Vaccines and immunostimulants
Jeffery Schlom, PhD  James L. Gulley, MD, PhD, FACP
James L. Gulley, MD, PhD, FACP  James W. Hodge, PhD, MBA
James W. Hodge, PhD, MBA
Overview
The U.S. Food and Drug Administration approvals of the prostate cancer therapeutic vaccine sipuleucel-T, the checkpoint inhibitor anti-CTLA-4 and anti-PD-L1/PD-1 monoclonal antibodies, along with the results of recent clinical studies involving cancer vaccines and other immunotherapeutics, have placed immunotherapy into the mainstream of the management of many cancer types. This chapter reviews the various cancer vaccine platforms now under clinical evaluation, the range of potential tumor-associated antigen vaccine targets, and the use of cancer vaccines in combination with so-called nonimmune standard-of-care therapies as well as other immunotherapeutics. The distinction in the clinical evaluation of cancer vaccines versus more conventional cytotoxic therapies is also discussed.
The U.S. Food and Drug Administration (FDA) approvals of (1) the prostate cancer therapeutic vaccine sipuleucel-T, (2) anti-CTLA-4 monoclonal antibody (MAb) ipilimumab for metastatic melanoma, and (3) the MAbs directed against the programmed death ligand 1 (PD-L1)/PD-1 immunosuppressive axis for the therapy of metastatic melanoma and lung carcinoma, along with the results of recent clinical studies involving immunotherapeutics, have placed immunotherapy into the mainstream of the management of many cancer types.
This chapter reviews the various cancer vaccine platforms now under clinical evaluation, the range of potential tumor-associated antigen (TAA) vaccine targets, and the use of cancer vaccines with so-called nonimmune standard-of-care therapies as well as other immunotherapeutics. The distinction in the clinical evaluation of cancer vaccines versus more conventional cytotoxic therapies is also discussed.
Targets for vaccine therapy
Many potential TAA targets for cancer immunotherapy have been identified. Targets of vaccine therapy need not be cell-surface proteins. When a molecule is a target for vaccine therapy, the activated T cells induced by vaccination recognize complexes of tumor antigen peptide and major histocompatibility complex (MHC) molecules on the cell surface. TAA vaccine targets can be grouped into several major categories (Table 1).
Tumor-specific antigens
Tumor-specific antigens (TSAs) comprise gene products that are uniquely expressed in tumors, such as point-mutated ras oncogenes, p53 mutations, and products of ribonucleic acid (RNA) splice variants and gene translocations. Three mutations at codon 12 represent the vast majority of ras mutations, which are found in approximately 20% to 30% of some human tumors.1 Although the ras protein is not found on the cell surface, one can envision vaccine therapy directed against peptide–MHC complexes on the cell surface. Indeed, there have been clinical trials in pancreatic carcinoma that target ras.2, 3 B-cell lymphomas overexpress a single immunoglobulin (Ig) variant on their cell surface; therefore, each B-cell lymphoma displays a unique target for immunotherapy.4–6 The gene products of RNA splice variants and deoxyribonucleic acid (DNA) translocations also represent unique fusion proteins that can be specific targets for immunotherapy, including c-erb-B2 RNA splice variants and the bcr/abl product of DNA translocation of chronic myelogenous leukemia.
Several viruses are associated with the etiology of some cancers. An excellent example of this is the connection between human papillomavirus (HPV) and cervical cancer. This has led to FDA approval of the HPV vaccine for prevention of cervical cancer.7
Tumor-associated antigens
TAAs can be categorized into three major groups: oncofetal antigens, oncogene products, and tissue-lineage antigens (Table 1). Oncofetal antigens, normally found during fetal development, are greatly downregulated after birth. This class of antigens, which includes prostate-specific membrane antigen (PSMA), carcinoembryonic antigen (CEA), and the cancer mucin MUC-1, are often overexpressed in tumors compared with normal tissues. The MUC-1 TAA is overexpressed in the majority of human carcinomas and several hematologic malignancies. Much attention has been paid to the hypoglycosylated variable number of tandem repeats (VNTR) region of the N-terminus of MUC-1 as a vaccine target. While previous studies have described MUC-1 as a tumor-associated tissue differentiation antigen, studies have now determined that the C-terminus of MUC1 is an oncoprotein, and its expression is an indication of poor prognosis in numerous tumor types.8
Table 1 Spectrum of current and potential therapeutic cancer vaccine targets
| Target type | Examples |
| Tumor-associated antigens | |
| Oncofetal antigen | CEA, MUC-1 |
| Stem cell/EMT | Brachyury, SOX-2, OCT-4, TERT, CD44high/CD24lo, CD133+ |
| Oncogene | MUC-1 C terminus, p53, EGFR, HER2/neu, WT1 |
| Cancer—testes | MAGE-A3, BAGE, SEREX-defined, NY-ESO, survivin |
| Tissue lineage | PAP, PSA, gp100, tyrosinase, glioma antigen |
| Glycopeptides | STn-KLH |
| Anti-angiogenic | VEGF-R |
| Tumor-specific antigens | |
| Oncogene | point mutated: ras, B-raf, frame shift mutations, undefined unique tumor mutations |
| Viral | HPV, HCV |
| B-cell lymphoma | Anti-id |
BAGE, B melanoma antigen; CEA, carcinoembryonic antigen; EMT, epithelial–mesenchymal transition; gp100, glycoprotein 100; HCV, hepatitis C virus; HPV, human papillomavirus; MAGE-A3, melanoma-associated antigen-A3; MUC-1, mucin 1; NY-ESO, New York esophageal carcinoma antigen 1; OCT-4, octamer-binding transcription factor 4; PAP, prostatic acid phosphatase; PSA, prostate-specific antigen; SOX-2, (sex-determining region Y)-box-2; STn-KLH, sialyl-Tn-keyhole limpet hemocyanin; TERT, telomerase reverse transcriptase; VEGF-R, vascular endothelial growth factor receptor; WT-1, wild-type 1.
Oncogene and suppressor gene products, such as nonmutated HER2/neu and p53, are analogous to oncofetal antigens in that they can be overexpressed in tumors and may be expressed in some fetal and normal tissues. Similarly, telomerase, an enzyme important in cellular replication and chromosomal stability, is overexpressed in malignant cells as compared with most normal cells. Epitopes derived from human telomerase have been reported and presumably may be overexpressed by neoplastic cells.9
Tissue-lineage antigens such as prostate-specific antigen (PSA) and the melanocyte antigens MART-1/Melan A, tyrosinase, gp100, and TRP-1/gp75 are usually expressed in a tumor of a given type and the normal tissue from which it is derived. Tissue-lineage antigens are potentially useful targets for immunotherapy if the normal organ/tissue in which they are expressed is not essential, such as the prostate, breast, or melanocyte.
Antigens associated with driving EMT, the metastatic process and drug resistance
The most provocative potential targets for vaccine therapy are those molecules associated with cancer “stem cells” and/or the epithelial-mesenchymal transition (EMT) process, both of which are associated with drug resistance. Drivers of EMT are also associated with tumor cell extravasation and intravasation to the metastatic site. Recent studies have described the plasticity of the so-called cancer stem cells and the similarities between cells undergoing EMT and the acquisition of “stemness” characteristics.10 Gene products associated with EMT and cancer “stemness” are SOX-2, OCT-4, and carcinoma cells with the phenotype CD44high/CD24lo, and/or are CD133+.11–15 Each of these gene products is currently being evaluated for immunogenicity in terms of generating human T-cell responses in vitro, but some also have a relatively broad range of expression in some normal adult tissues. The T-box transcription factor brachyury has recently been identified as a major driver of EMT.16 It has been shown to be selectively expressed on both primary and metastatic lesions of several carcinoma types. Vaccines that have the ability to generate human T cells capable of lysing a range of human carcinoma cells have been developed.17 Vaccines are currently in clinical trials to target gene products associated with EMT and cancer cells with “stemness” characteristics.18–20
Types of vaccines
Numerous vaccine-delivery platforms have been analyzed in experimental models and many of these are now being evaluated in the clinic (Table 2). Each of these platforms has advantages and disadvantages. Some of these modalities may eventually prove to be most beneficial when used in combination or in tandem.
Table 2 Spectrum of current vaccine platforms in phase II/III clinical studies
| Vaccine platform | Example | Cancer type |
| Peptides/proteins | ||
| Peptide | gp100, MUC-1, HER2/neu | Melanoma, lung |
| Protein | MAGE-A3, NY-ESO | Melanoma |
| Antibody | Anti-idiotype | Lymphoma |
| Glycoproteins | sTn-KLH | Melanoma |
| Recombinant vectors | ||
| Adenoviruses | Adeno-CEA, alpha-CEA | Carcinoma |
| Poxvirus | Vaccinia virus, MVA, fowlpox, (PROSTVAC) | Prostate |
| Saccharomyces cerevisiae (yeast) | Yeast-CEA | Pancreatic |
| Listeria | Listeria-mesothelin | Pancreatic |
| Tumor cells | ||
| Allogeneic | GVAX (+ GM-CSF) | Pancreatic |
| Dendritic cell/autologous tumor cell fusions | Myeloma | |
| Autologous | Adeno-CD40L, colon (BCG) | Chronic lymphocytic leukemia, colon, melanoma |
| Dendritic cells/APCs | ||
| Dendritic cell–peptide | Glioma peptides | Glioma, melanoma |
| Dendritic cells–vector infected | rV, rF-CEA-MUC1-TRICOM (Panvac-DC) | Colorectal |
| APC-protein | Sipuleucel-T (PAP-GM-CSF) | Prostate |
APC, antigen-presenting cell; BCG, Bacillus Calmette–Guerin adjuvant; CD40L, CD40 ligand; CEA, carcinoembryonic antigen; gp100, glycoprotein 100; GM-CSF, granulocyte macrophage colony-stimulating factor; MAGE-A3, melanoma-associated antigen 3; MUC-1, mucin 1; NY-ESO, New York esophageal carcinoma antigen 1; PAP, prostatic acid phosphatase; PSA, prostate-specific antigen; rF, recombinant fowlpox; rV, recombinant vaccinia; STn-KLH, sialyl-Tn-keyhole limpet hemocyanin.
