INTRODUCTION
SUMMARY
The quantum physicist Erwin Schrodinger surmised in his monograph “What is Life?” that the organized matter known as life needs to feed on “negative entropy” to avoid decay.1 He concluded that this feeding on negative entropy is achieved through metabolism, a term derived from Greek that describes an exchange of materials. Because of this centrality to life, metabolism’s core pathways—glycolysis and respiration—evolved early in Earth’s history and are highly conserved. At every stage of life, metabolism provides the needed nutrients, energy, and building blocks. Embryogenesis, for instance, requires metabolism of maternally derived nutrients to support cellular repair, growth, division, and differentiation. In particular, cell replication requires that the instructions emanating from the DNA sequence, modulated by the epigenome, couple with the import of nutrients and metabolic pathways to produce the components and energy necessary to build two copies of a cell and maintain high replication fidelity of the genome. During growth and development, and especially during adulthood, metabolism also plays the important role of providing bioenergetics for cellular and organismal homeostasis. Metabolism can also feature prominently in disease, and this chapter discusses how the metabolic pathways central to life and normal biology can be subverted in cancer to fuel abnormal growth.
Food, through metabolism, provides the nutrients necessary for homeostasis, repair, and reproduction of many organisms. To align supply and demand, mammalian metabolism is linked to sleep cycles through the central circadian clock that senses light and dark phases of the day via the eye and central nervous system. The central regulation of feeding and sleeping cycles coordinates nutrient availability from food with the circadian oscillation of metabolism of individual cells, which all have a molecular clock comprised of a network of transcription factors that regulates cell metabolism.2
Food is digested, absorbed through the gastrointestinal tract, and in part processed or stored in the liver, which is a key metabolic organ.3 Processed lipids in the form of lipoproteins are synthesized in the liver and disseminated throughout to supply the needs of various organs. Amino acids, with glutamine circulating at the highest level (0.5 mM), supply cells with building blocks for proteins. Some amino acids (nonessential) are synthesized by humans, but essential amino acids must be available from the diet. Complex carbohydrates are broken down and circulate as glucose, a vital nutrient for virtually all mammalian cells. In this regard, an endocrine system (insulin and glucagon) has evolved to control the circulating levels of this precious bioenergetic molecule. When in excess, amino acids and sugars contribute to lipogenesis, and the extra energy is stored as fat depots in adipose tissues. Excess glucose is stored as glycogen, which is deployed to release glucose in starved conditions. It is believed that, during our evolution, periods of gorging and feeding were separated by significant durations of starvation; hence, we have evolved mechanisms to survive starvation.
In contrast to the fed state, when insulin level increases in response to rising glucose to trigger cellular glucose uptake and storage, the starved state triggers glucagon secretion from the pancreas, which mobilizes cellular glycogen stores as nutrient. Prolonged starvation depletes liver and muscle glycogen stores—the only major glycogen stores—and prompts the mobilization of fat stores. The released fatty acids provide glycerol as a substrate for making glucose through gluconeogenic pathways and fatty acids for mitochondrial oxidation. Further prolonged starvation triggers the liver to convert fatty acids to ketone bodies, which can cross the blood–brain barrier to feed the brain. How whole organisms initiate autophagy (self-eating) during markedly prolonged starvation has not been thoroughly studied. But autophagy is clearly necessary for mammalian development, particularly upon birth when deletion of specific autophagic regulators results in death.4 Although autophagy plays an important role in mitochondrial and organellar homeostasis and cancer metabolism, it is discussed in Chap. 15; this chapter focuses primarily on intermediary metabolism.
