewed in Ref. 114).
Members of the Hox family of transcription factors are important regulators of hematopoietic cell decisions, at least at the level of self-renewal/expansion, based on (1) a similar role in multiple organ systems115; (2) their lineage- and differentiation-stage-specific expression pattern in hematopoietic cells116; (3) disruption of their usual level or pattern of expression that leads to hematologic expansion or malignancies117; and (4) their elimination,118 or elimination of the gene(s) that regulate them,119 which leads to significant defects in hematopoiesis. In addition, members of the extradenticle family of homeodomain-containing proteins serve as cofactors for Hox proteins, altering their cellular localization, DNA-binding affinities, and specificities. Like Hox genes, genetic elimination of some of these cofactor proteins can lead to HSC defects. For example, Pbx1 null mice display greatly reduced numbers of CMPs,120 and overexpression or altered expression of MEIS1 is associated with hematologic malignancy.121
The Ikaros gene encodes a family of lymphoid-restricted zinc-finger transcription factors related to the Drosophila hunchback gene.122 All isoforms of Ikaros contain a highly conserved carboxyl-terminal activation domain and two zinc-finger domains that mediate their dimerization. However, only isoforms 1 to 3 of the six known alternately spliced forms contain more than three of the four N-terminal zinc fingers required for DNA binding to the consensus DNA core motif GGGA.123 The PU.1 gene is 1 of approximately 30 members of the Ets family of transcription factors that bind to the purine-rich sequence 5′-GGAA-3′.122 Genetic elimination of the Ikaros and PU.1 genes have established their critical role in commitment of HSCs to the lymphoid lineage; fetal stem cells in Ikaros −/− mice fail to generate any definitive T- or B-lymphocyte precursors,124 and although thymocyte precursors can be identified postnatally, they undergo aberrant differentiation or fail to develop into the CD4, dendritic, and some γδT-cell subsets in adult mice. Consequently, Ikaros is essential for all of lymphopoiesis early during ontogeny, and for several subsets of lymphocytes later in life. In a similar fashion, PU.1-deficient mice also lack any definitive T- and B-cell precursors in their lymphoid organs at birth (and myeloid cells; see “HSC to CMP Commitment” below),125 and if knockout mice are maintained on antibiotics and survive the first 48 hours of life, they begin to develop normal-appearing T cells 3 to 5 days later. In contrast, mature B cells and macrophages remain undetectable in the older mice, indicating absolute tissue dependence for this lineage.
The SCL (stem cell leukemia) gene encodes one of the transcription factors responsible for the initial stages of myeloid development, a gene first identified at the site of chromosomal rearrangement in a patient with SCL.126 SCL belongs to the helix-loop-helix family of transcription factors, which form dimers and bind DNA at consensus E-box motifs (CANNTG).127 Although initially identified as a gene rearranged in T-cell acute lymphocytic leukemia, an essential role for SCL in hematopoietic development was established by gene ablation studies, which revealed a complete absence of primitive blood cells and lethality in scl−/− embryos at day 9.5 postcoitum.128 Consistent with this panhematopoietic phenotype, previous studies showed that SCL is downregulated in differentiating granulocytic and monocytic progenitor cells and that forced expression of the gene in hematopoietic cell lines inhibits cytokine-induced granulocytic and monocytic differentiation.129,130 Consistent with their respective roles in promoting stem cell and mature cell survival and proliferation, SCF sustains SCL expression in primary CD34+ cells, maintaining them in an undifferentiated state, whereas granulocyte-monocyte colony-stimulating factor (GM-CSF) downregulates SCL levels and favors granulocyte and monocyte differentiation.130,131 Together, these results suggest that SCL expression is required for HSC and CMP maintenance, and that down-modulation of the transcription factor is essential for myeloid differentiation.