Whole-tumor-cell vaccines
The major advantage of using whole-tumor-cell vaccines is that tumor-cell-mutated TSAs and TAAs, some identified and most as yet undefined, may be present in the vaccine preparation. The two types of whole-tumor-cell vaccine platforms being examined clinically are (1) autologous (i.e., manipulating the tumor from a patient into a vaccine for that patient) and (2) allogeneic, in which tumor cells from other patients, usually from established tumor cell lines, are used. Preparing an autologous tumor cell vaccine requires an enormous amount of effort, as fresh tumor needs to be obtained at surgery and prepared in a similar manner for each patient. Due to the considerable variability of tumors among patients, and costs associated with custom vaccine production, most current whole-tumor-cell vaccine approaches have centered on the use of allogeneic cell lines and cell banks.21–26 Moreover, the cell lines used in the preparations can be infected with vectors that express cytokine genes such as granulocyte-macrophage colony-stimulating factor (GM-CSF)27 or other immunostimulatory transgenes28 to enhance the immunogenicity of the tumor cell. Studies employing tumor cell lines transfected to express GM-CSF have met with some success in treating pancreatic cancer and other tumor types.24–27, 29, 30
Direct injection of cytokine genes or costimulatory molecule genes into tumor (in situ autologous whole tumor cell vaccines)
Two signals are required for the efficient activation of T cells. The first signal is mediated through a peptide–MHC complex on the cell surface of the antigen-presenting cell (APC), which binds to the T-cell receptor (TCR) on the surface of the T cell. The second signal involves the interaction of a T-cell costimulatory molecule on the surface of the APC with its ligand on the surface of the T cell. To date, the most studied of the T-cell costimulatory molecules is B7-1, which interacts with two ligands on the T-cell surface: CD28 for upregulation of T-cell function and CTLA-4 for downregulation of T-cell function. Numerous preclinical studies show that the addition of B7-1 to a weakly immunogenic tumor will make it more immunogenic.28 This phenomenon has also occurred when other costimulatory molecules, such as intercellular adhesion molecule (ICAM)-1 and lymphocyte function-associated antigen (LFA)-3, have been added to tumors. Thus, one can envision the direct injection of a vector expressing one or more costimulatory molecules into a tumor mass to induce an antitumor immune response. The advantage of this direct-injection approach is that the “vaccine” is the patient’s own tumor, which may express unique mutated TSAs or TAAs. In clinical studies, vaccinia- and avipox-based recombinants expressing the B7-1 costimulatory molecule have been directly injected into melanoma or carcinoma lesions.31 A clinical study has been reported with interesting findings in which recombinant vectors containing a triad of costimulatory molecules (B7-1, ICAM-1, and LFA-3; designated TRICOM) were directly injected into melanoma tumor lesions.32, 33 Cytokines can also be introduced into the tumor mass using vectors as delivery vehicles, as in the case of a recombinant vaccinia virus expressing GM-CSF directly injected into melanoma lesions.34
Peptides
Most peptides used to induce an immune response are from TSAs or TAAs and are approximately 8 to 11 amino acids in length. These peptides can bind with the appropriate class of the MHC molecule on the APC surface. These MHC-restricted responses are thus effective only if the appropriate MHC allele is present in a patient. The most studied MHC restriction element in the human population is the MHC class I allele, human leukocyte antigen (HLA)–A2, which is found in approximately 50% of the Caucasian population.
Using peptides as immunogens has some advantages: (1) preparation is relatively easy and affordable; (2) because the immunogen is extremely well-defined, the immune response can be analyzed in several ways and quantitated; and (3) peptides can be modified to be more immunogenic in the form of peptide agonists. The same property that makes a peptide vaccine attractive—its specificity—can also be a disadvantage. For example, a CTL (cytotoxic T-lymphocyte) epitope peptide may induce a CTL response that is short lived because of the lack of help provided by helper peptides. Moreover, peptides are useful only in the vaccination of patients who have that specific allele. Some peptide vaccines under study include HPV,35 ras,36, 37 HER-2/neu,38 MAGE,39 MART-1, tyrosinase,40 gp100,41, 42 CEA,43 MUC-1,44 and PSMA,45, 46 among others.47
Agonist peptides
Peptide agonists fall into two general categories. In the first category, amino acids of the peptide that bind to the MHC are modified. More vigorous MHC binding (i.e., higher affinity for the MHC molecule) often leads to the generation of a more vigorous T-cell response. Examples of this are the alterations in the brachyury, gp100 melanoma, and MUC-1 peptides.8, 41, 48 The second category of agonist has been termed a TCR agonist. In this case, an amino acid of the peptide that interacts with the TCR on the T cell is modified; this can also result in a more vigorous induction of the T-cell response. An example of this is the generation of the TCR agonist for CEA.49
Anti-idiotypes
The idiotypic network is involved in the control of immune regulation. B-cell lymphomas present unique Igs on their cell surface, which make exquisite targets for immunotherapy. For this malignancy, anti-idiotype (Id) MAb vaccines have been quite successful clinically.4, 5, 50–55 But the specificity inherent in these vaccine strategies is both an advantage and a disadvantage; different B-cell lymphomas will display unique Ids on their cell surface, making their preparation for each patient labor intensive.56
Vectors
Vectors are among the more flexible means of vaccine delivery. Several major platforms of both viral and bacterial vectors are now in use in the clinic (Table 2). Each vector category and type has its own potential advantages and disadvantages. Review articles have been published on the potential merits of these vectors.57–63 In general, the advantage of a vector-based vaccine is that (1) multiple genes (including genes for costimulatory molecules and cytokines) can be inserted into some types of vectors; (2) the relative cost of production is low compared to the preparation and purification of proteins or whole-tumor-cell vaccines; (3) many vectors have the ability to infect professional APCs so that the antigens they express can be processed; and (4) viral and bacterial vectors are composed of prokaryotic proteins that act as natural vaccine adjuvants in stimulating the host immune response. The disadvantage of some, but not all, vectors is the development of host-induced immunity to the vector itself, thus limiting its continued use.
Viral vectors
One of the most studied groups of vaccine vectors is the poxvirus group. Vaccinia virus, which was derived from a benign pox disease in cows, has been administered to more than 1 billion people and is responsible for the worldwide eradication of smallpox.64 As a result, smallpox vaccinations in the United States and most Western countries were halted approximately 40 years ago. However, most cancer patients are older than 40 and, therefore, have some level of preexisting immunity to vaccinia virus. For this reason, recombinant vaccinia viruses cannot be given multiple times in vaccine protocols. The poxvirus family contains the replication-incompetent modified vaccinia Ankara (MVA), a derivative of vaccinia virus.57 MVA is thought of as a safer alternative to the smallpox vaccine because it can infect mammalian cells but cannot replicate.65 Other replication-defective members of the poxvirus family are the avipox vectors (fowlpox and canarypox/ALVAC).58 These avipox vectors infect human cells and express their transgenes for 2 to 3 weeks before cell death; they are incapable of reinfecting cells. Clinical studies have shown that avipox-based CEA vectors can be given to patients numerous times with a resulting increase in CEA-specific T-cell responses.47, 66 Preclinical and clinical studies47, 67–69 show that optimal use of recombinant vaccinia or MVA may be to prime the immune response, followed by boost vaccinations with other vectors such as replication-defective pox vectors, peptides, or DNA. Advantages of using vaccinia virus or the replication-incompetent MVA avipox are (1) large amounts of foreign DNA can be inserted into the vector and (2) proteins expressed in poxviruses are more immunogenic than the native protein, which is most likely a result of the inflammatory responses triggered against highly immunogenic poxvirus proteins. Several clinical trials with recombinant poxviruses containing TAAs such as CEA, MUC-1, PSA, and HPV have been completed and others are ongoing.
Adenovirus has also been proposed as a vector in recombinant vaccine design because its viral genome can be altered to accept foreign genes that are stably integrated. To produce recombinant adenovectors, endogenous viral DNA sequences are typically deleted from replication-competent regions, which results in an attenuated form of the virus with potentially improved safety. A new viral vector gene delivery platform, adenovirus serotype-5 (Ad5) [E1-, E2b-],70–79 has been described. The platform consists of a replication-defective Ad5 in which portions of the early 1 (E1), early 2 (E2b), and early 3 (E3) gene regions have been deleted; the deletions have been reported to result in a dramatic decrease of late gene expression, which results in a marked reduction in host inflammatory responses and anti-Ad host responses. Human cells transfected with these adenovirus constructs were shown to express the encoded transgene(s) for prolonged times in vivo compared to other adeno vector platforms. In a Phase I/II clinical trial, cohorts of patients with metastatic colorectal cancer (mCRC) were vaccinated with escalating doses of the Adeno platform carrying a gene for CEA. CEA-directed T-cell responses were induced and patients exhibited evidence of a favorable survival probability.80
Yeast and bacterial vectors
One advantage of recombinant Saccharomyces cerevisiae as a vaccine vehicle is its lack of toxicity. Besides being inherently nonpathogenic, this particular species of yeast can be heat killed before administration and has been shown to be safe in humans in several clinical trials, with maximum tolerated dose not reached.63, 81, 82 S. cerevisiae can be easily engineered to express one or more antigens in large quantities, can be propagated and purified rapidly, and is very stable.83 In addition, recombinant yeast has been shown to induce a robust host immune response to non-self-antigens.19, 63, 83–86
Bacterial vectors, such as Salmonella87, 88 and Listeria,62 have the advantage of tropism for professional APCs, such as macrophages. While these vectors are potentially virulent in humans, several attenuated strains have been developed for human use. From a clinical perspective, both recombinant Salmonella and recombinant Listeria monocytogenes-based vectors may be administered orally.
Plasmid DNA
Polynucleotide vaccines are easy to prepare, but the mode of action of these vectors in inducing an immune response is not fully understood because most studies have involved intramuscular inoculations, and it is not known how antigens get to professional APCs. Many preclinical and clinical studies using infectious disease agents and cancer antigens have been carried out with DNA vectors.89–92
Dendritic cell vaccines
The dendritic cell (DC) is considered the most potent APC and, therefore, one of the most attractive means of immunization.93, 94 DCs can be employed by (1) loading with a peptide, protein, or anti-Id Ab, (2) infecting with a viral vector, (3) loading with apoptotic bodies from tumor cells, or (4) fusing with a tumor cell.95–98 The major disadvantages of this approach are the great cost and effort involved. Large amounts of peripheral blood mononuclear cells (PBMCs) must be obtained from patients via leukapheresis. The PBMCs must then be cultured for several days in the presence of cytokines such as GM-CSF, IL-4, and/or TNF-α, and then reinfused into the patient. This must be done for each patient. A successful strategy, approved by the FDA, involves immunization with autologous DCs loaded ex vivo with a recombinant fusion protein consisting of the tumor antigen prostatic acid phosphatase (PAP) linked to GM-CSF for patients with metastatic prostate cancer, as is discussed below.
Combination of vaccines
Although each of the various methods of immunization described has advantages and disadvantages, the most effective immunization protocol may involve priming with one type of immunogen and boosting with another. This method may be advantageous because (1) two different arms of the immune system may be enhanced by using two different modalities (i.e., CD4+ and then CD8+ T cells); (2) one methodology may be more effective in priming naive cells while another modality may be more effective in enhancing memory cell function; and (3) some of the most effective methods of immunization, like the use of recombinant vaccinia virus or adenoviruses, can be used only a limited number of times because of host anti-vector responses. These vectors may be most effective when used as priming agents, followed by boosts with other agents. Numerous preclinical and clinical studies demonstrate the advantages of diversified prime-and-boost protocols.47, 66, 67, 99–103
Non-specific immune stimulants
TAAs and TSAs are, by definition, weak immunogens because tumors develop, persist, and grow in the presence of an intact immune system. Conventional adjuvants such as incomplete Freund’s adjuvant (IFA) will allow the immunogen to be maintained at the injection site (the so-called depot effect) so that infiltrating APCs and effector cells can initiate a more vigorous immune response. Immunostimulants also work by crossing membrane barriers. In this case, proteins must be taken up by professional APCs and presented in the context of MHC molecules for effective presentation to the immune system. Adjuvants with lipophilic components and liposomes104 facilitate this process.