Although most of our cells are differentiated and do not proliferate, stem cell compartments are ubiquitous among tissues, allowing for replacement of used or damaged cells. The hematopoietic stem cell and hematopoiesis constitute probably the best-studied stem cell system.5 Cytokines, growth factors, and the extracellular matrix provide the cues for stem cells to maintain their quiescent state or to awaken and differentiate to replenish lost cells. In response to growth factors in the presence of nutrient-replete states, stem cells self-renew, proliferate, and then differentiate. In nutrient-deprived states, normal metabolic checkpoints forbid growth factor stimulated cells from proliferating.6 All of the mechanisms used by normal cells during fed and starved states are potentially exploitable by neoplastic cells for their survival. Genetic mutations drive neoplastic cells to grow and proliferate regardless of the availability of nutrients; in contrast, normal cells sense nutrients and do not proliferate under starved conditions. In this chapter, basic cell metabolism is covered along with discussions about growth signaling and its intersection with metabolism. Alterations in metabolism found in hematologic neoplastic cells are discussed in the context of therapeutic opportunities that are rapidly emerging from the latest basic research and translational efforts.
Acronyms and Abbreviations:
ABC, activated B-cell type; ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; AMPK, adenosine monophosphate kinase; ASCT2, ASC amino-acid transporter 2; ATRA, all-trans retinoic acid; BPTES, a glutaminase inhibitor; CDK, cyclin-dependent kinase; CL, cardiolipin; COO, cell of origin; DFMO, α-difluoromethylornithine; DLBCL, diffuse large B-cell lymphoma; eIF5A, eukaryotic translation initiation factor; ERK, extracellular regulated kinase; ETC, mitochondrial electron transport chain; Ets, E26, E twenty-six; F1,6BP, fructose 1,6 biphosphate; F2,6BP, fructose 2,6 biphosphate; FAO, fatty acid oxidation; FDG-PET, fluorodeoxyglucose positron emission tomography; FH, fumarate hydratase; FOS, a protooncogene; G3P, glycerol 3-phosphate; G6PD, glucose-6-phosphate dehydrogenase; GAP, glyceraldehyde 3-phosphate; GCB, germinal center B-cell type; GDP, guanosine-5′-diphosphate; GLS, glutaminase; GLUT, glucose transporter; GM-CSF, granulocyte-macrophage colony-stimulating factor; GOT, glutamate oxaloacetate transaminase; GPI, glucose phosphate isomerase; GPT, glutamate pyruvate transaminase; GTP, guanosine-5’-triphosphate; 2-HG, 2-hydroxyglutarate; HIF, hypoxia-inducible factor; HSC, hematopoietic stem cell; IDH, isocitrate dehydrogenase; IMPDH, inosine monophosphate dehydrogenase; LDHA, lactate dehydrogenase A; LKB1, Liver kinase B1; LSC, leukemic stem cell; Max, Myc-associated factor X; MDM2, mouse double minute 2 homolog; Miz-1, Myc-interacting zinc finger protein 1; MLL2, mixed-lineage leukemia protein 2; Mlx, Max-like protein X; MondoA, member of the MYC network of transcription factors that upregulates glycolysis (known as MLXIP, MLX interacting protein); mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; MYC, a protooncogene that is a major regulator of cell growth and metabolism; NADH, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; NAMPT, nicotinamide phosphoribosyltransferase; NRF2, nuclear respiratory factor-2; NTP, nucleotide triphosphate; ODC, ornithine decarboxylase; OGDH, oxoglutarate dehydrogenase; OXPHOS, oxidative phosphorylation; P53, activates oxidative phosphorylation and inhibits glycolysis; PC, phosphatidyl choline; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PE, phosphatidyl ethanolamine; PEP, phosphoenol pyruvate; PFK, phosphofructokinase; PG, glycerophosphoglycerol; PGC1α, an activator of mitochondrial biogenesis; PI, phosphatidyl inositol; PI3K, phosphoinositol 3′-kinase; PML, promyelocytic leukemia; PRPS, 5-phosphoribosyl-pyrophosphate synthetase; PS, phosphatidyl serine; PTEN, phosphatase and tensin homologue deleted on chromosome 10, an antioncogene; RAS, name given to a family of related proteins belonging to small GTPase involved in signal transduction; ROS, reactive oxygen species; rRNA, ribosomal RNA; SAM, S-adenosylmethionine; SCO2, cytochrome c oxidase; SDH, succinate dehydrogenase; SLC1A5, ASC amino-acid transporter 2; SOD, superoxide dismutase; TCA, tricarboxylic acid; TET, family of dioxygenases that catalyze conversion of 5-methylcytosine to 5-hydroxymethylcytosine; TFEB, transcription factor EB; THF, tetrahydrofolate; VHL, von Hippel-Lindau protein.