The GATA transcription factor family contains six members possessing a highly related DNA-binding domain composed of two conserved zinc-finger motifs.132 GATA1 and GATA2 are present in hematopoietic cells, GATA2 is found in the same cells as SCL, with GATA1 expression restricted to latter stages of erythroid/megakaryocytic (MEP) differentiation. Because genetic elimination of GATA2 is lethal as a result of numerous nonhematopoietic defects, and because individual hematopoietic lineage-specific knockouts have not yet been engineered, the role of GATA2 in early hematopoiesis is uncertain. However, like SCL, elimination of GATA2 expression is required for hematopoietic cell maturation.133
As noted above, numerous lines of evidence indicate that HSCs express the TPO receptor, c-Mpl, as best exemplified by its expression on all AA4+/Sca+ cells that are capable of long-term hematopoietic repopulation.87 Several investigators have shown that the 5′ flanking region of the c-mpl gene contains a functionally important GATA site and that GATA1 transactivates the gene in hematopoietic cell lines.134 Because GATA1 does not appear in hematopoietic cells until they have lost their repopulating capacity, it is possible that GATA2 fulfills this role in HSCs, although there is no evidence yet available establishing that this protein can transactivate the c-mpl GATA site.135
While blood cell counts do not change substantially in elderly mammals, a number of alterations are demonstrable in HSCs derived from older individuals. In aged mice, the HSC compartment expands, although each HSC has reduced capacity to expand.136 Upon transplantation, aged marrow HSCs display a myeloid skewing, generating reduced numbers of T- and B-lymphoid precursors, that along with thymic involution helps to explain the immune depletion seen in older adults. Similar findings were reported when aged human stem cells were transplanted into immunocompromised mice.137 This topic has been reviewed.138
THE HEMATOPOIETIC MICROENVIRONMENT
It has been estimated that the concentration of cells within the marrow is 109/mL; as a result, multiple cell–cell and cell–matrix interactions occur. A major advance in experimental hematology has been the capacity to grow hematopoietic cells in long-term culture. When high concentrations of marrow cells are placed in serum-containing cultures, a stromal cell layer and extracellular proteinaceous matrix form, and when subsequently recharged with fresh marrow cells, these long-term cultures (LTCs) are capable of supporting hematopoiesis for months with simple demidepletion and replacement of culture medium. It is assumed that the cell–cell and cell–matrix interactions that develop in such cultures more closely resemble those found in vivo, helping to explain the longevity of such cultures and their capacity to maintain hematopoietic stem and primitive progenitor cells far longer ex vivo than do nonstromal cell-containing cultures. The molecular basis for the improved hematopoietic environment of LTCs is thought to rely on stromal cell surface molecules that promote cell–cell contact, prevent programmed cell death, and regulate growth.
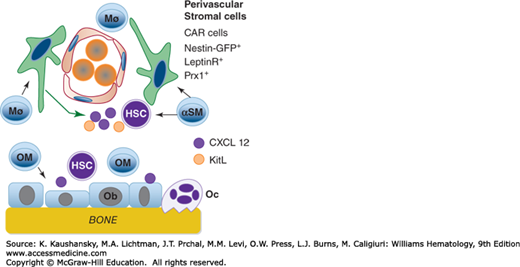
The microenvironmental effects on HSCs have far reaching clinical implications as well; our ability to mobilize marrow stem cells for transplantation has greatly changed the way we treat hematologic and other malignancies, and ultimate success in the efforts of experimental hematologists to expand HSCs ex vivo with cocktails of cytokines and stromal cells for applications in gene therapy and regenerative medicine will undoubtedly derive only from a thorough understanding of the molecular bases for the interaction of HSCs with their microenvironment (Fig. 18–2).
Figure 18–2.
The figure depicts multiple elements of the hematopoietic microenvironment. The niche has two major regions in the marrow, supported by osteoblasts or vascular cells. Several cell types provide cytokines that maintain osteoblasts (Ob), which, in turn, support hematopoietic stem cells (HSC) by secreting CXCL12 and other cytokines. Osteoclasts (Oc) are also shown but are of lesser importance in HSC maintenance, and may inhibit HSC survival/proliferation. Macrophages that express α smooth muscle actin (αSM) support perivascular cells, including the CXCL12 abundant reticular (CAR) cells, that, in turn, provide CXCL12 and SCF (here termed c-kit ligand [KitL]) to HSCs. In addition to paracrine support, direct perivascular cell–stem cell contact, through integrins, also support HSCs. (Reproduced with permission from Calvi LM, Link DC: Cellular complexity of the bone marrow hematopoietic stem cell niche. Calcif Tissue Int 2014 Jan;94(1):112-24.)