Microbial products such as those present in certain adjuvants (i.e., IFA), bacillus Calmette–Guerin adjuvant (BCG), and even viral vectors also are potent activators of DCs, partly because they contain agonists of toll-like receptors (TLRs). TLRs are expressed by DCs and other innate immune cell types and comprise a family of approximately 10 to 15 receptors.105, 106 Activation of DC via their TLRs augments expression of adhesion, chemokine, and chemokine receptor molecules, which, in turn, regulate cellular trafficking to sites of inflammation and pathogenic encounters. Thus, the biological consequences of TLR engagement lead to inflammation, characterized by the recruitment of key immune and nonimmune effector cells to mediate pathogen destruction. In regard to TLR agonists, CpG motifs constitute the most studied of these sequences.107–110
In addition, certain cytokines and chemokines have been shown to enhance the level of APC and/or effector cell function either locally or systemically. For example, GM-CSF has been reported to enhance antigen-specific T-cell responses, delayed-type hypersensitivity reactions, and antitumor responses.111–120 Clinical studies using GM-CSF along with protein immunogens have shown an enhancement of the antigen-specific immune response.50, 121 Flt-3L administration has been shown to enhance the number of DCs systemically, both in animal models and clinically.122, 123 Finally, several cytokines and chemokines have been shown to play a critical role in enhancing T-cell function.124–128 The most studied of these is IL-2.129, 130 When used as a single agent, IL-2 has been shown to have antitumor effects in melanoma and renal cell carcinoma (RCC) patients129, 131 and, as recently reported, to enhance the effectiveness of melanoma-based peptide vaccines.41 Other cytokines, such as IL-7, IL-12, and IL-15, have been shown to enhance T-cell responses and antitumor activity in experimental models and may have more potential clinical utility than IL-2.125, 132–134
One strategy for improving the safety of proinflammatory cytokines, such as IL-2 and IL-12, is to direct their delivery to tumors via fusion to a tumor-targeting antibody. Such antibody-cytokine fusion proteins, or “immunocytokines,” have previously demonstrated the ability to enhance antitumor immunity in preclinical models.135 To maximize immunocytokine tolerability, the antibody selected as a vehicle must bind specifically to an antigen found in tumors. A tumor-targeted IL-12 immunocytokine, called NHS-IL12, was engineered by genetically fusing two human IL-12 heterodimers to the C-termini of the heavy chains of the NHS76 antibody. NHS-IL12 was superior to recombinant IL-12 when evaluated as an antitumor agent in three murine tumor models. In preclinical studies combining NHS-IL12 treatment with a cancer vaccine, radiation, or chemotherapy resulted in greater antitumor effects than each individual therapy alone.136
Vaccine clinical trials
Below is a representative description of prior and ongoing vaccine clinical trials.
Prostate cancer
PAP, which is expressed on over 95% of prostate cancer cells, has been used as a target of the vaccine sipuleucel-T, also called Provenge® (PAP-GM-CSF-pulsed APCs). After early clinical trials of the sipuleucel-T vaccine demonstrated safety, larger trials were conducted in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer (mCRPC). A Phase III trial137 was conducted with overall survival (OS) as the endpoint, enrolling more than 500 patients. No change in time to progression (TTP) was seen, but OS was improved in the vaccine arm (25.8 months vs 21.7 months; p = 0.032). In April 2010, the FDA approved sipuleucel-T for the treatment of minimal or nonsymptomatic mCRPC. A second prostate cancer vaccine has also been evaluated in this same prostate cancer population. This “off-the-shelf” platform (PROSTVAC) consists of a recombinant vaccinia prime and multiple fowlpox booster vaccinations. Each vector contains transgenes for PSA and three costimulatory molecules (B7-1, ICAM-1 and LFA-3, designated TRICOM).138, 139 A randomized placebo-controlled 43-center Phase II trial enrolled 125 minimally symptomatic mCRPC patients.140 Similar to the experience with sipuleucel-T, PROSTVAC did not alter TTP, yet improved median OS relative to placebo (control vector), 25.1 versus 16.6 months. Patients receiving PROSTVAC also had a 44% reduction in death rate compared to the control cohort (p = 0.0061).140 The median OS in a second single-arm Phase II study141 was 26.6 months, similar to the larger PROSTVAC randomized trial. A global Phase III placebo-controlled trial of PROSTVAC in patients with minimally symptomatic prostate cancer (n = 1298) has now completed accrual. The endpoint is OS.
A recent review142 analyzed the immune impact induced by PROSTVAC (PSA-TRICOM) in prostate cancer patients. Of 104 patients tested for T-cell responses, 57% demonstrated an increase in PSA-specific T cells 4 weeks after vaccine, and 68% of patients mounted post-vaccine immune responses to TAAs not present in the vaccine (antigen spreading/antigen cascade). Measurements of systemic immune response to PSA may underestimate the true therapeutic immune response (as this does not account for cells that have trafficked to the tumor) and does not include antigen spreading/antigen cascade (as described below). Furthermore, although the entire PSA gene is the vaccine, only one epitope of PSA is evaluated in the T-cell responses. Because this vaccine is directed at generating a cellular/Th1 immune response, less than 0.6% of patients tested had evidence of PSA antibody induction following vaccine. Active surveillance is becoming an increasingly utilized approach in men diagnosed with “low-risk” prostate cancer. A randomized, multicenter, placebo-controlled study of PROSTVAC vaccination versus active surveillance in patients with newly diagnosed clinically localized prostate cancer is under way. The endpoints of this study will be the changes in immune infiltrate and tumor extent at the 12-month biopsy versus initial biopsy.
GVAX, a GM-CSF-secreting vaccine, is an admixture of two prostate cancer cell lines that have been transduced with a replication-defective retrovirus for GM-CSF expression. In two separate multicenter Phase II trials, patients with asymptomatic CRPC given GVAX had a median survival of 26.2–35.0 months.24, 143 By estimating predicted survival based on a commonly used nomogram,144 patients in both trials exceeded anticipated survival by > 6 months.145
Nineteen HLA-A2-positive patients with rising PSA without detectable metastatic disease or local recurrence received a vaccine containing 11 HLA-A*0201-restricted and two HLA class II synthetic peptides derived from prostate carcinoma tumor antigens subcutaneous (s.c.) for 18 months or until PSA progression. PSA doubling time of 4/19 patients (21%) increased from 4.9 to 25.8 months during vaccination.146 A Phase II adenovirus/PSA vaccine trial was conducted in patients with recurrent or hormone refractory prostate cancer. The majority of the patients demonstrated anti-PSA T-cell responses above pre-injection levels. Sixty-four percent of the patients demonstrated an increase in PSA doubling time.147 Clinical trials have also been carried out using peptide-pulsed DCs loaded with peptides of human PSMA,148–150 as well as DCs transfected with PSA tumor RNA.151 In these studies, antigen-specific immune responses have been observed and some decreases in serum PSA have also been noted.
Melanoma
The vast majority of vaccine clinical trials have been conducted in patients with melanoma because (1) interferon (IFN) and IL-2 have both shown clinical responses in melanoma; (2) melanoma lesions often are readily accessible and thus can be studied for immune infiltrate and cells from melanoma lesions can be grown in culture to obtain both tumor cells and tumor-infiltrating lymphocytes (TILs); and (3) numerous melanoma-associated antigens have been identified.
Clinical trials have been conducted using various peptides derived from melanoma-associated antigens, including tyrosinase, MART-1, gp100, Melan-A (a modified gp100 epitope152), and members of the MAGE family.152–157 Vaccination-induced immune responses as well as clinical benefits were reported. Newer approaches have combined these peptides with GM-CSF emulsified in Montanide ISA-51.158 Another peptide approach is vaccination with autologous tumor-derived heat shock protein/peptide complexes.159 Immune responses and clinical responses have also been observed using melanoma peptide vaccination in conjunction with anti-CTLA-4,160 melanoma peptide-pulsed DCs,94, 154 tumor RNA-transfected DCs,161 and intratumoral administration of autologous DCs.162 A randomized Phase III study showed no benefit using an autologous peptide-pulsed DC vaccine compared to dacarbazine in first-line treatment of patients with metastatic melanoma.163
A randomized Phase III trial was conducted involving 185 patients with stage IV or locally advanced stage III cutaneous melanoma, expression of HLA*A0201, and suitability for high-dose IL-2 therapy. Patients were randomly assigned to receive IL-2 alone or gp100 plus IFA, followed by IL-2. The vaccine–IL-2 group, as compared with the IL-2-only group, had a significant improvement in overall clinical response (16% vs 6%, p = 0.03).The median OS was also longer in the vaccine–IL-2 group than in the IL-2-only group (17.8 months vs 11.1 months; p = 0.06).164 In a multicenter randomized trial, 175 patients with stage IV melanoma were enrolled into four treatment groups. Best clinical response was partial response in 7/148 evaluable patients (4.7%) without significant difference among study arms.165 In another trial, three HLA class I peptides were administered in a factorial 2 × 2 design: peptide vaccine alone, or combined with GM-CSF, or combined with IFN-α2b, or combined with both. At a median follow-up of 25.4 months, the median OS of patients with vaccine immune response was significantly longer than that of patients with no immune response (21.3 months vs 13.4 months; p = 0.046).166
Vitespen is an autologous, tumor-derived heat shock protein gp96 peptide vaccine. Patients (n = 322) with stage IV melanoma were randomly assigned 2 : 1 to receive vitespen or physician’s choice of a treatment. OS in the vitespen arm was statistically indistinguishable from that in the physician’s choice arm.167 A randomized trial provided evidence that a DC vaccine is associated with longer survival compared with a tumor cell vaccine, and is consistent with previous data suggesting a survival benefit from this patient-specific immunotherapy.168 A Phase II trial of a DC vaccine for metastatic melanoma patients with mainly the HLA-A24 genotype was investigated; OS analysis revealed a significant survival prolongation effect in patients receiving the DC vaccine.169 Studies were also carried out to detect a pretreatment gene expression signature predictive of response to MAGE-A3 vaccine in patients with metastatic melanoma. An expression signature was associated with the patient’s clinical response.170 A Phase II trial investigated a peptide vaccination against survivin, a protein crucial for the survival of tumor cells, in patients with treatment-refractory stage IV metastatic melanoma. Survivin-specific T-cell reactivities strongly correlated with tumor response and patient survival.171