CELL GROWTH AND METABOLISM
The canonical cell has significant bioenergetic needs for maintenance and homeostasis.6 Protein synthesis and maintenance of cellular membrane potentials consume most of the ATP produced under homeostatic conditions.7 In many differentiated or quiescent cells, it is believed that fatty acid oxidation provides the bulk of the energy, followed by the use of glucose. In this regard, mitochondrial respiration is essential for adult tissues and cells. It is notable, however, that specialized cell functions in the various organs could require different metabolic pathways. Glucocorticoid hormone-producing cells, for example, express specialized metabolic pathways. Although cardiac muscle cells depend heavily on fatty acid oxidation, skeletal muscle cells use glucose. The brain depends largely on glucose, but it can feed on ketone bodies under starved states. For differentiated cells, which are the bulk of cells in mammals, homeostasis drives the demand for nutrients, to “the availability of which are determined by feeding and interorgan (liver, muscle, and endocrine tissues) metabolic interplays. For example, lactate produced by exercising muscle circulates back to the liver and is processed via the Cori cycle to produce glucose.3 Glucose plasma level is tightly controlled by the pancreas, which produces insulin and glucagon, and by the liver (as well as kidney) that can produce glucose through gluconeogenesis.
Normal cell growth and proliferation are triggered by extracellular cues. Yeast cells, for example, only require the presence of nutrients to initiate cell growth or an increase in cell size.6 During this growth phase, nutrients are imported and channeled into biomass, which is largely comprised of ribosomes. It is estimated that ribosomes constitute more than 50 percent of cellular dry mass, and hence ribosome biogenesis is highly regulated and vitally important for cell growth and proliferation. Once a critical cell size (mass) is reached with a balanced nucleotide pool, DNA synthesis begins. Yeast cells sense nutrients, particularly glucose and glutamine, through pathways involving RAS and target of rapamycin complex 1 (TORC1), which silence transcriptional repressors of ribosome biogenesis.8 With nutrient deprivation, activation of these transcriptional repressors provides a metabolic checkpoint that restrains cells from growing in the absence of adequate bioenergetic support. Hence, the normal feedback loops couple nutrient availability with cell growth: no nutrients, no growth. The normal feedback loops can be artificially disrupted by deletion of transcriptional repressors of ribosomal biogenesis, rendering yeast mutants constitutively activated for growth. These mutants resemble mammalian cancer cells, which have mutations that drive autonomous cell growth with disregard for nutrient availability. The severance of nutrient sensing from growth signaling causes addiction of these yeast mutants to nutrients, such that deprivation of glucose or glutamine results in nonviable mutants. Similarly, cancer cells are addicted to nutrients.6
Cell growth in multicellular organisms requires additional cues in addition to the availability of nutrients. Mammalian cells live in a community of cells and are constantly bathed in nutrients derived from the circulation, but they do not proliferate unless there are appropriate cues from growth factors and the extracellular matrix. Mammalian cells can be envisioned as bioreactors that require at least two signals to grow: (1) growth factor and (2) nutrients.6 Cell growth is arrested in the absence of either growth factor or nutrients. Similar to yeast cells, metabolic checkpoints are critical to the growth of normal mammalian cells.