Marrow stromal cells influence hematopoiesis in a number of ways, by producing several cytokines that positively or negatively affect hematopoietic cell growth,139,140,141,142 including some, like SCF, that are expressed on their cell surfaces, resulting in enhanced biologic activity.143 Stromal cells are the origin of a number of extracellular matrix proteins that either directly affect hematopoietic cells, or do so indirectly by binding growth factors and presenting them in a functional context.144 They also bear the Jagged/Delta family ligands that stimulate Notch proteins to undergo cleavage and translocation into the nucleus, events that are critical mediators of cell fate decision making,145,146 including for hematopoietic cells.147 Cell–cell interactions mediated by integrins present on hematopoietic cells and counterreceptors on stromal cells are also very important for hematopoiesis.65,72 In addition to bringing hematopoietic cells into close proximity to cells producing soluble or cell-bound cytokines, and hence raising the local concentration of these growth promoting proteins, integrin engagement leads to intracellular signaling, usually promoting entry into the cell cycle and preventing programmed cell death.148 Reflecting the vital and sometimes lineage specific roles of the hematopoietic microenvironment, the extracellular matrix and stromal cells reside in a highly organized structure (Chap. 5).
Hematopoiesis is highly compartmentalized within areas of red marrow, with erythropoiesis occurring in clusters surrounding a central macrophage,149 granulocyte development associated with stromal cells,150 and megakaryopoiesis occurring adjacent to the endothelial sinusoidal cells.151 In the adult marrow, the specialized niche in which HSCs develop into differentiated progeny has been termed the hematon by Peault, a structure that includes Str01+ mesenchymal cells, desmin-positive perivascular lipocytes, Flk1+ endothelial cells, macrophages, and hematopoietic progenitors.152 From these structures can be derived all lineages of committed colony-forming cells (e.g., CFU-GM and BFU-E) and primitive cells that score positive in CAFC assays, LTC-IC, and high proliferative potential colony-forming cell assays (Chap. 5).
One consequence (or perhaps cause) of this anatomical arrangement is that the stem cell microenvironment is quite hypoxia. It is estimated that the O2 level of the stem cell niche is approximately 5 percent. The HSC response to hypoxia is discussed in “Metabolic Characteristics” above.
The marrow microenvironment is composed of multiple cell types. Fibroblasts are perhaps the best-studied of the marrow stromal cells, and can bind to primitive hematopoietic cells by engaging cell-surface integrins.153 Marrow endothelial cells also support primitive hematopoietic cells, including LTC-IC.154 The CXCL12–abundant reticular (CAR) cells, which surround the sinusoidal endothelial cells in vivo, are also likely to play the critical niche function of the vascular wall.155,156 However, based on their ability to increase the number of HSCs when experimentally increased, osteoblasts, which line trabecular bone and reside adjacent to primitive hematopoietic cells,157 are thought to provide a critical role in serving as the HSC supportive niche.158 The origin of all of these cell types is thought to reside in the mesenchymal stromal cell (MSC), a functionally defined entity that under specific conditions can be induced to form fibroblasts, endothelial cells, CAR cells, and osteoblasts, amongst others,159 and hold promise to therapeutically manipulate hematopoiesis.160 MSCs are discussed more extensively in Chap. 30.
Marrow stromal cells affect HSCs in multiple ways. Each of these cells is known to produce a number of cytokines critical for primitive and mature hematopoietic cell development. For example, although a number of organs produce TPO constitutively,161 marrow stromal cells are induced to produce the hormone in states of thrombocytopenia.162,163 Stromal cells produce SCF constitutively in both soluble and membrane bound forms,76 and FLT-3 ligand (FL) is produced both constitutively by stromal cells and lymphocytes and can be induced to high levels in the presence of pancytopenia.164
Besides growth factor production, stromal cells are also known to display counterreceptors for the integrins present on hematopoietic cells, including VCAM-1,165 interactions that promote cell survival and proliferation in several ways.166 Osteoblast-derived annexin II serves as an adhesion molecule for HSCs.167 Stromal cells also elaborate extracellular matrix components, including collagen, laminin, FN, heparins, hyaluronan, and tenascin, which display important effects on HSCs (see “Matrix Proteins” later). These substances, in turn, engage a number of HSC integrins and other cell-surface molecules, and form a solid matrix on which hematopoietic cells firmly attach. Of considerable clinical interest, it appears that interference with cell–matrix interactions,168 or digestion of the extracellular matrix itself,169 is involved in mobilizing HSCs by some agents such as granulocyte colony-stimulating factor (G-CSF) and IL-8.