Several clinical studies have employed autologous melanoma cells as vaccine. Clinical responses were observed when these melanoma-cell vaccines were preceded by cyclophosphamide.172, 173 Clinical responses were also observed when autologous melanoma cells were transduced with GM-CSF and given as vaccine.174, 175 In addition, intratumoral administration of recombinant vaccinia encoding GM-CSF in patients with cutaneous melanoma has been reported to induce antitumor immune responses.31, 32, 176 Several randomized Phase II trials have been carried out using either autologous177, 178 or allogeneic179, 180 melanoma vaccine in patients with resected melanoma. In these trials, there is evidence of improved disease-free survival (DFS) in the vaccine arm versus control or no-treatment groups. Recent research indicates that immunization with hybrids of tumor cells and APCs can induce protective immunity and rejection of established tumors in various rodent models.95, 181, 182 Novel clinical trial strategies incorporating DC-based vaccines have recently been reported.183–185 Purified hybrids from the fusion of DCs and tumor cells (dendritomas) have been shown to be safe and to induce both immunological and clinical responses when combined with low-dose IL-2.184
T-VEC is an intralesional oncolytic immunotherapy comprising an HSV-type 1 engineered to make GM-CSF. In the randomized Phase III trial, 436 patients with stage IIIB-IV melanoma were randomized 2 : 1 to receive intralesional T-VEC versus s.c. GM-CSF. T-VEC significantly improved the proportion of patients with duration of response ≥ 6 months (16% vs 2%) and an overall response rate (26% vs 6%) versus s.c. GM-CSF. Many patients remained in remission with a median follow-up of over 18.4 months.186, 187
Colorectal carcinoma
A prospective randomized-controlled Phase III clinical trial was carried out in 254 patients with stage II and stage III colon cancer using autologous tumor cell vaccine with BCG postsurgery. A 5.3-year median follow-up showed 40 recurrences in the control group and 25 in the vaccine group. The vaccine showed a statistically significant recurrence-free survival (RFS) in patients with surgically resected stage II colon cancer, but not stage III colon cancer.188 Further follow-up demonstrated a significant survival benefit in the vaccine arm over surgery alone.189
Several clinical trials have been conducted employing CEA-based vaccines. Results of vaccination in patients with recombinant vaccinia virus,69 recombinant avipox virus,66, 190, 191 or a prime with recombinant vaccinia (rV)-CEA and multiple boosts with avipox-CEA47 have demonstrated the generation of CEA-specific immune responses in patients with advanced gastrointestinal (GI) carcinomas and other CEA-expressing carcinomas. A trial using both vaccinia- and avipox-CEA vaccines containing TRICOM has suggested improved survival in patients receiving the prime-and-boost regimen plus GM-CSF; 40% had stable disease (SDs) for at least 4 months.47, 79, 192, 193 In a multicenter Phase II study, colorectal cancer patients received PANVAC vaccine (rV-, rF-CEA-MUC1-TRICOM) post-metastasectomy for liver and lung metastases.192 Patients received either PANVAC or PANVAC-infected DCs and there was no statistical difference in clinical outcome in the two arms. Vaccinated patients (n = 74) were compared to a contemporary “control” (not randomized) group of colorectal cancer patients (n = 161) undergoing metastasectomy and receiving standard-of-care therapies. Both this control group and the PANVAC-vaccinated groups had similar prognostic characteristics and TTP for both groups was virtually identical with a median TTP of approximately 24 months.20, 192 However, the OS of PANVAC-vaccinated patients was 95% at 2 years and 90% at 40 months, compared to the contemporary control group with an OS of 75% at 2 years, and 47% at 40 months. The OS values of the control group are similar to 3–5 year survival data in this population from five other clinical studies.194–199 This is yet another example (as seen with ipilimumab, sipuleucel-T, and PROSTVAC) of no increase in TTP versus control with an increase in OS.
CEA peptide-pulsed DCs have also been employed in clinical studies using a modified CEA agonist peptide.200 Two of 12 patients with advanced colorectal cancers experienced tumor regression, one patient had a mixed response, and two had SD. Clinical responses correlated with the expansion of CEA-specific T cells. Twenty-six patients who had undergone resection of colorectal metastases were treated with intranodal injections of an autologous tumor lysate- and control protein-pulsed DC vaccine. Patients were randomized to receive DCs that had either been activated or not activated with CD40L. All patients were followed up for a minimum of 5.5 years. Patients with evidence of a vaccine-induced, tumor-specific T-cell-proliferative or IFN-γ response 1 week after vaccination had a markedly better RFS at 5 years (63% vs 18%, p = 0.037) than nonresponders.201
Clinical studies have also been carried out using anti-Id antibodies directed against anti-CEA MAbs. These studies have demonstrated the induction of immune responses,202, 203 slowed disease progression, and prolonged survival.204 MVA containing the gene for an oncofetal antigen found on many tumors (5T4) has been tested in a variety of cancers (TroVax, Oxford, UK). In a trial of 22 patients with mCRC, immune responses appeared to correlate with disease control, which was seen up to 18 months in patients with immune responses.205–207
Pancreatic carcinoma
Allogeneic whole-tumor-cell vaccines modified to secrete GM-CSF have also been employed in patients with pancreatic cancer. Evidence of vaccine-induced immune responses were observed as measured by delayed-type hypersensitivity.25, 208 A 60-patient Phase II study of the same vaccine in the adjuvant setting showed that post-chemotherapy induction of mesothelin-specific CD8 cells correlated with progression-free survival.209 The median OS was about 26 months compared with 21 months for chemotherapy alone in this same patient population at the same institution.210 GVAX and CRS-207 are cancer vaccines that have been evaluated in pancreatic ductal adenocarcinoma (PDA). GVAX is composed of two irradiated GM-CSF-secreting allogeneic PDA cell lines administered 24 h after treatment with low-dose cyclophosphamide (Cy) to inhibit regulatory T cells. CRS-207 is recombinant live-attenuated, double-deleted Listeria monocytogenes, engineered to express mesothelin. GVAX induces T cells against a broad array of PDA antigens, and mesothelin-specific T-cell responses have been shown to correlate with survival. Mesothelin is a TAA overexpressed in most PDAs. In a prior study, patients with previously treated advanced PDA who received Cy/GVAX had better induction of mesothelin-specific CD8+ T cells than those treated with GVAX alone. Median survival was 4.3 and 2.3 months, respectively. Previously treated patients with metastatic pancreatic adenocarcinoma were randomly assigned at a ratio of 2 : 1 to two doses of Cy/GVAX followed by four doses of CRS-207 or Cy/GVAX alone. In a prespecified per-protocol analysis of patients who received at least three doses (two doses of Cy/GVAX plus one of CRS-207 or three of Cy/GVAX), OS was 9.7 months versus 4.6 months. Enhanced mesothelin-specific CD8+ T-cell responses were associated with longer OS, regardless of treatment arm.211
Breast cancer
Sialy1-Tn (STn) is a glycopeptide contained in carcinoma-associated mucin. Patients were randomized to receive vaccine [STn coupled to keyhole limpet hemocyanin (KLH)] with or without cyclophosphamide. Patients treated with vaccine and cyclophosphamide were reported to have lived significantly longer212 than those treated with the same vaccine without cyclophosphamide. In addition, STn KLH vaccine has been administered to breast and ovarian cancer patients after an autologous stem cell transplant following high-dose chemotherapy. Vaccinated patients appeared more likely to survive and less likely to relapse, but further studies are required.213 A multicenter, double-blinded, randomized Phase III trial of STn conjugated to KLH vaccine was completed in 1028 women with metastatic breast cancer who had nonprogressive disease or no evidence of disease after first-line chemotherapy. The women treated with concomitant endocrine therapy and STn-KLH had longer TTP and OS than the control group of women who received KLH alone. Moreover, of the women who received endocrine therapy, those who had a median or greater antibody response to the STn-KLH vaccine had significantly longer median OS than those who had a below-median antibody response.214
Several clinical trials have been carried out using MUC-1 as a target, including the use of MUC-1 peptides,215–217 recombinant vaccinia virus encoding MUC-1 and IL-2,218 MVA expressing human MUC-1,219 and mannan–MUC-1 fusion protein.220, 221 In studies carried out with the HER2/neu peptides with or without GM-CSF, patients with breast cancer were able to generate HER2-specific immune responses.222–224 A Phase I/II clinical trial was carried out vaccinating breast cancer patients with E75, an HLA-A2/A3-restricted HER2/neu peptide, and GM-CSF. The 24-month analysis DFS was 94.3% in the vaccinated group and 86.8% in the control group (p = .08). In subset analyses, patients who benefited most from vaccination had lymph-node-positive, HER2 IHC 1+-2+, or grade 1 or 2 tumors and were optimally dosed.225 Fusion-cell (DCs and tumor cells) vaccination of patients with metastatic breast and renal cancer was shown to induce both immunological and clinical responses.226 An anti-Id antibody that mimics the human milk-fat globule antigen has also been utilized as a breast cancer vaccine in conjunction with autologous stem cell transplantation (ASCT). In that study, the 3-year OS rate was 48%, whereas the progression-free survival rate was 32%.227 A HER2 peptide-based vaccine (nelipepimut-S) has been tested in a Phase II trial of 195 patients with localized breast cancer at high risk of recurrence. HLA-A2-/A3-positive patients received vaccine with GM-CSF and those that were not HLA-A2 or -A3 positive were controls. The 5-year DFS was 89.7% in vaccinated patients versus 80.2% in nonvaccinated patients. The 5-year DFS was 94.5% in optimally dosed patients. A Phase III study has been initiated.228
Lung cancer
Three Phase III studies in patients with non–small cell lung cancer (NSCLC) failed to meet their primary endpoints. A MAGE-A3 peptide-based vaccine was evaluated in stages IB, 2 and 3A tumor-resected NSCLC patients whose tumors expressed the MAGE-A3 gene.229, 230 The L-BLP25 vaccine consists of a peptide of the tandem repeat region of the MUC-1 gene. A Phase III trial employing this vaccine after chemoradiation in patients with unresectable stage 3 NSCLC did not meet its primary endpoint of increased OS.231 Lucanix is a vaccine composed of four TGF-β2 antisense modified, irradiated allogeneic NSCLC cell lines. In a Phase III trial in stages IIIA, IIIB, and IV, for those NSCLC patients who did not progress after front-line chemotherapy, a considerable increase in OS was not observed in the vaccine arm; however, the time elapsed between randomization and the end of front-line chemotherapy had a significant impact on survival (p = 0.002).232, 233 Other vaccines in patients with NSCLC are also being evaluated. A modified vaccinia virus (MVA) encoding the MUC-1 and IL-2 genes was assessed in combination with first-line chemotherapy in patients (n = 74) with advanced NSCLC. Six-month PFS was 43.2% in the vaccine plus chemotherapy group versus 31.5% in the chemotherapy-alone group.234 A Phase II study of a telomerase peptide vaccine in NSCLC patients (n = 23) after chemoradiation has also been evaluated.235 Immune responders recorded a median PFS of 371 days versus 182 days for nonresponders.