Growing cells largely depend on glucose, glutamine and other amino acids.9 Indeed, the core metabolic pathways including glycolysis, glutaminolysis, and the tricarboxylic acid (TCA) cycle link amino acid and glucose metabolism to lipogenesis and nucleotide synthesis (Fig. 14–1). Glycolysis starts with the transport of glucose into cells through several transporters, known as GLUTs, with SLC2A1 (GLUT1) being one that is coupled with cell growth stimulation. Once inside the cell, glucose is phosphorylated in an ATP-consuming step by hexokinases (HK) with HKII being stimulated by many growth signals and directly regulated by the MYC oncogene or the hypoxia inducible factor 1 alpha (HIF-1α). Glucose-6-phosphate (G6P) is used by the pentose phosphate pathway to produce ribose for nucleotide synthesis (Fig. 14–1) or converted to fructose phosphate via an isomerization reaction catalyzed by glucose phosphate isomerase (GPI).9 Fructose-6-phosphate is further phosphorylated with consumption of a second ATP through a rate-limiting step catalyzed by phosphofructokinase (PFK) to fructose-1,6-bisphosphate (F1,6BP). F1,6BP is converted by aldolase and an isomerase to the three-carbon phosphorylated molecule, glyceraldehyde 3-phosphate (GAP), which is oxidized and phosphorylated using inorganic phosphate by the dehydrogenase, GAPDH, to 1,3-bisphosphoglycerate. The energy gained by nicotinamide adenine dinucleotide (NAD+)-mediated oxidation and phosphorylation is released from 1,3-bisphosphoglycerate by phosphoglycerate kinase, which transfers the high-energy phosphate bond to adenosine diphosphate (ADP) to form ATP. The resulting 3-phosphoglycerate (3-PG) provides a substrate for serine and glycine synthesis, for the production of glycerol, or for the production of phosphoenol pyruvate (PEP) in glycolysis. Pyruvate kinase mediates the transfer of the high-energy phosphate bond from PEP to ADP producing ATP and pyruvate, which is the terminal substrate of glycolysis. Collectively, glycolysis uses ATP to charge up several intermediates for their transformations and uses NAD+ to oxidize intermediates and generate energy through new high-energy phosphate bonds with inorganic phosphate. Each glucose molecule results in the production of a net two ATP molecules from ADP through glycolysis.
Figure 14–1.
Central metabolic pathways involving glycolysis, glutaminolysis, and the tricarboxylic acid (TCA) cycle. Glucose is shown metabolized to pyruvate, which can be converted to lactate, alanine, or acetyl-coenzyme A (acetyl-CoA). Upstream of pyruvate, glucose carbons are shunted toward the pentose phosphate pathway for ribose synthesis, glycine, and glycerol synthesis. Acetyl-CoA (2-carbon unit) combines with oxaloacetate (4-carbon) to form citrate (6-carbon), which is subsequently metabolized in the TCA cycle to generate isocitrate, α-ketoglutarate, and other intermediates as depicted. Glutamine is shown to enter the TCA cycle via α-ketoglutarate, after being converted to glutamate. Glucose gives rise to glycerol and citrate, which contributes 2-carbon units for fatty acid synthesis, and contributes to lipid synthesis. Glutamine and glucose are depicted to contribute to nucleotide synthesis.