It has long been known that the marrow is innervated by the autonomic nervous system,170 which influences HSCs in several ways, such as directing HSC trafficking by acting on nestin-positive microenvironmental cells.171 One or more of these functions appear to be critical for HSC homeostasis, as marrow nerve injury impairs hematologic recovery following chemotherapy-induced injury.172
The regulation of stem cell survival, proliferation, and differentiation has been difficult to address because of the rarity of stem cells and the requirement that they be assessed using cumbersome transplantation assays. Several cytokines are able to exert effects on HSCs. The pursuit of the cytokines that affect HSCs is of more than pure physiologic interest, as the availability of the right combination of such proteins could allow expansion of the cells for therapeutic use without sacrificing their pluripotent and self-renewal capacities. Three proteins—SCF, FL, and TPO—and their corresponding receptors (c-Kit, Flt3, and c-Mpl, respectively) exert important effects on the number and/or growth of HSCs both in vitro and in vivo (Table 18–1).
| Cytokine | Principal Activities |
|---|---|
| IL-1 | Induces production of other cytokines from many cells, works in synergy with other cytokines on primitive hematopoietic cells |
| IL-2 | T-cell growth factor |
| IL-3 | Stimulates the growth of multiple myeloid cell types, involved in delayed type hypersensitivity |
| IL-4 | Stimulates B cell growth and modulates the immune response by affecting immunoglobulin class switching |
| IL-5* | Eosinophil growth factor and affects mature cell function |
| IL-6 | Stimulates B lymphocyte growth; works in synergy with other cytokines on megakaryocytic progenitors |
| IL-7* | Principal regulator of early lymphocyte growth |
| IL-9 | Produced by Th2 lymphocytes; costimulates the growth of multiple myeloid cell types |
| IL-11 | Shares activities with IL-11; also affects the gut mucosa |
| IL-15* | Modulates T lymphocyte activity and stimulates natural killer cell proliferation |
| IL-21 | Affects growth and maturation of B, T, and natural killer cells |
| SCF* | Affects primitive hematopoietic cells of all lineages and the growth of basophils and mast. Also termed c-Kit Ligand |
| EPO* | Stimulates the proliferation of erythroid progenitors |
| M-CSF* | Promotes the proliferation of monocytic progenitors |
| G-CSF* | Stimulates growth of neutrophilic progenitors, acts in synergy with IL-3 on primitive myeloid cells and activates mature neutrophils |
| GM-CSF | Affects granulocyte and macrophage progenitors and activates macrophages |
| TPO* | Affects hematopoietic stem cells and megakaryocytic progenitors |
| CXCL12 | Chemokine that attracts HSCs by binding to the CXC4 receptor |
The molecule termed SCF, steel factor, mast cell growth factor, or c-Kit ligand was cloned by several groups based on its binding to a cell surface receptor encoded by the protooncogene c-Kit,76 previously identified as responsible for the severe defects in hematopoiesis, pigmentation, and gametogenesis in W mice. As the phenotype of mice bearing alleles of W was quite similar to those of steel (Sl), but in transplantation studies one strain displayed a stem cell autonomous defect (W) while the other was not (Sl), it had been hypothesized that the two genes represented the receptor for a growth factor and the cytokine itself, respectively, a tenet proven true with the cloning of SCF.
The extracellular domain of c-Kit is composed of five immunoglobulin-like domains, which leads through a typical transmembrane domain to the intracellular domain that bears a split domain-type tyrosine kinase. A single molecule of SCF binds to the first three immunoglobulin domains (D1D2D3) of two c-Kit receptors. The two D4 domains of a dimeric c-Kit receptor display substantial electrostatic repulsion toward each other, precluding the juxtaposing of the two transmembrane domains, and by extension, the two intracellular kinase domains. Once SCF binds to c-Kit the affinity for dimer formation overcomes the D4 electrostatic repulsion and the two kinase domains are brought together to initiate signaling.173 The intracellular mediators activated by SCF binding to c-Kit include phosphoinositol 3′-kinase (PI3K), mitogen-activated protein kinases (MAPKs), phospholipase C gamma (PLCγ), and c-Src (Chap. 17; reviewed in Ref. 174).