Ovarian/cervical cancer
Clinical findings have also been reported in patients with advanced ovarian cancer using a vaccine consisting of an MAb directed against the tumor antigen CA125.236 A Phase II study examining an anti-idiotypic antibody vaccine that functionally imitated the CA125 antigen reported survival benefit; median OS of the vaccine group was 19.4 months versus 4.9 months for the control group.237
The purpose of another Phase II single-arm clinical trial was to evaluate whether pretreatment with low-dose cyclophosphamide improves immunogenicity of a p53-synthetic long peptide vaccine in patients with recurrent ovarian cancer. SD was observed in 20.0% (2/10) of patients.238 Two independent consecutive studies were conducted of combinatorial immunotherapy comprising DC-based autologous whole-tumor-antigen vaccination in combination with anti-angiogenesis therapy. Thirty-one patients had recurrent progressive stage III and IV ovarian cancer. Vaccination was well tolerated and elicited tumor-specific T-cell responses against various ovarian tumor antigens in both studies, and a clinical benefit of 65% correlated with the immune response with some experiencing prolonged PFS.239 A Phase II clinical trial of recombinant vaccinia-NY-ESO-1 (rV-NY-ESO-1) followed by booster vaccinations with recombinant fowlpox-NY-ESO-1 (rF-NY-ESO-1) in 22 epithelial ovarian cancer (EOC) patients with advanced disease who were at high risk for recurrence/progression was conducted. Median PFS was 21 months, and median OS was 48 months. CD8+ T cells derived from vaccinated patients were shown to lyse NY-ESO-1-expressing tumor targets. These data provide preliminary evidence of clinically meaningful benefit for diversified prime and boost recombinant poxviral-based vaccines in ovarian cancer.240
Studies were undertaken to determine the toxicity, safety, and immunogenicity of a human papillomavirus 16 (HPV16) E6 and E7 long peptide vaccine administered to end-stage cervical cancer patients. The HPV16 E6 and E7 long peptide-based vaccine was well tolerated and capable of inducing a broad IFN-γ-associated T-cell response even in end-stage cervical cancer patients.241 Vulvar intraepithelial neoplasia is a chronic disorder caused by high-risk types of HPV, most commonly HPV type 16 (HPV-16). Twenty women with HPV-16-positive, grade 3 vulvar intraepithelial neoplasia were vaccinated with a mix of long peptides from the HPV-16 viral oncoproteins E6 and E7.242 One-half of a group of 20 patients with HPV16-induced vulvar intraepithelial neoplasia grade 3 displayed a complete regression after therapeutic vaccination. There was a strong correlation between a defined set of vaccine-prompted specific immune responses and the clinical efficacy of therapeutic vaccination.243
Leukemia/lymphoma
The variable regions of B-cell receptor Igs on lymphomas are excellent targets for vaccines. These Ids have now been used successfully in the treatment of B-cell lymphomas.50, 244–247 An Id protein vaccine was used in 25 patients after a chemotherapy-induced second complete clinical remission in follicular lymphoma (FL). After vaccination, tumor-specific humoral or cellular immune responses were found in 20 of 25 patients.246 All of the responders with enough follow-up maintained a second CR longer than the first CR, whereas the five patients who did not mount an immune response had a second CR that was, as expected, shorter than the first CR. Vaccination with hybridoma-derived autologous tumor Ig Id conjugated to KLH and administered with GM-CSF induces FL-specific immune responses. A double-blind multicenter controlled Phase III trial of this vaccine was conducted. Of 234 patients enrolled, 177 (81%) achieved CR/CRu after chemotherapy and were randomly assigned. For the 177 randomly assigned patients, median DFS between Id-vaccine and control arms was 23.0 months versus 20.6 months, respectively (p = 0.256). For patients who received Id vaccine, median DFS after randomization was 44.2 months for Id-vaccine arm versus 30.6 months for control arm. Vaccination with patient-specific hybridoma-derived Id vaccine after chemotherapy-induced CR/CRu may prolong DFS in patients with FL.248
A multiple myeloma vaccine has been developed whereby patient-derived tumor cells are fused with autologous DCs, creating a hybridoma that stimulates a broad antitumor response. A Phase II trial was conducted in which patients underwent vaccination following ASCT to target minimal residual disease. Seventy-eight percent of patients achieved a best response of CR or PR and 47% achieved a CR/near CR (nCR). Remarkably, 24% of patients who achieved a partial response following transplant were converted to CR/nCR after vaccination and at more than 3 months post transplant, consistent with a vaccine-mediated effect on residual disease. The post-transplant period for patients with multiple myeloma provides a unique platform for cellular immunotherapy in which vaccination with DC/myeloma fusions resulted in the marked expansion of myeloma-specific T cells and cytoreduction of minimal residual disease.249 Clinical responses have also been observed using Id vaccine in patients with multiple myeloma.52, 250
For patients with chronic myelogenous leukemia, vaccination with bcr-abl oncogene breakpoint fusion peptides has generated specific immune responses.251 A study investigated the immunogenicity of Wilms tumor gene product 1 (WT1)-peptide vaccination in WT1-expressing acute myeloid leukemia (AML). Vaccination consisted of GM-CSF and WT1 peptide and KLH. AML patients (n = 17) received a median of 11 vaccinations. Objective responses in AML patients were 10 SDs including four SDs with more than 50% blast reduction and two with hematologic improvement. An additional four patients had clinical benefit after initial progression, including one complete remission and three SDs.252 A Phase I/II trial investigated the effect of autologous DC vaccination in 10 patients with AML. Two patients in partial remission after chemotherapy were brought into complete remission after intradermal administration of full-length WT1 mRNA-electroporated DCs. In these two patients and three other patients who were in complete remission, the AML-associated tumor marker returned to normal after DC vaccination, compatible with the induction of molecular remission. Clinical responses were correlated with vaccine-associated increases in WT1-specific CD8+ T cell frequencies.253
Other tumor types
Intravesical BCG has been used successfully in the treatment of bladder cancer, implicating immune mechanisms.254 Intravesical administration of wild-type vaccinia virus has now been shown to be safe, and three of four patients treated were disease free at 4-year follow-up.255 The response of renal carcinoma to high-dose IL-2 and IFN-alpha also implicates immune mechanisms in therapeutic responses.256 An autologous RCC tumor-cell vaccine that was genetically modified to overexpress B7-1 to provide costimulation to tumor-reactive T cells was employed.257 In this single-arm Phase II study, 66 patients enrolled and 39 received at least one dose of vaccine and low-dose IL-2. Best responses were CR (3%), PR (5%), SD (64%), and PD (28%). A post hoc analysis suggested that lymphocytic infiltration of the vaccine site determined by biopsy directly correlated with survival (28.4 months versus 17.8 months, p = 0.045). A Phase I trial was conducted to evaluate the feasibility, safety, and efficacy of a DC-based vaccination in patients (n = 21) with metastatic RCC. Autologous mature DCs were pulsed with HLA-A2-binding MUC1 peptides. In six patients, regression of the metastatic sites was induced after vaccinations with three patients achieving an objective response (one complete response, two partial responses, two mixed responses, and one SD). An additional four patients were stable during the treatment for up to 14 months.258
Vaccination of pediatric solid tumor patients with tumor-lysate-pulsed DCs has been shown to expand specific T cells and mediated some tumor regression.259 In addition to DC fusion vaccination of patients with metastatic breast and renal cancer, clinical trials have examined vaccination of glioma patients with DC/glioma fusions.226, 260 Early clinical trials in patients with primary brain tumors have shown immune responses and objective responses with DC vaccines,261 autologous formalin-fixed262 or TGF-β-modified263 whole-tumor-cell vaccines. A pilot study of s.c. vaccinations with glioma-associated antigen (GAA) epitope peptides in HLA-A2-positive children with newly diagnosed brainstem gliomas (BSGs) and high-grade gliomas (HGGs) was undertaken. Twenty-six children were enrolled, 14 with newly diagnosed BSGs treated with irradiation and 12 with newly diagnosed BSGs or HGGs treated with irradiation and concurrent chemotherapy. Five children had symptomatic pseudoprogression, which responded to dexamethasone and was associated with prolonged survival. Only two patients had progressive disease during the first two vaccine courses; 19 had SD, two had partial responses, one had a minor response, and two had prolonged disease-free status after surgery.264
Considerations in the analysis of vaccine clinical trial results
It has now been demonstrated that appropriate vaccine delivery systems and vaccine strategies can elicit immune responses to TAAs in patients with a range of cancers and, in some cases, can mediate prolonged survival, prolonged disease-free interval, drops in serum tumor markers, and/or regression of metastatic disease. Patients with advanced cancers are perhaps the least appropriate population to demonstrate the efficacy of a cancer vaccine; thus, future clinical trials should be carried out in patients with less or with low tumor burden metastatic disease or earlier in the disease process. Optimal use of vaccines should occur immediately prior to, during, or immediately following adjuvant therapy. Several such studies are ongoing. The overall goal is the use of vaccine regimens in combination with front-line cancer therapies, thereby reducing the interval between disease diagnosis and the initiation of vaccine; this would bring the use of cancer vaccines closer to their original intention—use in patients with minimal disease.
Combination therapies
There are numerous ways in which vaccine therapy can be used in combination with the so-called non-immune-based therapies (Table 3) as well as with other immunotherapies. One of the major advantages of cancer vaccines is their reduced toxicity. One of the conventional thoughts now being reevaluated is that cancer vaccines cannot be used in combination with chemotherapy or radiation therapy. One should not confuse the potential reduced efficacy of a vaccine given to patients who have failed multiple rounds of prior chemotherapy versus administration of vaccine with or prior to chemotherapy. Preclinical studies, and now several clinical studies, have shown that patients can mount an immune response to a cancer vaccine when given in combination with certain cytotoxic drugs, hormones, or local radiation of tumor. Studies have shown that when tumor cells are exposed to sublethal doses of radiation or certain chemotherapeutic agents, the phenotype of tumor cells is actually modulated to make them more susceptible to T-cell-mediated killing (Figure 1). Indeed, the era of the use of therapeutic cancer vaccines in combination with other immunotherapeutics as well as the so-called nonimmune conventional cancer therapeutics is still in its early stage of development and full clinical potential.
Table 3 Vaccines in combination with other therapeutic modalities
| Modality | Mechanism of action to enhance vaccine efficacy |
| Radiation | Alteration in tumor cell phenotype |
| Chemotherapy | “Immunologic” tumor cell death Alteration in tumor cell phenotype Enriched effector: regulator cell ratios |
| Hormonal therapy | Thymic regeneration and induction of naive T cells Alteration of tumor cell phenotype |
| Small-molecule-targeted therapeutics | Modulate tumor microenvironment Alteration in tumor cell phenotype |
| Monoclonal antibodies | Enhanced antibody-dependent cell-mediated cytotoxicity Immune checkpoint modulation |
| Imidsa (lenalidomide) | Stimulate T-cell proliferation |
a Imids are a novel class of immunomodulators.