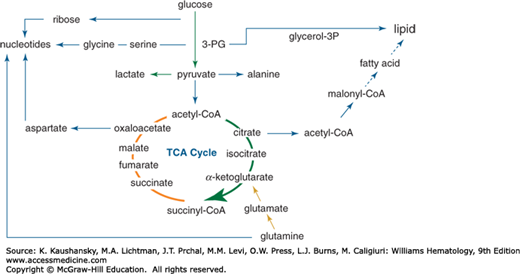
Pyruvate, derived from glucose through glycolysis, from malate through malic enzyme or from alanine through transamination, could enter the mitochondria through specific transporters and be converted to acetyl-coenzyme A (CoA) by pyruvate dehydrogenase (PDH) (see Fig. 14–1).10 PDH activity can be attenuated by phosphorylation, mediated by PDH kinase (PDK), which is activated by hypoxia to divert glucose carbons away from the TCA cycle toward lactate production. Under aerobic conditions, acetyl-CoA combines with oxaloacetate coming from a complete turn of the TCA cycle to produce citrate, which can be extruded into the cytoplasm to participate in lipid synthesis or which can be converted to isocitrate in the TCA cycle. Isocitrate is further oxidized to α-ketoglutarate by isocitrate dehydrogenase (IDH) with the production of either nicotinamide adenine dinucleotide (NADH) or nicotinamide adenine dinucleotide phosphate (NADPH) and release of a carbon dioxide molecule. There are three IDH isozymes with IDH1 being located in the cytosol, while IDH2 and IDH3 are in the mitochondrion. NADH in the mitochondrion contributes to the high-energy electrons that drive production of ATP through the electron transport chain. NADPH produced by cytosolic IDH1 or mitochondrial IDH2 could participate in reductive biosynthesis of fatty acids or nucleobases.
In addition to being a key TCA cycle intermediate at the crossroads of several metabolic pathways, a-ketoglutarate (or oxoglutarate) serves as a cofactor for many important oxygenases, such as those involved in the hydroxylation and degradation of the hypoxia inducible factors (HIFs), modification of ribosomes, or those involved in demethylation of DNA and histones.11,12 Notably, glutamine can enter the TCA cycle at this junction. α-Ketoglutarate is further oxidized by oxoglutarate dehydrogenase (OGDH) to produce succinyl-CoA and carbon dioxide. Succinyl-CoA, which is also used for heme synthesis, is then converted to succinate with the production of a guanosine-5′-triphosphate (GTP) from guanosine-5′-diphosphate (GDP). Succinate is then converted to fumarate by succinate dehydrogenase (SDH), which is mutated in certain familial cancer syndromes. Fumarate hydratase (FH), which is also mutated in cancer syndromes, converts fumarate to malate that is, in turn, converted to oxaloacetate. Oxaloacetate can serve as a substrate for glutamate oxaloacetate transaminase (GOT) for the production of aspartate for nucleotide synthesis, or it can further cycle forward into the TCA cycle by combining with acetyl-CoA to form citrate, thus completing the TCA (citric acid or Krebs) cycle (see Fig. 14–1).
Glutamine also serves as a key metabolic substrate for growing cells (see Fig. 14–1). Glutamine is imported by membrane transporters, such as SLC1A5 or ASCT2.13,14 Once in the cytosol, glutamine can contribute to protein synthesis or glucosamine or nucleobase biosynthesis by donating its nitrogen. Glutamine is further imported into the mitochondrion and converted to glutamate by glutaminase (GLS) with the release of ammonia. Glutamate is converted to α-ketoglutarate by either glutamate dehydrogenase (primarily in nongrowth states) or aminotransferases (GOT or glutamate pyruvate transaminase [GPT]). In this manner, glutamine serves as a major growth substrate for growing cells. Hence, the TCA cycle is a metabolic roundabout that uses carbons from glucose, glutamine, and fatty acids to generate carbon skeletons for biosynthesis, NADH for the production of ATP, or α-ketoglutarate for catalyzing key oxygenase reactions.
Oxidation of glucose, glutamine, and fatty acids produces energy for growing cells. On the other hand, synthesis of fatty acids and other building blocks require the reductive power of NADPH for bond formation. NADPH is produced from several well-characterized pathways, including the pentose phosphate pathway, malic enzyme, IDH, and the folate pathway.15 Glucose-6-phosphate dehydrogenase (G6PD) is well-known for its role in oxidation of G6P to 6-phosphogluconolactone and the concurrent reduction of NADP+ to NADPH, which contribute to an antioxidant state through maintaining reduced glutathione. Specifically, loss of G6PD function is associated with severe hemolytic anemia in patients who inherit hypomorphic alleles of G6PD (see Chap. 47). Malic enzyme mediates the oxidation of malate to pyruvate using nicotinamide adenine dinucleotide phosphate (NADP+), which is reduced to NADPH. IDH1 oxidizes isocitrate to α-ketoglutarate with the production of NADPH from NADP+. Lastly, it was recently documented that the folate pathway plays a major role in NADPH production through the oxidation of methylene-tetrahydrofolate (THF) to formyl-THF.15 The largest consumer of NADPH, on the other hand, involves fatty acid synthesis with reduction of glutathione following closely behind. Thus production of NADPH is critical for both biosynthesis and for redox homeostasis.