SCF is synthesized by marrow fibroblasts and other cell types. Soluble SCF is a highly glycosylated 36-kDa protein released from its initial site on the cell membrane by proteolytic processing. An alternatively spliced form of SCF messenger RNA (mRNA), that does not encode the cleavage site, remains on the cell membrane, and is a more potent stimulus of c-Kit-receptor-bearing cells.140 The ratio of soluble to membrane encoding SCF mRNA varies widely in different tissues, ranging from 10:1 in the brain, to 4:1 in the marrow, to 0.4:1 in the testis.
The importance of SCF to hematopoiesis is easily demonstrated; although nullizygous mice (Sl/Sl) are embryonic lethal because of a number of developmental defects, the presence of a partially functional allele (Sld) allows compound heterozygotes (Sl/Sld) to survive into adulthood, albeit with severe anemia because of diminished numbers/quality of HSCs. In addition to its critical role in the development of embryonic and fetal hematopoiesis, treatment of adult mice with an antibody that neutralizes the SCF receptor, c-Kit, also results in severe pancytopenia, indicating an important hematopoietic role for the receptor/ligand pair throughout life.
When present in culture SCF alone can maintain the long-term repopulating ability of murine Sca-1+/Rhlo/Lin− hematopoietic cells, suggesting that the cytokine can promote the survival of HSCs in vitro.175 However, alone, SCF is only a weak stimulator of cell proliferation, primarily inducing the development of mast cells both in vitro and in vivo. Nevertheless, in the additional presence of IL-3, IL-6, IL-11, G-CSF, or TPO, SCF exerts profound effects on the generation of hematopoietic progenitor cells of all lineages,176,177,178 pointing to primitive hematopoietic cells as critical targets. The molecular mechanisms of such synergy are beginning to emerge.179 A physical association of c-Kit and EPOR has been detected following SCF stimulation of cells bearing both receptors, an event that is essential for their functional synergy.180
FL was cloned as the binding partner for the then newly identified novel orphan receptor Flt3,181 a protein most closely related to the receptors for macrophage colony-stimulating factor (M-CSF) (hence the term flt = fms-like tyrosine kinase), and c-Kit. FL is expressed by T lymphocytes and marrow stromal cells.164,181 The Flt3 receptor is a 160-kDa cell-surface molecule expressed primarily on primitive hematopoietic cells.182 Like c-Kit, activation of Flt-3 results in activation of several signaling mediators, including the p85 subunit of phosphatidylinositol 3-kinase, SHP, PLCγ, and a guanosine 5′-triphosphatase (GTPase)-activating protein, activating Ras.183 Normal Flt3 signaling also activates the MAPKs extracellular regulated kinase (ERK)-1 and ERK2 but leads to only weak phosphorylation of signal transducer and activator of transcription (STAT)-5, in contrast to an oncogenic form of the receptor, identified in 25 percent of patients with myelodysplastic syndromes or acute myelogenous leukemia.184,185 In the leukemic cells of such patients Flt3 bears an internal tandem duplication of the kinase domain, resulting in the constitutive activation of the receptor. Clinically, this is associated with reduced likelihood of patient survival; hence, this observation has led to an attempt to control the growth of such mutant-receptor-bearing cells with specific Flt3 kinase inhibitors,186 with some success (Chap. 88).
FL was initially cloned using a soluble form of the receptor to identify ligand-bearing cells.187 As their receptors bear a number of common structural features, it was not surprising to find that FL shares significant structural homology, as well as biologic properties with both M-CSF and SCF. Like the other two cytokines, FL displays a 4α-helix bundle tertiary structure and exists in both membrane-bound and soluble states, the result of alternate splicing of the primary transcript that does or does not include a cleavage site for its release from the cell membrane.188
Unlike SCF levels that remain relatively static regardless of blood cell counts,76 blood concentrations of FL can rise more than 25-fold in response to pancytopenia.189
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