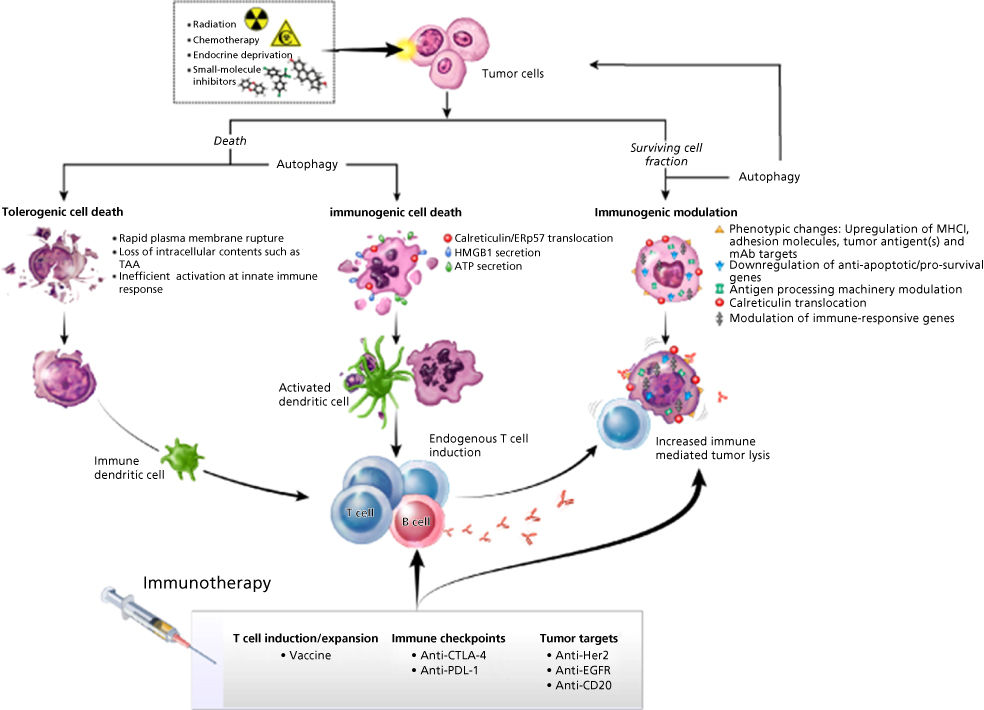
Figure 1 Multiple immunogenic consequences of radiation therapy, chemotherapy, endocrine deprivation, or small-molecule inhibitors that can be harnessed to promote synergy with immunotherapeutic regimens. These immunogenic consequences of anticancer therapy, ranging from immunogenic cell death to immunogenic modulation, can be exploited to achieve optimal synergy with therapeutic cancer vaccines.
Source: Adapted from Ref. 265.
Vaccination plus other immunotherapies
The recent clinical studies employing MAbs directed against the checkpoint inhibitor PD-L1/PD-1 immune suppressive axis have placed cancer immunotherapy into consideration as a mainstream component of cancer therapy. Results with checkpoint inhibitors have been most impressive in the treatment of the majority of patients with metastatic melanoma and subsets of patients with lung carcinoma.266–271 The use of the PD-L1/PD-1 checkpoint inhibitors has also shown some activity in a minority of patients with other tumor types (e.g., bladder cancer) and extremely low levels of activity in other cancers such as colorectal and prostate cancer. The major correlate of clinical response in virtually all of these studies with the PD-L1/PD-1 checkpoint inhibitors is the presence of T cells in the tumor. It would appear that these TILs are inducing PD-L1 on tumor cells and consequently becoming anergized, thus recapitulating the process of the body’s defense against autoimmunity. The administration of an anti-PD-L1/PD-1 agent would “release the brakes” on T cells by reducing or eliminating this immunosuppressive phenomenon, leading to the activation of the TILs and subsequent tumor cell destruction (Figure 2). One potential reason for the lack of success of some tumor vaccine trials could be due to this upregulation of PD-L1 on tumor cells in response to the presence of TILs. It would thus appear that the use of a therapeutic vaccine prior to or at the same time as the use of an anti-PD-L1/PD-1 agent would induce the presence of T cells in the tumor, which would subsequently be activated by inhibiting the PD-L1 expression on the tumor, leading to tumor cell lysis (Figure 2). Carefully controlled randomized clinical studies will be required to validate this hypothesis.
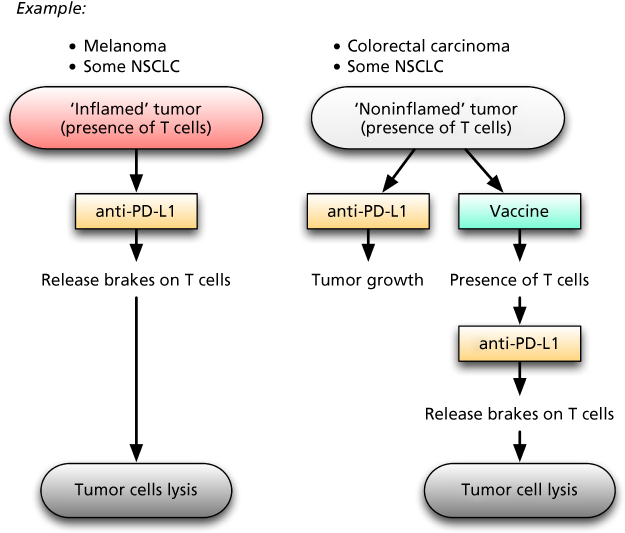
Figure 2 Working model for understanding T-cell-inflamed versus noninflamed tumor microenvironments with implications for vaccine therapy. The potential reasons for lack of spontaneous immune responses against a subset of tumors despite T-cell infiltration include failed immune activation (a) by extrinsic inhibition by PD-L1/PD-1 interactions. This local inhibition may be overcome by blockade with anti-PD-L1/PD-1 checkpoint inhibitor monoclonal antibodies, yielding clinical benefit. Other tumor types, termed “noninflamed,” typically do not respond to PD-L1 inhibition due to lack of potential infiltrating responding T cells. This model suggests that these tumors may be converted to an “inflamed” T-cell-infiltrated phenotype by vaccine regimens (b). These T cells may then be further activated by PD-L1/PD-1 blockade.
Preclinical studies have shown that vaccine plus checkpoint inhibition can act synergistically. The addition of anti-CTLA4 MAb to CEA-TRICOM vaccination was shown to increase the avidity of T-cell responses, resulting in enhanced antitumor activity of the vaccine.138, 272 When used in combination with GVAX vaccine, blockade of both PD-1 and CTLA-4 resulted in reversal of CD8+ TIL dysfunction and enhanced antitumor activity.273
In a Phase I clinical study,274 30 metastatic prostate cancer patients received PROSTVAC vaccine in combination with increasing doses of ipilimumab (anti-CTLA4). Fifty-eight percent of patients had PSA declines. While this trial was not randomized, median OS for patients receiving the combination was 34.4 months compared to 26.3 months for patients receiving PROSTVAC alone in another trial (Halabi-predicted survival144 was similar for patients in both trials at about 18 months). A recently completed trial in mCRPC patients using ipilimumab plus radiation showed a median OS of 17.3 months. Also, no additional toxicity above that observed with ipilimumab alone was seen in the vaccine plus ipilimumab-treated patients. In another study, a DNA/peptide vaccine induced T-cell responses in melanoma patients that were “boosted” following CTLA-4 blockade.275 CTLA-4 MAb was also infused into nine previously immunized advanced cancer patients276; evidence of increased tumor immunity was seen in some melanoma and metastatic ovarian cancer patients.
There are also several ways in which vaccine and MAbs can be used in combination. These include (1) the use of therapeutic antibodies such as herceptin or rituxan in combination with vaccine, in which each acts independently, (2) the use of antitumor antibody cytokine fusion proteins such as antitumor antibody/IL-12 to enhance T-cell activity at the site of tumor,277–281 and (3) the use of MAbs or fusion proteins directed against regulatory T cells.282–284 Vaccines can also be employed in combination with adoptive transfer of antigen-specific T cells. In preclinical studies, point-mutated ras-specific T cells were adoptively transferred into immunodepleted hosts. Donor antigen-specific T-cell responses were greatest in recipient mice that received peptide boosts (with or without IL-2). These results indicate that a vaccine administration after T-cell transfer was more obligatory than exogenous IL-2 to sustain adoptively transferred T cells.285
Immunogenic cell death and immunogenic modulation
Certain cancer therapeutic regimens have the ability to trigger cancer cell death, which leads to stimulating endogenous immune responses against the tumor; this is termed “immunogenic cell death” (Figure 1).286–293 The cardinal signs of immunogenic cell death are (1) calreticulin exposure on the surface of dying cells, (2) the release of high-mobility group box 1 (HMGB1), (3) the release of ATP and, most importantly, (4) cell death.289 Each of these molecules acts on DCs to facilitate the presentation of tumor antigens to the immune system.287 Calreticulin, a chaperone and calcium regulator, when exposed on the surface of a dying cell, serves as a phagocytic signal to DCs.291, 294 When HMGB1, a non-histone chromatin-binding protein, is released from dying cells, it engages TLR-4 on DCs, leading to DC maturation.289 The secretion of ATP by dying cells binds receptors on DCs, further supporting T-cell activation.288 In addition, tumor cells that survive therapy have been shown to alter their biology to render them more sensitive to immune mediated killing; this phenomenon is termed “immunogenic modulation” (Figure 1).295–302 Immunogenic modulation encompasses a spectrum of molecular alterations in the biology of the cancer cell that independently or collectively make the tumor more amenable to CTL-mediated destruction. These include (1) downregulation of anti-apoptotic/survival genes,297, 298, 303 (2) modulation of antigen-processing machinery (APM) components,299, 302 and (3) calreticulin translocation to the cell surface of the tumor (Figure 1).265, 299 One can envision that these immunogenic consequences of anticancer therapy, ranging from immunogenic cell death to immunogenic modulation, can thus be harnessed to achieve optimal synergy with therapeutic cancer vaccines and other immunotherapy regimens, thereby maximizing the clinical benefit for patients receiving combination therapies.