Growth factors and nutrients drive the growth and proliferation of cells (Fig. 14–2 and Chap. 17). Growth factor engagement of a (usually dimeric) growth factor receptor triggers allosteric alterations that lead to autophosphorylation, in the case of the receptor tyrosine kinase family, or phosphorylation by Janus kinase in the cases of the hematopoietic cytokine receptor family. The phosphorylated receptor then recruits adaptor molecules that initiate a cascade of phosphorylation events, which culminate in the activation of concurrent pathways through phosphoinositol 3′-kinase (PI3K)/PTEN/AKT and RAS-RAF-ERK (extracellular regulated kinase) (Fig. 14–2). These cascades relay signals to mTOR complex, which is a vital hub for metabolic sensing and short-term post-transcriptional control of cell growth.16 mTOR, potentially coupled with additional outputs of the RAS-RAF-ERK pathway, also activates a genomic response to the growth signal. In essence, the mTOR pathway provides an immediate response to growth stimulus and nutrients followed thereafter by a transcriptional response that provides an increase in specific mRNAs needed for the production of new building blocks for the growing cell. The initial growth response occurs in cells that have a basal number of ribosomes, which serve to translate delayed early response genes. The cascade down the ERK pathway also activates the expression of early response genes such as FOS and MYC. The activation of MYC is probably mediated through the ERK-activation of Ets transcription factors, whose regulatory motifs are found in the MYC gene.17 Furthermore, ERK phosphorylates and stabilizes the MYC protein, enhancing ERK’s ability to drive a transcriptional response to growth.18,19
Figure 14–2.
Canonical signal transduction pathways emanating from a receptor tyrosine kinase and their connections to metabolism. The phosphoinositol 3′-kinase (PI3K)-PTEN-Akt and Ras-Raf-ERK (extracellular regulated kinase) pathways are shown with connections to MYC and hypoxia-inducible factor 1 (HIF-1α), which trigger metabolic transcriptional programs. All of these pathways coordinate a response to growth factors and drive nutrients into the cell to generate ATP and building blocks for lipid, nucleotide, and protein synthesis. The net result is cell growth and generation of waste products, such as reactive oxygen species (ROS), lactate and carbon dioxide. Tumor suppressors (red octagons) and protooncogenes (green bursts) are highlighted.
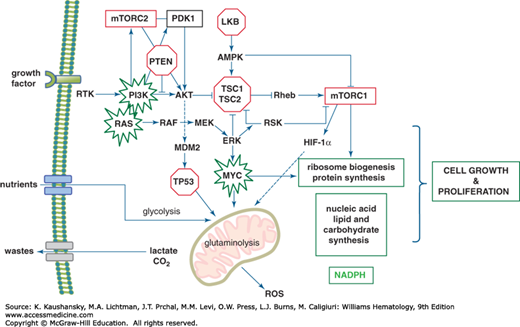
The immediate sensing of nutrients is mediated through the adenosine monophosphate kinase (AMPK) and mTOR pathways, which, in turn, modulate cellular responses through phosphorylation of regulatory proteins (see Fig. 14–2).9,16,20 In the presence of nutrients, the import of branched-chain amino acids is sensed through lysosomes to activate mTOR complex 1 (mTORC1). Import of glutamine, which is converted to glutamate, is thought to play an important role in activating mTOR through the antiporter, SLC7A1, which exports glutamate in exchange for leucine. Leucine is one of the most potent activators of mTORC1, which, in turn, phosphorylates key regulatory proteins to increase protein translation, mitochondrial biogenesis and respiration, glycolysis, and lipogenesis. Many of these effects are also mediated through mTORC1’s regulation of transcription factors such as PGC1α (mitochondrial biogenesis), HIF-1α (glycolysis), and SREBP (lipogenesis). mTORC1 also phosphorylates and inactivates the transcription factor TFEB, which is a master positive regulator of lysosome biogenesis.16 Presumably, inhibition of TFEB would also diminish the machinery involved in autophagy. Although mTORC1 also stimulates ribosome biogenesis, the mechanism by which this occurs is not yet known. Activation of the mTORC2 complex is less well defined, but mTORC2 is responsible for the subsequent activation of AKT that plays a critical role in activating glycolysis. Thus, growth factor signaling stimulates nutrient uptake that in turn activates mTOR to stimulate cell growth.
In nutrient-deprived states, the AMPK pathway regulates cellular responses that optimize energy production and diminish energy utilization pathways (see Fig. 14–2).20,21 AMPK is allosterically altered by binding to adenosine monophosphate (AMP), which makes AMPK permissive for phosphorylation and activation by the tumor suppressor LKB1. The phosphorylated AMPK in turn phosphorylates and regulates many pathways involved in energy regulation. One of the earliest discoveries was that AMPK phosphorylates and inactivates acetyl-CoA carboxylase, which is involved in fatty acid synthesis. Thus, by diminishing lipogenesis, AMPK is able to inhibit an energy consuming process as well as inhibit cell growth by curbing lipogenesis. AMPK also attenuates protein synthesis through phosphorylation of RNA polymerase I, which is required for ribosome biogenesis. On the other hand, AMPK increases energy yield by stimulating glycolysis through phosphorylation and activation of PFK-2. AMPK stimulates mitochondrial biogenesis through phosphorylation of PGC1α and increases autophagy to recycle energy by phosphorylating ULK-1.21 Thus, increased AMPK activity conserves energy and maximizes energy production.
Together with posttranscriptional responses to growth signaling and nutrients, the nuclear transcriptional response is necessary to sustain the growth program through production of components of the ribosome and mRNAs that give rise to all other components of the cell. mTOR through its direct activation of specific transcription factors contributes to lipogenesis and mitochondrial biogenesis. Growth signaling also activates the MYC protooncogene, that regulates gene expression broadly to support cell growth and proliferation (see Figs. 14–2 and 14–3).19 Loss of function of Drosophila dMYC results in decreased cell and body size, a phenotype that underscores MYC’s role in cell growth.22 This phenotype mimics the loss of ribosome protein gene function in a group of mutant flies termed Minutes. Hence, Drosophila genetics links MYC to cell growth control. Furthermore, MYC is the only transcription factor capable of stimulating the activity of RNA polymerases I, II, and III, all of which are involved in ribosome biogenesis.
Figure 14–3.
Diagram depicting fates of growth factor-stimulated normal cells and MYC transforming cell. Upon engaging a growth factor (pink square), the stimulated cell reacts as a bioreactor, which grows and duplicates itself. MYC overexpression triggers checkpoints via p53, which induces cell death. With loss of p53 MYC’s full transforming potential is unleashed through its transcriptional activities.