Vaccine plus radiation
Radiation therapy is standard-of-care for multiple malignancies, aimed at direct tumor destruction. However, systemic disease, treatment resistance, or the need for sublethal dosing to minimize normal tissue toxicity, often translates into surviving tumor cell populations and disease progression.304, 305 Radiation can induce a continuum of immunogenic alterations in dying and/or surviving tumor cells (Figure 1). Lethal irradiation has been reported to induce immunogenic cell death.306 Although immune responses in cancer patients receiving radiation alone are often weak and rarely translate into protective immunity, the immunogenic effects of radiation therapy can be exploited to promote synergistic clinical benefit for patients receiving combination regimens with immunotherapy.304, 305, 307 It has been demonstrated that the use of relatively low doses of external beam radiation, insufficient to kill tumors, induces immunogenic modulation, altering those tumor cells to render them more susceptible to T-cell-mediated lysis. Cell-surface expression of MHC class I molecules and calreticulin on tumors was increased in a radiation-dose-dependent manner, ultimately rendering tumor cells more susceptible to T-cell-mediated killing.265, 298, 302 It has been demonstrated in preclinical studies that radiation combined with a cancer vaccine elicits greater tumor antigen-specific CD8+ T-cell responses and/or reduction in tumor burden than either modality alone.298, 308–310 Importantly, the induction of CD8+ T cells specific for multiple TAAs not encoded by the vaccine was observed after the combination therapy (antigen cascade). This polyclonal T-cell response functionally mediated the regression of TAA-negative metastases at distal s.c. or pulmonary sites.310
These findings have translated into promising clinical findings for patients receiving radiation therapy plus immunotherapy.304, 305, 307, 311, 312 A Phase I study evaluated the combination of radiation therapy plus injection of an autologous DC vaccine in 14 patients with advanced-stage/metastatic hepatoma.311 Patients received a single fraction of radiation followed by two intratumoral injections of autologous immature DCs. This combination resulted in tumor-specific immune responses in 7/10 assessable patients, and six patients had objective clinical responses. Utilizing poxviral vaccines, patients in a randomized Phase II study with localized prostate cancer who were treated with radiation and a poxviral-based vaccine had a significant increase in PSA-specific T-cell responses compared to patients receiving radiation alone.312 In a multicenter Phase II study, patients with mCRPC (n = 44) were randomized to receive 153Sm-EDTMP (Quadramet®, an FDA-approved radiopharmaceutical targeting bone metastasis) alone or in combination with PSA-TRICOM vaccine. TTP significantly improved (p = 0.03) with combination therapy (3.7 months) compared to 153Sm-EDTMP alone (1.7 months).313 In addition, several ongoing clinical trials are investigating this combination strategy,314 including a Phase II study of sipuleucel-T plus radiation therapy,62 a Phase II study of sipuleucel-T plus stereotactic ablative body radiation,63 and a pilot study of sipuleucel-T plus high-dose single-fraction radiation64 in hormone-refractory prostate cancer.
Vaccine plus chemotherapy
If cancer vaccines are to be used early in the disease process, they would most likely need to be used in combination with certain chemotherapeutic regimens. While counterintuitive, it has recently been shown that vaccine therapy may not only be compatible with certain chemotherapies, but also may actually be synergistic (Figure 1).296, 315 Various chemotherapy agents have been shown to induce immunogenic modulation in tumors of diverse origin by upregulating various immune-relevant proteins on the surface of cancer cells, including multiple TAAs, calreticulin, adhesion molecules such as ICAM-1, and MHC class I proteins.295, 298–300, 303, 316, 317 These phenotypic changes translated into increased murine and/or human tumor sensitivity to CTL-mediated lysis in vitro after exposure to sublethal doses of chemotherapy with cisplatin/5-FU317 docetaxel,299 paclitaxel299 and cisplatin plus vinorelbine.317 These preclinical findings and others have translated into various hypothesis-generating clinical trials, now with encouraging preliminary data. In a randomized Phase II trial, metastatic breast cancer patients who received docetaxel combined with the therapeutic cancer vaccine PANVAC (poxvirus-based vaccine encoding the tumor antigens CEA and MUC-1, along with three T-cell costimulatory molecules; TRICOM) attained a superior PFS relative to those receiving docetaxel alone (6.6 months vs 3.8 months).318 A recent report319 demonstrated that the administration of docetaxel to patients with metastatic prostate or breast cancer, as well as that of cisplatin plus vinorelbine to NSCLC patients, significantly increased the ratio between effector T cells and Tregs and reduced the immunosuppressive activity of the latter in the majority of patients. These studies provide the rationale for the selective use of active immunotherapy regimens in combination with specific standard-of-care therapies to achieve the most beneficial clinical outcome among carcinoma patients. Several things are important in considering the use of chemotherapy with vaccine: (1) the combined use of vaccine and chemotherapy early in the disease process should not be confused with the use of vaccines following multiple regimens of different chemotherapeutic agents in the advanced disease setting, where the immune system would most likely be impaired, (2) not all chemotherapeutic agents will be compatible with vaccine, and (3) dose scheduling of vaccine when used with chemotherapy may be extremely important. Obviously, future studies will be required to optimize the combined use of vaccine and chemotherapy. A recent review on combining antineoplastic drugs with tumor vaccines has been published.320
Vaccine plus hormone therapy
The use of vaccines with anti-androgen therapy represents an intriguing possibility for the treatment of prostate cancer. It has previously been shown that androgen-ablative therapy induces T-cell infiltration in the prostate. T-cell infiltration was readily apparent after 1 to 3 weeks of therapy.321 The immunogenic modulation potential of androgen deprivation has been described with a second-generation androgen-receptor antagonist (ARA), enzalutamide, using the murine TRAMP model of prostate carcinoma.322 In vitro exposure of TRAMP-C2 prostate tumor cells to enzalutamide significantly enhanced cell-surface expression of Fas and MHC class I, consequently improving their sensitivity to immune-mediated lysis. These immunomodulatory properties of enzalutamide were exploited upon combination with a therapeutic cancer vaccine targeting a transcription factor associated with the metastatic process. Combination treatment significantly increased antigen-specific T-cell proliferation and OS compared to the levels observed in mice receiving no treatment or enzalutamide alone. In addition, enzalutamide or abiraterone, an inhibitor of androgen biogenesis, were both able to render androgen receptor positive LNCaP human prostate tumor cells more sensitive to T-cell-mediated lysis.316 Findings from these and other studies provided a rationale for the use of ADT in combination with active immunotherapy. A clinical trial has been conducted in prostate cancer patients who had received prior hormonal therapy and had increasing serum PSA without radiographic evidence of metastases. The trial was randomized for a second-line hormone therapy (nilutamide) versus vaccine with a crossover at disease progression so that each arm would receive combination vaccine plus hormone therapy. Time to treatment failure and survival at 4+ years323 was prolonged in patients initially receiving vaccine alone and then receiving vaccine plus nilutamide. Further studies are obviously needed to validate such observations and are under way (NCT01867333, NCT01875250).
Vaccine plus small-molecule inhibitors
A number of recent studies have indicated that antiangiogenic tyrosine kinase inhibitors (TKIs) target multiple components of the tumor microenvironment and are an ideal class of agents for synergizing with cancer vaccines.324 TKIs are well known to modulate tumor endothelial cells, leading to vascular normalization; however, these agents have also been recently shown to decrease tumor compactness and tight junctions, thereby reducing solid tumor pressure and allowing for improved perfusion of collapsed vessels and increased tumor oxygenation.325 In addition, select TKIs are capable of inducing immunogenic modulation (Figure 1). The alteration of the immune landscape, direct modification of tumor cells, and improved vascular perfusion lead to improved antitumor efficacy when antiangiogenic TKIs are combined with immunotherapy. These data support the clinical combination of multitargeted antiangiogenic TKIs, including, but not limited to, cabozantinib,326 sunitinib,325, 327–329 and sorafenib,325 as well as to other antiangiogenic therapies, such as the anti-VEGF antibody bevacizumab, with cancer vaccines for improved treatment of solid tumors.
Dose scheduling of vaccine with other therapies
Arguably the most unique feature of cancer vaccine therapy is a vaccine’s ability to initiate a dynamic process of host immune responses that can be exploited in subsequent therapies (Table 4). In a Phase I study, patients with advanced-stage progressive cancer received a vaccine directed against cytochrome P4501B1. Most patients who developed immunity to vaccine but required salvage therapy on progression showed marked responses to their next treatment regimen; most of these responses lasted >1 year.331 In another study in patients with extensive-stage small cell lung cancer,332 a high rate of objective clinical responses to chemotherapy immediately followed vaccine therapy. These clinical responses were also closely associated with induction or augmentation of immune response to vaccine.
Table 4 Therapeutic vaccines versus conventional therapy
| Therapeutic vaccines | Conventional therapy | |
| Target | Immune system | Tumor or its microenvironment |
| Pharmacodynamics | Delayed (adaptable, may get better over time) | Often immediate action |
| Memory response | Yes | No |
| Tumor evolution/new mutations | New immunogenic targets | Resistance to therapy |
| Limitations | Requires adequate immune system function (both systemically and at tumor site) | Toxicity |
Source: Adapted from Ref. 330.
Three randomized clinical trials in prostate cancer provided further evidence of this phenomenon. In the first trial, patients with metastatic CRPC were randomized to receive vaccine alone or vaccine plus weekly docetaxel.333 Patients on the vaccine-alone arm were allowed to cross over to receive docetaxel at time of progression. After vaccine, median PFS on docetaxel was 6.1 months compared with a PFS of 3.7 months with the same docetaxel regimen and patient population at the same institution. Similar findings were observed using the sipuleucel vaccine.334 In a randomized multicenter study, patients in both the vaccine and placebo arms received docetaxel at progression. There was a statistically significant (p = 0.023) increase in OS with docetaxel in patients who had prior vaccine versus placebo.
In a Phase II trial,335 patients with nonmetastatic CRPC and rising serum PSA were randomized to receive either vaccine or nilutamide, an ARA. After 6 months, patients with rising PSA were allowed to cross over to a combination of both therapies. Median time to treatment failure was similar in the vaccine and ARA arms. However, for patients who received vaccine first and then received vaccine plus ARA, time to treatment failure was 13.9 months from the initiation of ARA and the time to treatment failure from the initiation of any therapy was 25.9 months. Of the initial randomized population, for those patients who first received nilutamide alone or nilutamide and then vaccine, 5-year OS was 38% versus a median OS of 59% for patients who first received vaccine alone or nilutamide plus vaccine.336
With traditional cytotoxic agents, it is widely believed that improved TTP is a prerequisite for improved OS. A recent study337 evaluated tumor regression and growth rates in four chemotherapy trials and one vaccine trial in patients with mCRPC. Cytotoxic agents affect the tumor only during the period of administration; soon after the drug is discontinued, due to drug resistance or toxicity, antitumor activity ceases and the growth rate of the tumor increases (Figure 3). The mechanism of action and kinetics of clinical response with vaccine therapy appears to be quite different (Table 4).337 Therapeutic vaccines do not directly target the tumor, but rather target the immune system. Immune responses often take time to develop and can potentially be enhanced by continued booster vaccinations; any resulting tumor-cell lysis can lead to cross-priming of additional TAAs, thus broadening the immune repertoire via a phenomenon known as antigen cascade or epitope spreading (Figure 4). This broader, and perhaps more relevant, immune response may also take some time to develop. Although a vaccine may not induce any significant reduction in tumor burden, vaccines as monotherapy have the potential to apply antitumor activity over a long period of time, resulting in a slower tumor growth rate (Figure 3). This deceleration in growth rate may continue for months or years and, more importantly, through subsequent therapies. This process can thus lead to clinically significant improved OS, often with little or no difference in TTP and a low rate of or lack of objective response340; thus, treating patients with lower tumor burden with vaccine may result in far better outcomes. It is hypothesized that the combined use of vaccine and cytotoxic therapy may result in both tumor regression (via the cytotoxic therapy) and reduced tumor growth rate (via vaccine therapy) (Figure 3).337, 338 Early clinical trials with vaccine may thus well have been terminated prematurely with the observance of tumor progression before sufficient vaccine boosts could be administered. This phenomenon has actually led to modifications in how vaccine clinical trials are now designed and to “new immune response criteria” for immunotherapy.341
Figure 3 Tumor growth rates in patients with metastatic prostate cancer, from four trials with chemotherapy and one with PROSTVAC (PSA-TRICOM) (adapted from data in Refs ). (a) Tumor growth rate with no therapy. (b, blue line) Chemotherapy-induced initial tumor reduction, but tumor growth rate at cessation of therapy due to toxicity or drug resistance was similar to pretreatment tumor growth rate. (c, red line) PROSTVAC vaccine reduced tumor growth rate following therapy. Thus, patients who received vaccine showed little if any tumor reduction (and virtually no increase in time to progression), but showed an increase in overall survival. (d, green line) Predictions of enhanced overall survival for patients treated with both vaccine and cytotoxic therapy. Combination therapy couples the reduced tumor burden from cytotoxic therapy with the reduced growth rate from immunotherapy, culminating in both objective clinical responses and increased overall survival. This phenomenon could potentially be further enhanced if vaccine were initiated earlier in the disease process or in patients with low tumor burden. Arrows indicate the initiation of treatment; crosses indicate time of death from cancer.