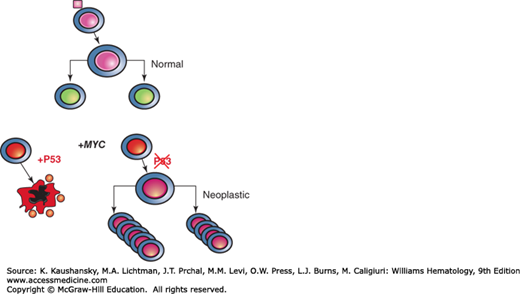
MYC dimerizes with its partner Max to bind a specific DNA sequence, termed E-box (CACGTG), and activate transcription.23 It can also inhibit transcription partly through direct binding to Miz-1 and diminishing the expression of Miz-1 target genes, including the cyclin-dependent kinase (CDK) inhibitor p21 and genes involved in autophagy. Upon MYC activation, it is binding to proximal promoters accounts for most of it is transcriptional function in normal cells. When MYC is experimentally expressed at levels comparable to those found in cancers,19 excess MYC triggers p53-dependent checkpoints (see Fig. 14–3) that cause cell growth arrest or apoptosis. In multistep tumorigenesis, therefore, p53 is often lost, unleashing MYC’s full oncogenic potential. A high, unchecked level of MYC allows it to alter the transcriptome by amplifying selected target genes.24 MYC was first shown to directly regulate genes involved in glycolysis, thereby linking an oncogenic transcription factor to metabolism.6,19 Since these initial observations, high-throughput methods have mapped MYC to a broad swath of metabolic enzyme genes involved in glycolysis, glutaminolysis, and lipogenesis. MYC also directly regulates genes involved in mitochondrial biogenesis and the production of ribosomes. Specifically, genes highly induced by MYC include those involved in nucleolar function and ribosome biogenesis, such as Ncl, NPM1, fibrillarin, and NOP52. Collectively, these studies uncover MYC’s role as a central regulator of cell growth through coupling of energy metabolism with cellular biosynthetic processes.
Ribosome biogenesis is a critically important process for cell growth or cell mass accumulation.25,26 Ribosomes are produced through RNAs that are transcribed by RNA polymerases I (rRNA [ribosomal RNA]), II (mRNA), and III (tRNAs [transfer RNAs] and small RNAs). rRNA is synthesized in the nucleolus from high copy numbers of rDNA, whose chromatin structure and transcription depends on nutrient availability. Under nutrient limitation, rDNA chromatin becomes less accessible, thereby restricting ribosome biogenesis.26 Ribosomal proteins produced from mRNAs reenter the nucleolus, where components of ribosomes are assembled into mature ribosomal particles, which are exported to the cytosol. The production of rRNAs and proteins also provides an opportunity for bioenergetic sensing of adequate nutrients to support nucleic acid and protein synthesis required for growth. In this regard, specific ribosomal protein subunits (RPL5, RPL11, and others) can bind and inhibit MDM2 (mouse double minute 2 homolog), which binds to and mediates the degradation of p53.25 Thus, it is surmised that ribosomal proteins in excess of rRNAs would activate p53, triggering checkpoints that block progression through the cell cycle, presumably in response to nutrient limitation sensed as an imbalance in rRNA and ribosomal protein synthesis.
In addition to sensing ribosome biogenesis, p53 also responds to genotoxic stresses by directly regulating metabolism (see Fig. 14–2). P53, in general, activates oxidative phosphorylation and inhibits glycolysis.27 P53 can activate HK, which phosphorylates glucose in the first step of glycolysis, and stimulate TIGAR that shunts glucose to the pentose phosphate pathway through decreasing the levels of fructose-2,6-bisphosphate (F2,6BP), which allosterically activates PFK. P53 also increases the efficiency of mitochondrial function through induction of cytochrome c oxidase (SCO2).28 Overall, it appears that the normal function of p53 is to rewire metabolism to mitigate oxidative stress through increased production of NADPH and the antioxidant glutathione. Gain of p53 function through specific mutations, on the other hand, appears to alter metabolism through specific target genes that are involved in cholesterol biosynthesis or phospholipase function.29,30
Other tumor suppressors are also involved in metabolism (see Fig. 14–2). PTEN negatively modulates PI3K, and hence its loss stimulates the PI3K pathway that is a potent regulator of cell metabolism through stimulation of glycolysis and activation of mTOR, AKT, MYC, and HIF.31 The tumor suppressor Lkb1, which is lost in some lung cancers, normally activates the AMPK pathway and diminishes lipogenesis.21
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