Source: Adapted from Gulley 2011338 and Madan 2010.339
Figure 4 Antigen cascade. Multiple antigens are released from dying tumor cells that can activate an immune response. (a) Cancer therapy induces death of cancer cells. (b) As these tumor cells die, they release a range of tumor protein antigens (including mutated proteins) that are taken up by scavenger cells called antigen-presenting cells. (c) These antigen-presenting cells then travel to regional lymph nodes where they present these multiple antigens to T cells, initiating or potentiating an antitumor immune response. (d) Activated polyclonal tumor-specific T cells can then traffic to areas of tumor to participate in immune-mediated tumor killing, destroying more cancer cells and further potentiating the cascade.
Mechanisms involved in vaccine activity
Recent studies have shown that a range of effector cells can be involved in antitumor effects. In most preclinical studies, the CD8+ CTL is involved in antitumor effects and the CD4+ helper T cell is important for supplying help via cytokines to further activate the CTL. Other effector cells, however, can be involved, including macrophages and natural killer (NK) cells, and studies have also shown that in certain cases the generation of antitumor antibodies can mediate antitumor effects via antibody-dependent cell-mediated cytotoxicity.342 T-cell activation has been shown to be a complex phenomenon involving the interaction of the peptide–MHC complex on the APC with the TCR on the T cell; for weak antigens such as TAAs, however, accessory molecules are necessary for efficient T-cell activation. These have been termed T-cell costimulatory molecules. At this time, over a dozen such costimulatory molecules have been identified. Costimulatory molecules are found on professional APCs, such as DCs, but are not found on the vast majority of solid tumors. It has also recently been shown that combinations of these costimulatory molecules act synergistically to further enhance T-cell activation.139 Recent studies have also shown that it is not necessarily the quantity of T cells generated that is essential, but their quality, or avidity.138, 343, 344 This is measured by the ability of different populations of T cells to actually kill targets. It has recently been shown that the use of vaccines with T-cell costimulation and certain cytokines can actually enhance T-cell avidity.138
Several different types of regulatory T cells have now been identified.345–348 The evolutionary reason for their existence is most likely to reduce autoimmune phenomena. Since TAAs for the most part are self-antigens, regulatory T cells appear to have a major role in reducing immune responses to TAAs. Several types of regulatory T cells that have now been identified are CD4+CD25+FoxP3+ (Tregs), immature macrophages, and CD4+ NKT+ cells.347, 348 Studies are ongoing employing reagents that can potentially reduce the activity of these regulatory T cells, including the fusion protein Ontak,349 cyclophosphamide,212, 221, 350 and anti-CD25 Abs.351, 352 The inhibition of these cells will most likely be a part of cancer vaccine therapy in the future.
Antigen cascade
The phenomenon of antigen cascade (Figure 4) is now evolving as an important facet in the evaluation of cancer vaccines. This was first defined using the term epitope spreading, in which a given peptide was used as a vaccine and the host immune response post vaccination was not only directed to that epitope, but also to other epitopes of the same tumor antigen.308, 352, 353 This phenomenon has now been expanded using the term antigen cascade, in which a given antigen is used as a vaccine and the host immune response post vaccination is not only directed against the antigen in the vaccine but also to other antigens in the tumor. The explanation for these phenomena is that as a consequence of some tumor-cell destruction induced by the vaccine, APCs will engulf tumor fragments and then cross-present those tumor fragments to T cells, thus initiating the antigen cascade phenomenon. It has also been shown that 1/8 prostate cancer patients receiving radiation developed antibodies to an array of TAAs, while 15/33 patients receiving PROSTVAC plus radiation developed such antibodies.354, 355 Antigen cascade has now been observed in preclinical models356 and several trials have shown that antigen cascade correlated with antitumor activity.142, 357–361 This phenomenon of antigen cascade is also a mechanism by which the host will direct an immune response to the many mutated gene products in the tumor. Some studies have also suggested improved clinical outcomes for patients who demonstrated a broadened immune response to epitopes not found in the vaccine.359, 362–364
Immune monitoring
One is always asked for immune correlates of patient benefit in therapeutic cancer vaccine clinical studies. See Ref. 365 for an excellent review. The extreme complexity of the human immune system, the genetic diversity of the human population, and the diversity among cancer patients in terms of type and stage of disease make this a truly daunting task. In addition to the analysis of antigen-specific CD4 and CD8 immune cell subsets, numerous other immune subsets may play an important and/or interdependent role in patient responses; these include regulatory T cells and myeloid-derived suppressor cells as examples. Over 130 different human immune cell subsets have now been identified. Soluble factors produced by tumors or in the tumor microenvironment can also influence immune responses; these include, among others, TGF-β, IL-8, VEGF, IFN-γ, TNF, CD40L, and sCD27. Confounding factors other than those mentioned above include number and type of prior therapies, age and sex of the patient, and size of tumor lesions. Differences in immune cells in the periphery versus different locations within the tumor are of extreme importance. These types of data are more easily obtained in melanoma patients, but biopsies of metastatic lesions are not as easily obtained in most patients with solid tumors.
Newer techniques are currently being employed to investigate patients’ immune responses; these include, among others, gene chip RNA arrays of tumor and peripheral blood samples, analysis of changes in TCR clones pre- versus post vaccination, and changes in over 100 immune cell subsets via multilaser FACS analyses. In addition, analyses of numerous immune cell subsets of patients prior to therapy are emerging as a valuable tool in determining which patients are more likely to benefit from vaccine therapy.318, 366
Paradigm shifts
The evaluation of cancer vaccines in clinical trials may well necessitate new paradigms.20, 315 Cancer vaccines and cytotoxic drugs act differently in terms of mode of action dynamics, as well as limitations (Table 4, Figure 3). Cytotoxic drugs either kill or do not kill tumor cells. Lack of efficacy is due to either drug resistance by tumor cells, inadequate amounts of drug delivered to the tumor, and/or drug-induced toxicity in the host. Thus, if a patient’s disease is progressing on a cytotoxic drug, that drug is immediately withdrawn. The use of a cancer vaccine involves the initiation of a dynamic process in which the patient’s own immune system is activated. It is clear from decades of preclinical studies, and now clinical trials, that to maximize the host immune response, one must not only give a primary vaccination but also multiple booster vaccinations over a period of weeks or months to further enhance the level of the immune response. Multiple vaccinations are especially important because factors that will blunt the immune response may be given off by the tumor. Thus, it is not unusual to see disease progression following early vaccinations, only to see disease stabilization or a reduction in tumor burden following multiple vaccinations. Thus, the paradigm of “drug withdrawal upon progression” should be revisited with the use of cancer vaccines.
Clinical trials with a new drug are traditionally evaluated in patients with advanced tumors (many times with a large tumor burden) who have failed prior cytotoxic therapies. Objective clinical responses are evaluated by RECIST criteria, looking for a sustained ≥ 30% reduction in the sum of the longest diameter of target lesions. The scenario is all too familiar where antitumor activity is observed with cytotoxic agents or small-molecule targeted therapies via a reduction of tumor burden by RECIST criteria, but no statistical difference in survival is seen. Unfortunately, there is still a “paradigm paralysis” by some who hold on to this paradigm for the evaluation of immunotherapy agents as the only means of evaluation of efficacy.
Since generation of an immune response to a vaccine is a dynamic process in which T cells can be continually generated upon booster vaccines, a more appropriate endpoint for evaluation should be TTP and, more importantly, survival. Since only a finite amount of T cells can be generated by a vaccine, it may be inappropriate to evaluate a cancer vaccine by its ability to drastically reduce a large tumor burden; this is especially true in patients whose immune system has been compromised due to numerous rounds of prior chemotherapy. A more appropriate setting would be the evaluation of cancer vaccines earlier in the disease setting (e.g., neoadjuvant or adjuvant setting), or in metastatic patients with a small tumor burden. This should particularly be considered in light of the relatively low level of toxicity seen in the use of cancer vaccines.
Summary
The U.S. Food and Drug Administration approvals of (1) the prostate cancer therapeutic vaccine sipuleucel-T, (2) anti-CTLA-4 monoclonal antibody (MAb) ipilimumab, and (3) the MAbs directed against the programmed death ligand 1 (PD-L1)/PD-1 immunosuppressive axis, along with the results of recent clinical studies involving cancer vaccines and other immunotherapeutics, have placed immunotherapy into the mainstream of the management of many cancer types. Many potential TAA targets and vaccine platforms for cancer immunotherapy have been identified. The evaluation of cancer vaccines in clinical trials may well necessitate new paradigms. Cancer vaccines and cytotoxic drugs act differently in terms of mode of action and dynamics. Cytotoxic drugs either kill or do not kill tumor cells; lack of efficacy is due to drug resistance by tumor cells, inadequate amounts of drug delivered to the tumor, and/or drug-induced toxicity in the host. The use of a cancer vaccine involves the initiation of a dynamic process in which the patient’s own immune system is activated. Thus, it is not unusual to see disease progression following early vaccinations, only to see disease stabilization or a reduction in tumor burden following multiple vaccinations. Thus, the paradigm of “drug withdrawal upon early progression” should be revisited with the use of cancer vaccines. Preclinical studies, and now several clinical studies, have shown that patients can mount an immune response to a cancer vaccine when given in combination with certain cytotoxic drugs, hormones, or local radiation of tumor. Studies have shown that when tumor cells are exposed to sublethal doses of radiation, certain chemotherapeutic agents, hormone manipulating therapeutics or small-molecule targeted therapeutics, the phenotype of tumor cells is actually modulated to make them more susceptible to vaccine-mediated T-cell-mediated killing. It would also appear that the use of a therapeutic vaccine prior to or at the same time as the use of an anti-PD-L1/PD-1 agent would induce the presence of T cells in the tumor, which would subsequently be activated by inhibiting the PD-L1 expression on tumor, leading to tumor cell lysis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


